









")

 типа болезнь Реклингаузена")











")


")





















 (Синдром Лежена)")
 (Синдром Лежена)")

 (46,ХХ4p-) Это расстройство развивается на фоне потери дистального конца короткого плеча")

 болезни")


 Медицина
МедицинаПохожие презентации:










Классификация наследственных заболеваний. Лекция 5
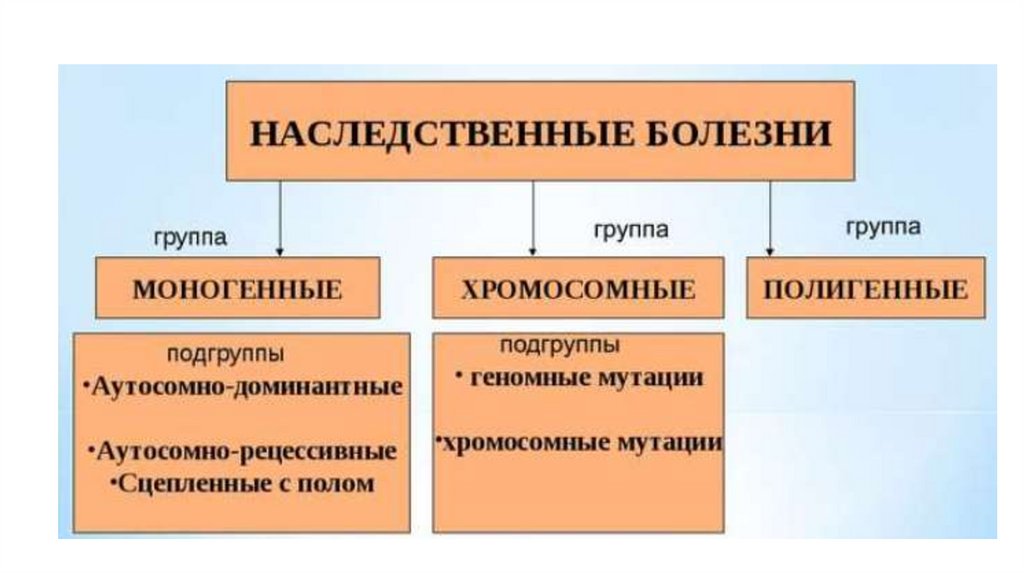
1. Классификация наследственных заболеваний
Лекция 52.
3.
4. Наследственные заболевания имеют общие клинические особенности
1.2.
3.
4.
5.
6.
Повторные случаи аналогичной патологии у членов
одной семьи
Врожденный характер заболевания. Большинство
пороков развития и хромосомных синдромов
обнаруживаются уже при рождении ребенка
Хроническое, рецидивирующее течение заболевания,
приводящее к постепенному ухудшению состояния
больного
Одновременное поражение у человека нескольких
органов или систем
Наличие специфических симптомов
Устойчивость к наиболее распространенным методам
терапии
5. Моногенные болезни
• Болезни, возникшие в результате структурных изменений водном гене.
Включают в себя:
– «менделевские болезни» (наследуемые согласно законам
Менделя, например, с аутосомной передачей или сцепленные с
полом),
– «неменделевские» , которые наследуются не по законам
Менделя (например, митохондриальные заболевания,
передающиеся по материнской линии).
6.
Моногенные болезни - это заболевания, протекающие спостоянным прогрессированием патологического
процесса.
Индивидуальный и популяционный риск возникновения моногенных
болезней существенно различаются из-за неравномерного
распространения обусловливающих их генов.
7. Моногенные болезни можно разделить по типу наследования
1)Аутосомно-доминантные2)Аутосомно-рецессивные
3)Х - Сцепленные доминантные
4)Х – Сцепленные рецессивные
8. Аутосомно-доминантные болезни: около 3700 заболеваний. 1. Могут возникать из-за появления мутации – когда проявляется дефект в
структурном белке, в результате чего становитсяневозможным нормальное функционирование организма.
2. Либо может нарушиться регуляция экспрессии генов –
изменяется баланс выработки необходимых организму веществ.
Поскольку набор аутосом одинаков у мужчин и женщин,
вероятность возникновения заболевания не зависит от пола. При
этом аутосомно-доминантные болезни обычно характеризуются
фенотипическим полиморфизмом, зависящим от степени
выраженности мутантного гена в фенотипе.
В большинстве случаев моногенные наследственные болезни
проявляются в виде синдромов, когда у одного больного
поражаются различные системы органов.
9. Синдром Марфана
Наследственное заболеваниесоединительной ткани , проявляющееся
изменениями скелета: высоким ростом с
относительно коротким туловищем ,
длинными паукообразными пальцами (
арахнодактилия), разболтаннностью
суставов , часто сколиозом , кифозом ,
деформациями грудной клетки ,
аркообразным небом . Характерны также
поражения глаз . В связи с аномалиями
сердечно-сосудистой системы средняя
продолжительность жизни сокращена.
10. Антуан Марфан
Синдром назван именемАнтуана Марфана,
французского педиатра
в 1896 году.
Причина синдрома Марфана мутации гена FBN1,
который отвечает за синтез
фибриллина .
11.
Известные люди ссиндромом Марфана
12. Брахидактилия (короткопалость)
• аномалия развития рук или ног, укорочениепальцев.
• для проявления аномалии у ребенка ген
брахидактилии должен быть унаследован от
одного из родителей. При отсутствии других
аномалий люди с брахидактилией могут жить
обычной, нормальной жизнью, однако
короткопалость накладывает ограничения на
профессиональный выбор. Брахидактилия с
одинаковой частотой встречается среди
представителей обоих полов;
распространенность в популяции - 1,5:100 000.
13. Полидактилия
– деформация конечности, характеризующаясяналичием дополнительных пальцев на кистях или
стопах; врожденное увеличение количества пальцев
на руке или ноге от шестипалости до многопалости.
Сведения о частоте полидактилии в популяции
разноречивы: по данным разных авторов
многопалость встречается у 1 из 630-3300
новорожденных; соотношение полов одинаковое.
Носителями гена многопалости являются отец или
мать, но при этом они могут быть здоровы.
Вероятность унаследования полидактилии детьми
составляет 50%.
14. Нейрофиброматоз I (первого) типа болезнь Реклингаузена
распространенное наследственное заболевание, главнымпризнаком которого является развитие опухоли нервной оболочки
— нейрофибромы. Помимо этого у пациентов наблюдаются
пигментные аномалии (пятна на коже цвета “кофе с молоком”) и
ряд других нарушений. В разных странах заболеваемость
составляет в среднем 1 случай на 2500–7800 человек.
15. Миотоническая дистрофия
• аутосомно-доминантное заболевание. Частота составляет от1:8000 до 1:40 000 населения. Ген локализован на 19-й
хромосоме. Признаки заболевания появляются преимущественно
в возрасте 16-20 лет (возможно от 5 до 35 лет) и складываются из
миотонического, миопатического синдромов и нарушений
деятельности сердечно-сосудистой, нейроэндокринной, костносуставной систем, а также висцеральных и глазных расстройств.
Миотонические симптомы проявляются в виде миотонических
спазмов (главным образом в сгибателях пальцев и жевательной
мускулатуре).
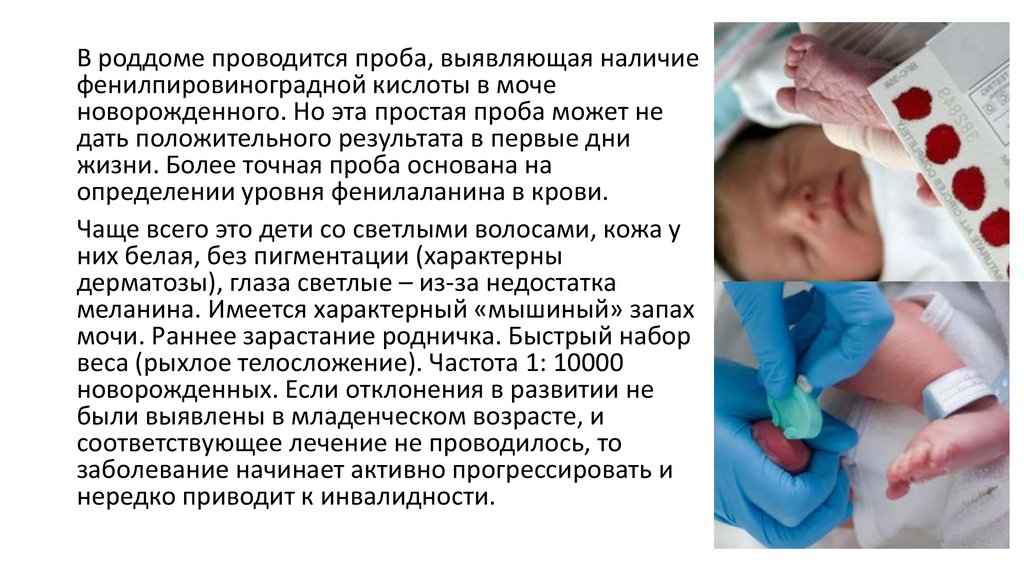
16. Фенилкетонурия
одно из наиболее распространенных аутосомнорецессивных наследственных заболеваний снарушением обмена аминокислоты
фенилаланина. Возникновение характерного
метаболического блока провоцирует активацию
побочных путей по обмену фенилаланина, что
приводит к накоплению токсичных производных от
его действия. К таким производным в частности
относятся фенилмолочная и
фенилпировиноградные кислоты, практически не
образующиеся при нормальном состоянии
организма.
17. Фенилкетонурия –обусловлена блокадой превращения аминокислоты – фенилаланина в тирозин.
18.
В роддоме проводится проба, выявляющая наличиефенилпировиноградной кислоты в моче
новорожденного. Но эта простая проба может не
дать положительного результата в первые дни
жизни. Более точная проба основана на
определении уровня фенилаланина в крови.
Чаще всего это дети со светлыми волосами, кожа у
них белая, без пигментации (характерны
дерматозы), глаза светлые – из-за недостатка
меланина. Имеется характерный «мышиный» запах
мочи. Раннее зарастание родничка. Быстрый набор
веса (рыхлое телосложение). Частота 1: 10000
новорожденных. Если отклонения в развитии не
были выявлены в младенческом возрасте, и
соответствующее лечение не проводилось, то
заболевание начинает активно прогрессировать и
нередко приводит к инвалидности.
19. Врожденный гипотиреоз
• группа моногенных и мультифакториальных заболеваний сосниженной функцией щитовидной железы или ее полным
отсутствием. При этом у девочек данная патология встречается в
2—2,5 раза чаще, чем у мальчиков.
• Если заболевание не распознано вовремя и лечение не начато
своевременно развивается кретинизм — необратимое
поражение головного мозга. Основная цель лечения —
устранение расстройств обмена веществ и компенсация
недостаточности щитовидной железы. Основной метод лечения
гипотиреоза — пожизненная заместительная терапия
препаратами тиреоидных гормонов.
20. Адреногенитальный синдром
• аутосомно-рецессивное наследственное заболевание снарушением биосинтеза глюко- и минералокортикоидов при
повышеной секреции андрогенов в коре надпочечников.
• Раннее развитие половой системы у мальчиков
• Мужские половые признаки у девочек
21. Галактоземия
аутосомно-рецессивное заболевание.Галактоза, поступающая с пищей в составе молочного сахара —
лактозы, подвергается превращению, но реакция превращения не
завершается в связи с наследственным дефектом ключевого
фермента.
При подтверждении диагноза
необходим перевод ребёнка на питание
с исключением молока и ряда других продуктов
22. Муковисцидоз
аутосомно-рецессивноезаболевание с
распространенным поражением
экзокринных желез
(бронхиальных, потовых,
слезных, слюнных, ушных,
кишечника, поджелудочной и
печени), характеризующееся
выделением вязкого секрета и
кистозным перерождением
желез кишечника,
поджелудочной железы,
дыхательных путей.
23. Благодаря развитию современной молекулярной биологии списки диагностируемых заболеваний постоянно расширяются:
• Диагностика моногенных болезней осуществляется какклиническими методами, с помощью, так называемого,
синдромологического подхода, так и специальными
биохимическими и молекулярно-генетическими методами.
• Для пренатальной диагностики (ПД), в первую очередь,
представляют интерес моногенные болезни, приводящие к
тяжелой, нередко смертельной патологии, в отношении которых
пока отсутствуют или все еще малодоступны методы
лекарственной терапии. Из более 6 000 заболеваний, известных
на сегодняшний день, доля заболеваний, безусловно
заслуживающих ПД, не превышает 1 %
24. Сравнительно частые и тяжелые болезни, для которых актуальна ПД:
• муковисцидоз,• миодистрофия Дюшенна,
• синдром ломкой (фрагильной) Х-хромосомы,
• гемофилия А и В,
• фенилкетонурия,
• атаксия Фридрейха,
• спинально-мышечная атрофия,
• врожденная вирилизующая гиперплазия коры надпочечников,
или адреногенитальный синдром
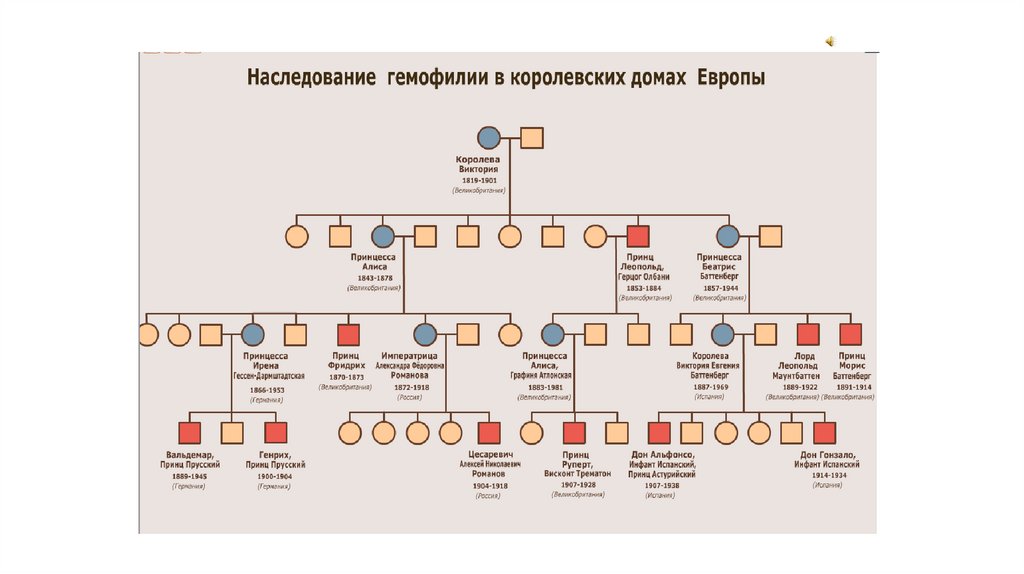
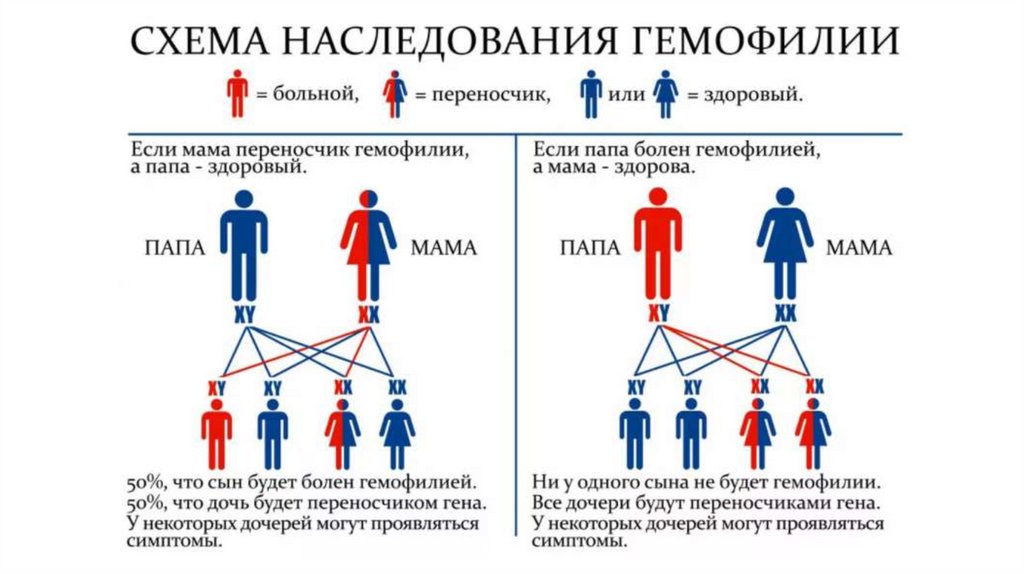
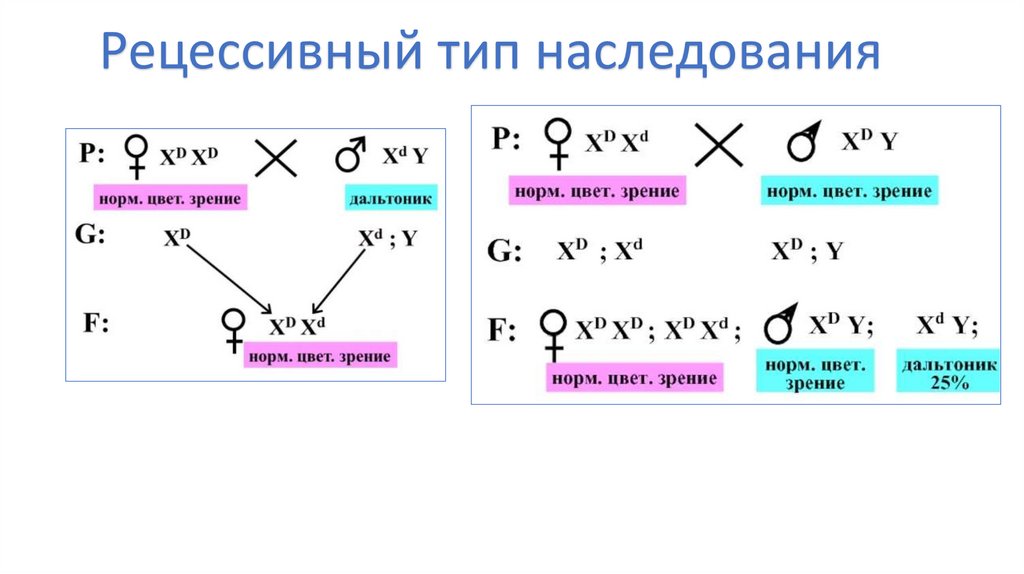
25. Х-сцепленный рецессивный тип наследования
Гемофилия — наследственное• Гемофилия
заболевание, характеризующееся
снижением или нарушением синтеза
факторов свертывания крови. Обычно
болезнью страдают мужчины, женщины
же выступают как носительницы
гемофилии.
Самой известной носительницей
гемофилии в истории была английская
королева Виктория, передавшая
дефектные гены двум дочерям и сыну
Леопольду, а в дальнейшем — внукам и
правнукам, включая российского
цесаревича Алексея Николаевича, мать
которого — царица Александра
Федоровна была носительницей гена
гемофилии.
26.
27.
28. Дальтонизм
• Передача дальтонизма по наследству связана с X-хромосомой ичасто передаётся от матери-носителя гена к сыну. У мужчин
дефект в единственной X-хромосоме не компенсируется, так как
«запасной» X-хромосомы нет, в результате чего дальтонизм в
двадцать раз чаще проявляется у мужчин, имеющих набор
половых хромосом XY. При этом все дочки мужчины дальтоника
являются носителями гена дальтонизма, но проявляется он у них
только в случае, если тот же ген имеется и во второй Xхромосоме, полученной ими от матери. В результате разной
степенью дальтонизма страдают 2—8 % мужчин, и только 0,8 %
женщин.
29.
Рецессивный тип наследования30. Синдром ломкой Х-хромосомы (Мартина-Белл)
• Наследственное Х-сцепленное (связанное с полом)заболевание, возникающее в результате
нарушений в гене FMR1. Синдром характеризуется
умственной отсталостью разной степени,
нарушением концентрации внимания, речевого
развития, а также может сопровождаться другими
клиническими проявлениями.
• Синдром ломкой X-хромосомы может встречаться
у обоих полов, но мужчины страдают этим
заболеванием чаще и тяжелее, так как имеют всего
одну X-хромосому.
31. Синдром Мартина-Белл из числа болезней экспансии числа нуклеотидных повторов
Истончение участка Х- хромосомы обусловлено патологическимувеличением специфических повторов аминокислотных остатков
— цитозин-гуанин-гуанин (Ц-Г-Г). Такая комбинация повторений
присутствует по несколько раз в других генах, однако важно,
собственно, количество этих повторений. В норме оно колеблется
от 29 до 31. В случае синдрома Мартина-Белла такое количество
является резко увеличенным и может встречаться от 230 до 4000
раз, что приводит к увеличению хромосомы и нарушенного
функционирования особого гена — FMR1, который отвечает за
правильное функционирование и развитие нервной системы.
32. Х-сцепленный доминантный тип наследования: Гипофосфатэмия
• редкое (орфанное), жизнеугрожающее, генетическиобусловленное, врождённое заболевание, проявляющееся
нарушением минерализации костей скелета и зубов, а также
системными осложнениями.
• Женщины передают патологический признак дочерям и
сыновьям c вероятностью 50%, мужчины — только дочерям с
вероятностью 100%.
• В настоящее время завершена разработка Асфотазы Альфа —
препарата ферментной заместительной терапии для лечения
ГФФ.
33. Х-сцепленный доминантный гипофосфатемический рахит («фосфат-диабет»)
• Не поддаётся лечению витамином Д – в чём принципиальноеотличие от «обычного» рахита.
34. ХРОМОСОМНЫЕ ЗАБОЛЕВАНИЯ ЧЕЛОВЕКА
Хромосомный наборздорового человека
– 46 хромосом:
22 пары аутосом и
1 пара половых хромосом
(женщина - ХХ,
мужчина - ХУ)
Число описанных типов хромосомных аномалий приближается к 1000, из них
более 100 форм клинически изучены и называются синдромами.
Распространенность хромосомных болезней одинакова во всех национальных
и этнических группах и составляет в среднем 7-8 больных на каждую 1 000
новорожденных. В Российской Федерации эта патология регистрируется
примерно у 12 000 новорожденных ежегодно.
35.
• Хромосомные синдромы (болезни) — это группа, врожденныхпатологических состояний, проявляющихся различными
аномалиями развития и обусловленных нарушениями числа или
структуры хромосом в соматических клетках (аутосомные синдромы) или половых клетках (гоносомные синдромы).
36. КЛАССИФИКАЦИЯ ХРОМОСОМНЫХ ЗАБОЛЕВАНИЙ
* Этиологический принцип, т.е. характеристика хромосомной илигеномной мутации
I. Хромосомные болезни, связанные с аномалиями числа хромосом при
сохранении их структуры.
1) Болезни: обусловленные числовыми аномалиями половых (Х и У) хромосом
(болезни Шерешевского-Тернера, Клайнфельтера).
2) Болезни, обусловленные числовыми аномалиями аутосом (синдромы Дауна,
Патау, Эдвардса).
3) Болезни, обусловленные увеличением кратности полного гаплоидного набора
хромосом – полиплоидии.
II. Хромосомные болезни, обусловленные структурными перестройками
хромосом. Виды хромосомных аберраций:
1) Транслокации – перенос участка хромосомы.
2) Инверсии – разворот участка хромосомы на 180 градусов.
3) Делеции – отрыв участка хромосомы.
4) Дупликации – удвоение участка или целой хромосомы.
37. Нарушения в гаметогенезе
38. Изменения в количестве половых хромосом
• Синдром Клайнфельтера (ХХУ)• Синдромом Шерешевского-Тернера (Х0)
Нерасхождение половых хромосом при мейозе, образование
сперматозоидов с лишней половой хромосомой и без половой
хромосомы
39. Синдром Клайнфельтера
• генотип (47,ХХУ)• встречается как 1 случай на 500-1000 новорожденных
• Описан в 1942 году
Тельце
Барра
40. Синдром Клайнфельтера
Симптомы:• высокий рост
• слабое оволосение тела
• яички маленького размера, бесплодие
• общий женоподобный вид и гинекомастия
• Снижение либидо
• Остеопороз (разрежение костной ткани)
• Венозная патология
• Аутоиммунные системные проявления
• Низкий уровень энергетики
• Обладают нормальным интеллектом
• Низкая самооценка
• Проблемы психического развития
• Трудности общения
Мальчик 6 лет
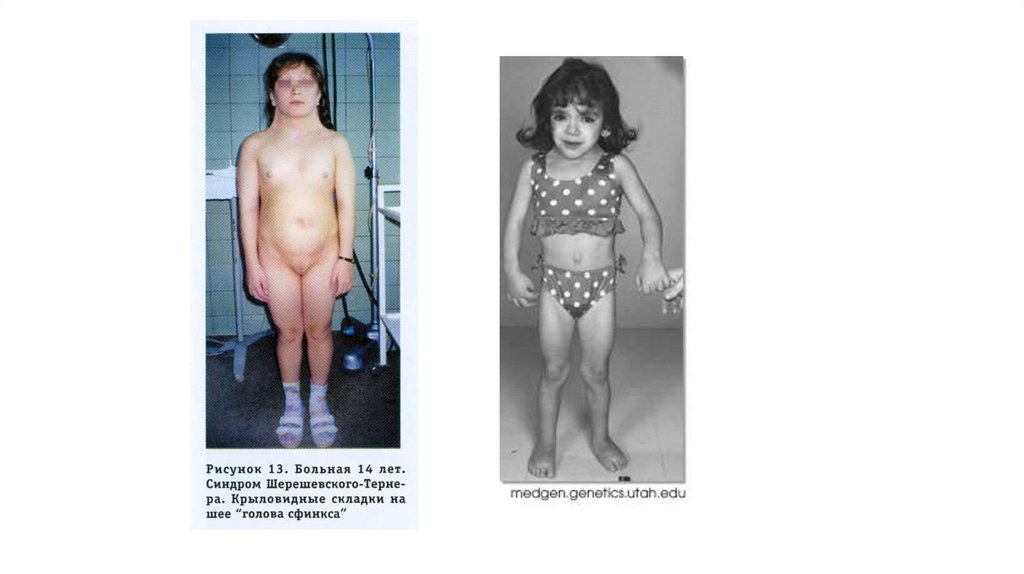
41. Синдромом Шерешевского-Тернера
• генотип (45,Х0)• Частота встречаемости: 1 на 3000
• Описан Н.А.Шерешевским в 1925 году и Г.Тернером в 1938 году
нет
Тельца
Барра
42. Синдромом Шерешевского-Тернера
Симптомы:• болеют женщины,
• низкий рост
• короткая шея, двусторонняя
перепонка шеи (весьма
характерный, но необязательный
симптом); рост волос на нижней
границе шеи;
• отсутствие большинства вторичных
половых признаков,
• бесплодие,
• умственная отсталость
43.
44. Изменения в количестве аутосом
• Синдром Дауна (трисомия 21)• Синдром Эдвардса (трисомия 18)
• Синдром Пата́у (трисомия 13)
45. Синдром Дауна
• Генотип47,ХУ, +21 или 47,ХХ, +21
• Частота встречаемости
1 на 650 новорожденных
• Описан Джоном Дауном в 1866
году
46. Типичные признаки больных с синдромом Дауна
• «плоское лицо» — 90 %• брахицефалия (аномальное укорочение
черепа) — 81 %
• кожная складка на шее у новорожденных —
81 %
• монголоидный разрез глаз — 80 %
• эпикант (вертикальная кожная складка,
прикрывающая медиальный угол глазной
щели) — 80 %
47. Типичные признаки больных с синдромом Дауна
• брахимезофалангия (укорочение всех пальцев засчет недоразвития средних фаланг) - 70 %
• открытый рот (в связи с низким тонусом мышц и
особым строением нёба) — 65 %
• плоская переносица — 52 %
• поперечная ладонная складка (называемая также
«обезьяньей») — 45 %
• короткая широкая шея — 45 %
• короткий нос — 40 %
48.
Большинство мужчин с синдромомДауна бесплодны. По крайней мере
50 % женщин с синдромом Дауна могут
иметь детей. 35-50 % детей, рождённых
от матерей с синдромом Дауна,
рождаются с синдромом Дауна или
другими отклонениями.
49. Синдром Эдвардса
• Генотип47,ХУ, +18 или 47,ХХ, +18
• Частота встречаемости
1 на 7000 новорожденных
• Описан в 1960 году Джоном
Эдвардсом
50. Синдром Эдвардса
Фенотипические проявления• Чаще всего возникают аномалии мозгового и лицевого черепа,
мозговой череп имеет долихоцефалическую форму (относительно
длинная и узкая форма головы человека).
• Глазные щели узкие и короткие.
• Ушные раковины деформированы
• Грудина короткая, грудная клетка шире и короче нормальной.
• Аномальное развитие стопы: пятка резко выступает, свод провисает
(стопа-качалка), большой палец утолщен и укорочен.
• Из дефектов внутренних органов наиболее часто отмечаются пороки
сердца и крупных сосудов: дефект межжелудочковой перегородки,
аплазии одной створки клапанов аорты и легочной артерии.
60 % детей умирают в возрасте до 3 мес, до года доживает лишь 5-10 %.
Основной причиной смерти служат остановка дыхания и нарушения
работы сердца. Оставшиеся в живых — глубокие олигофрены
51. Синдром Пата́у
• Генотип47,ХУ, +13 или 47,ХХ, +13
• Частота встречаемости
1 на 6000 новорожденных
• Описан Эразмусом Бартолином в 1957
и Клаусом Патау в 1960
52. Синдром Пата́у
Проявления заболевания• тяжелые врожденные пороки
• умеренная микроцефалия,
• нарушение развития различных отделов ЦНС,
• низкий скошенный лоб,
• суженные глазные щели, расстояние между
которыми уменьшено, микрофтальмия (одно или оба глазных яблока
уменьшены в размере), помутнение роговицы,
• запавшая переносица, широкое основание носа,
• деформированные ушные раковины,
• расщелина верхней губы и нёба, полидактилия,
• флексорное положение кистей (см.рис), короткая шея.
• характерна задержка умственного развития.
В связи с тяжелыми врожденными пороками развития большинство
детей с синдромом Патау умирают в первые недели или месяцы
(95% - до 1 года)
53. Синдром Пата́у
54. СИНДРОМЫ ЧАСТИЧНЫХ АНЕУПЛОИДИЙ
• Синдром трисомии по короткому плечу 9-й хромосомы (9р+)• Синдром кошачьего крика (5р-)
• Синдром Орбели (13q-)
• Синдром Вольфа-Хиршхорна (4р-)
и др.
55. Синдром трисомии по короткому плечу 9-й хромосомы
• Для больных с трисомией 9р+ характерны умственная отсталость, задержкароста, микроцефалия, антимонголоидный разрез глазных щелей, глубоко
посаженные глаза, опущенные уголки рта, нос с характерным округлым
кончиком, низко расположенные оттопыренные ушные раковины,
недоразвитие ногтей и дистальных фаланг пальцев рук. Часто наблюдаются
выступающие лобные кости, пятна цвета кофе с молоком на коже, эпикант,
косоглазие, высокое нёбо, короткая шея, сколиоз, частичная синдактилия
(сращивание) пальцев стоп. Примерно в четверти случаев обнаруживаются
врожденные пороки сердца.
• Прогноз для жизни сравнительно благоприятный (при отсутствии патологии
внутренних органов) - описаны больные, достигшие преклонного возраста.
• По частоте встречаемости среди детей-олигофренов занимает 2 место после
болезни Дауна.
56. Синдром кошачьего крика (5р-) (Синдром Лежена)
• обусловлен делецией (потеря участка)короткого плеча 5-й хромосомы
• Формула кариограммы: 46,ХХ,del(5р-)
• Популяционная частота синдрома примерно 1:45 000
Продолжительность жизни у больных с
этим синдромом значительно снижена,
только около 14% из них переживают
возраст 10 лет
57. Синдром кошачьего крика (5р-) (Синдром Лежена)
Проявления заболеванияспецифический плач, напоминающий кошачье
мяуканье,
лунообразное лицо,
мышечная гипотония,
умственное и физическое недоразвитие,
Микроцефалия - значительное уменьшение
размеров черепа и, соответственно, головного мозга ,
низко расположенные, иногда деформированные
ушные раковины,
эпикант,
антимонголоидный разрез глазных щелей,
косоглазие.
Иногда наблюдаются атрофия зрительного нерва и
очаги депигментации сетчатки.
пороки сердца.
58. Синдром Орбели
• обусловлен делецией длинного плеча 13-й хромосомы.Популяционная частота не установлена.
• Характерны микроцефалия, отсутствие носовой вырезки
(лоб непосредственно переходит в нос), эпикант,
антимонголоидный разрез глаз, высокое нёбо, низко
расположенные деформированные ушные раковины.
Отмечаются поражения глаз (микрофтальмия, косоглазие,
катаракта,
ретинобластома),
опорно-двигательного
аппарата (короткая шея, синдактилии кистей и стоп),
атрезии (зарощения) прямой кишки. Часты пороки
развития сердца, почек, головного мозга. Для всех детей с
синдромом Орбели характерна глубокая олигофрения.
• Большинство больных с синдромом 13q- погибают на 1-м
году жизни.
59. Синдром Вольфа-Хиршхорна (46,ХУ4p-) (46,ХХ4p-) Это расстройство развивается на фоне потери дистального конца короткого плеча
хромосомы 4.60. Синдром Вольфа-Хиршхорна
• Популяционная частота 1:100000.• обусловлен делецией
сегмента короткого плеча
хромосомы 4.
Симптомы: задержка роста,
расщелина губы и неба,
широкий или клювовидный
нос; асимметрия костей
черепа, низко
расположенные
деформированные ушные
раковины; гематомы кожи
небольших размеров в
области лица.
• Жизнеспособность детей
резко снижена. Большинство
умирают в возрасте до 1 года.
61. Мультифакториальные (полигенные) болезни
(наследственно предрасположенные, много-факторные, «Сomplexgenetic disorders») - это большая и нозологически разнообразная группа
болезней, развитие которых определяется взаимодействием
определенных наследственных факторов (мутаций или сочетаний
аллелей) и факторов среды. Этиология и патогенез данных болезней
сложны, многоступенчаты и во многом еще неясны и, естественно,
разные для каждой болезни.
Болезни с наследственной предрасположенностью возникают у лиц с
соответствующим генотипом (сочетание «предрасполагающих»
аллелей) при провоцирующем действии факторов среды
С определенной долей условности мультифакториальные болезни
можно разделить на:
1) врожденные пороки развития,
2) распространенные психические и нервные болезни,
3) распространенные болезни «среднего» возраста.
62. Полигенные болезни
связаны с действием многих генов, к мультифакториальнымболезням относятся: ревматоидный артрит, ишемическая болезнь
сердца, гипертоническая и язвенная болезни, цирроз печени,
сахарный диабет, бронхиальная астма, псориаз, шизофрения и др.