







































































































 Медицина
МедицинаПохожие презентации:










Введение в патологию
1.
«Как философическое познание объемлетсущность, причину и следствие вещей,
общая патология есть часть врачебной
науки, показывающая сущность,
происхождение и следствие болезней
вообще» Ф.К. Гартман, 1825 (цит. по Д.С.
Саркисову, 1993).
ВВЕДЕНИЕ В ПАТОЛОГИЮ
2.
Патология - учение о структурнофункциональных проявленияхболезней и патологических процессов.
Общепринято подразделять
патологию на патологическую анатомию
и патологическую физиологию.
3.
Предмет патологической анатомии –морфологический субстрат болезней и
патологических процессов.
Патологическая анатомия ставит перед собой большой
круг вопросов при изучении болезней человека и
позволяет дать представление о
- причинах заболевания
- механизмах его развития, или патогенезе
- структурных проявлениях этих механизмов, или
морфогенезе
- осложнениях болезни, а в случае смерти – определить
причину смерти и механизм умирания (танатогенез).
4.
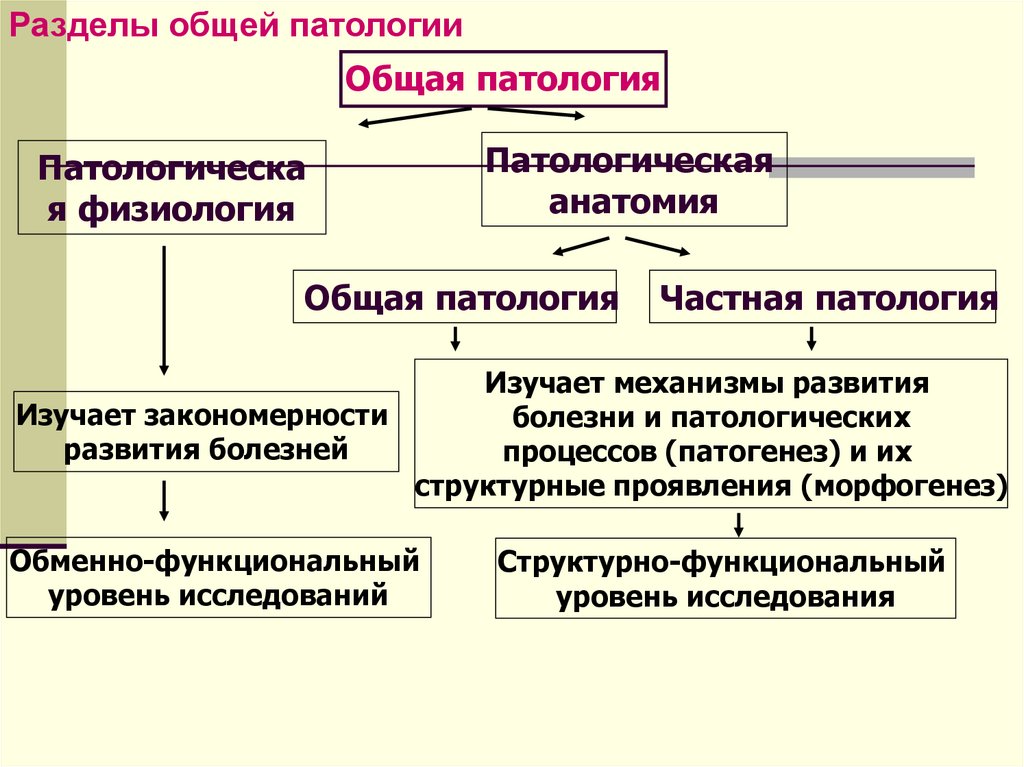
Разделы общей патологииОбщая патология
Патологическа
я физиология
Патологическая
анатомия
Общая патология
Частная патология
Изучает механизмы развития
Изучает закономерности
болезни и патологических
развития болезней
процессов (патогенез) и их
структурные проявления (морфогенез)
Обменно-функциональный
уровень исследований
Структурно-функциональный
уровень исследования
5.
1.2.
Представления о том, что происходит в организме,
когда он болен, исторически развивались по двум
направлениям. Это
Учение о нозологии – существовании конкретных
болезней, каждая из которых имеет причину
возникновения, механизм развития и присущую
только ей клинико-анатомическую картину
Общепатологический подход к изучению болезней,
базирующийся на изучении типовых
патологических процессов, различные сочетания
которых и составляют суть самых разных
заболеваний.
6.
Важнейшие общепатологические процессы:Воспаление
Дистрофия
Атрофия
Некроз
Нарушение кровообращения
Склерозирование
Нарушения регенерации (дисрегенерация)
7.
Особенности общепатологического подхода1. базируется на учении об общепатологических
(типовых, стереотипных) процессах, или
проявлениях различных заболеваний, сформировавшихся в ходе эволюции и генетически детерминированных; реализующихся у человека и животных
преимущественно в соматической сфере и высоко
воспроизводимых в эксперименте;
2. ассоциируется с такими категориями, как
- патология клетки
- патогенез
- общепатологические процессы
- этиология
- морфогенез
- нозология
3. объединяет результаты морфологических,
клинических, физиологических, биохимических и
прочих методов исследования для достижения
основной цели – познание сущности болезни
8.
Таким образом, комплексный подход кизучению болезней и патологических
процессов, лежащий в основе общей
патологии, служит как теории, так и
клинической практике, поэтому общая
патология является научно-прикладной
дисциплиной и составляет фундамент всей
медицины.
9.
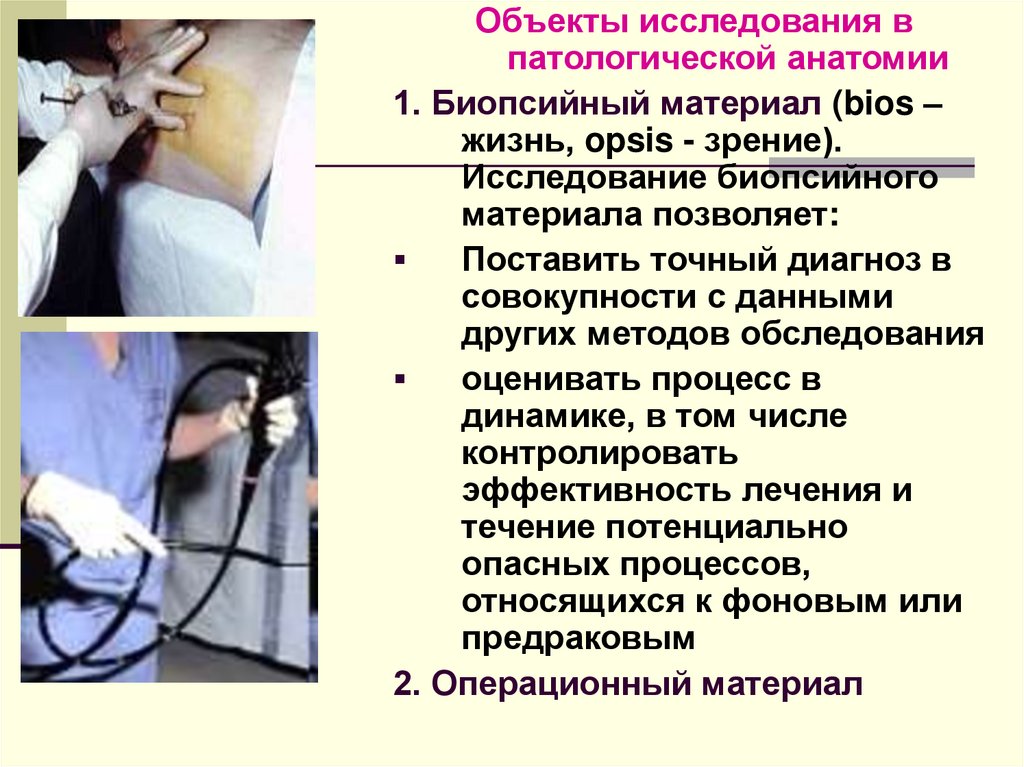
Объекты исследования впатологической анатомии
1. Биопсийный материал (bios –
жизнь, opsis - зрение).
Исследование биопсийного
материала позволяет:
Поставить точный диагноз в
совокупности с данными
других методов обследования
оценивать процесс в
динамике, в том числе
контролировать
эффективность лечения и
течение потенциально
опасных процессов,
относящихся к фоновым или
предраковым
2. Операционный материал
10.
3. Цитологический материал – аспирационные пунктаты,мазки-отпечатки, соскобы, промывные воды
4. Аутопсийный материал (autopsia – видение
собственными глазами) – материал, получаемый при
вскрытии трупов.
5. Экспериментальный материал. Любой патологический
процесс (от воспаления до канцерогенеза) может быть
смоделирован в эксперименте, а так как эксперимент
позволяет исследовать процесс в динамике, изучать
отдельные тонкие механизмы патогенеза,
исследование экспериментального материала может
иметь в определенных случаях большое значение для
накопления знаний.
11.
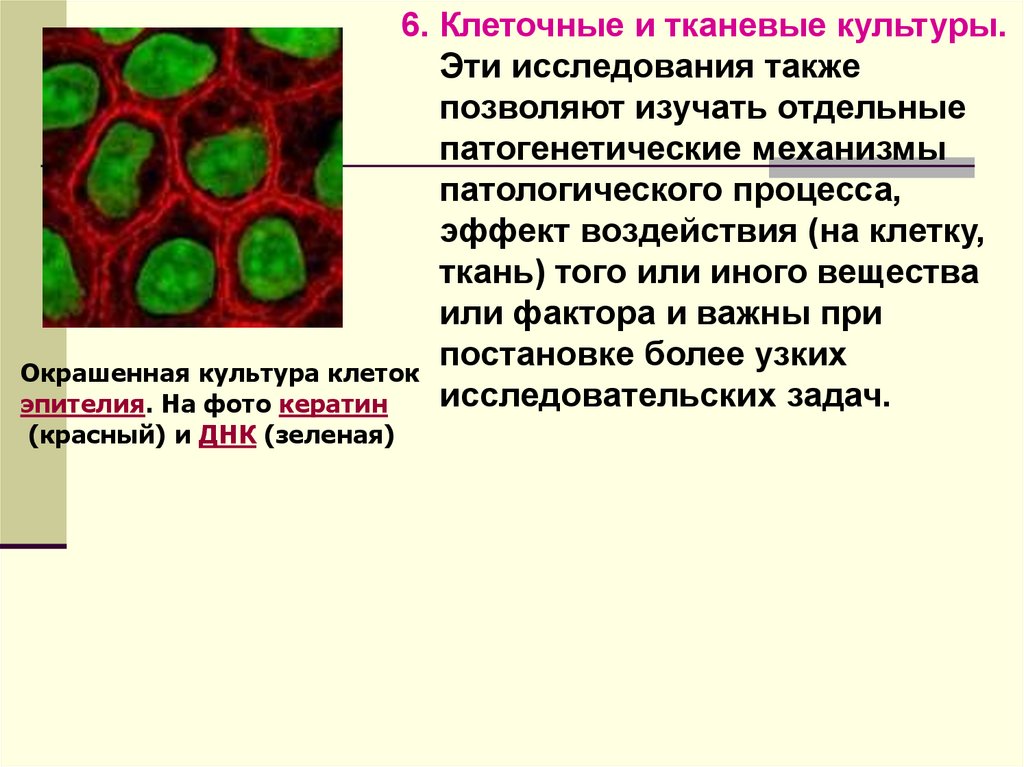
6. Клеточные и тканевые культуры.Эти исследования также
позволяют изучать отдельные
патогенетические механизмы
патологического процесса,
эффект воздействия (на клетку,
ткань) того или иного вещества
или фактора и важны при
постановке более узких
Окрашенная культура клеток
исследовательских задач.
эпителия. На фото кератин
(красный) и ДНК (зеленая)
12.
Методы исследования в патологииМакроскопическое исследование органов и
тканей (при аутопсии, а также различных
эндоскопических методах)
Гистологическое, цитологическое
исследование (световая микроскопия)
Ультраструктурное исследование
(электронная микроскопия)
Радиоавтография (применение меченых
изотопами продуктов синтеза, используемых
клеткой)
13.
Радиоавтография14.
Иммуногистохимическое исследование(инкубация образцов с раствором антител к
искомым антигенам с последующей световой
или электронной микроскопией). Помимо
диагностической ценности во многих областях
патологии, особенно онкологии, этот метод
имеет большое значение для понимания
патогенеза многих заболеваний.
15.
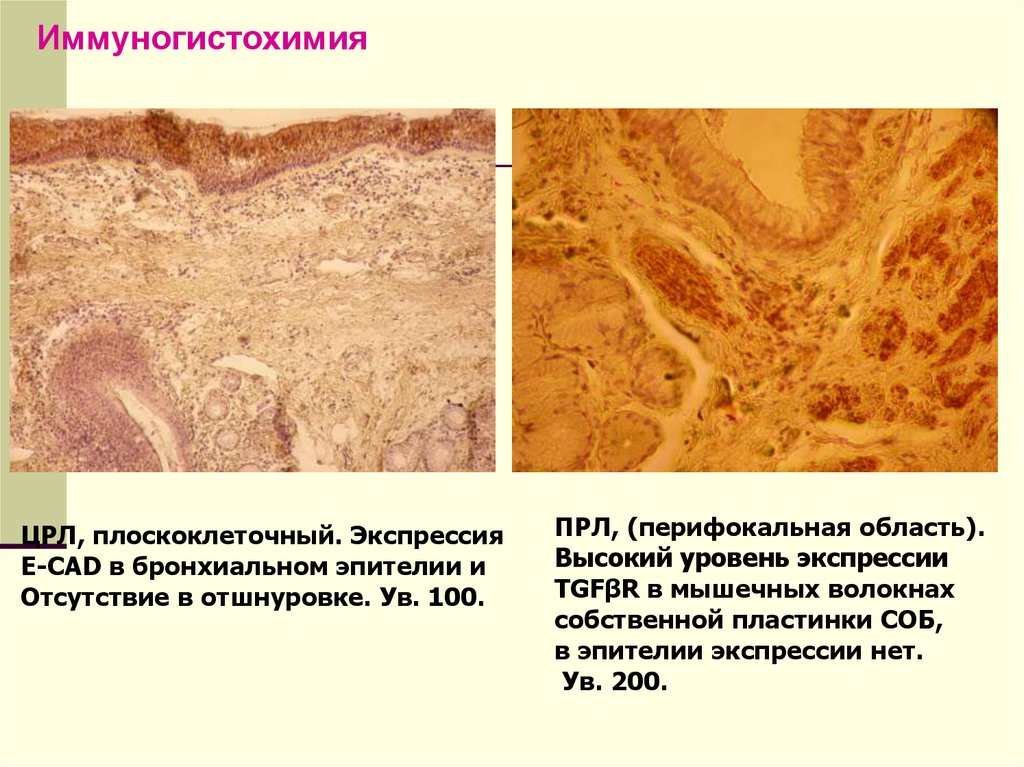
ИммуногистохимияЦРЛ, плоскоклеточный. Экспрессия
E-CAD в бронхиальном эпителии и
Отсутствие в отшнуровке. Ув. 100.
ПРЛ, (перифокальная область).
Высокий уровень экспрессии
TGFβR в мышечных волокнах
собственной пластинки СОБ,
в эпителии экспрессии нет.
Ув. 200.
16.
Морфометрическое исследование (сюда же может бытьотнесена проточная цитометрия, позволяющая проводить
количественный анализ содержания в клетках ДНК)
Метод клеточных и тканевых культур
Методы молекулярной и клеточной биологиии
метод полимеразной цепной реакции (PCR) позволяет
определять даже одну молекулу или фрагмент ДНК или РНК,
широко используется для определения генов, их структуры и
уровня экспрессии
метод гибридизации in situ обеспечивает возможность точной
локализации специфической нуклеотидной последовательности в
отдельных клетках, но ограничен (по сравнению с PCR) низкой
чувствительностью. Использование молекулярной техники
позволило скомбинировать оба метода
Новый метод получил название «in situ PCR», он
используется для определения вирусной или чужеродной
последовательности нуклеиновых кислот, изучения эндогенных
последовательностей ДНК
17.
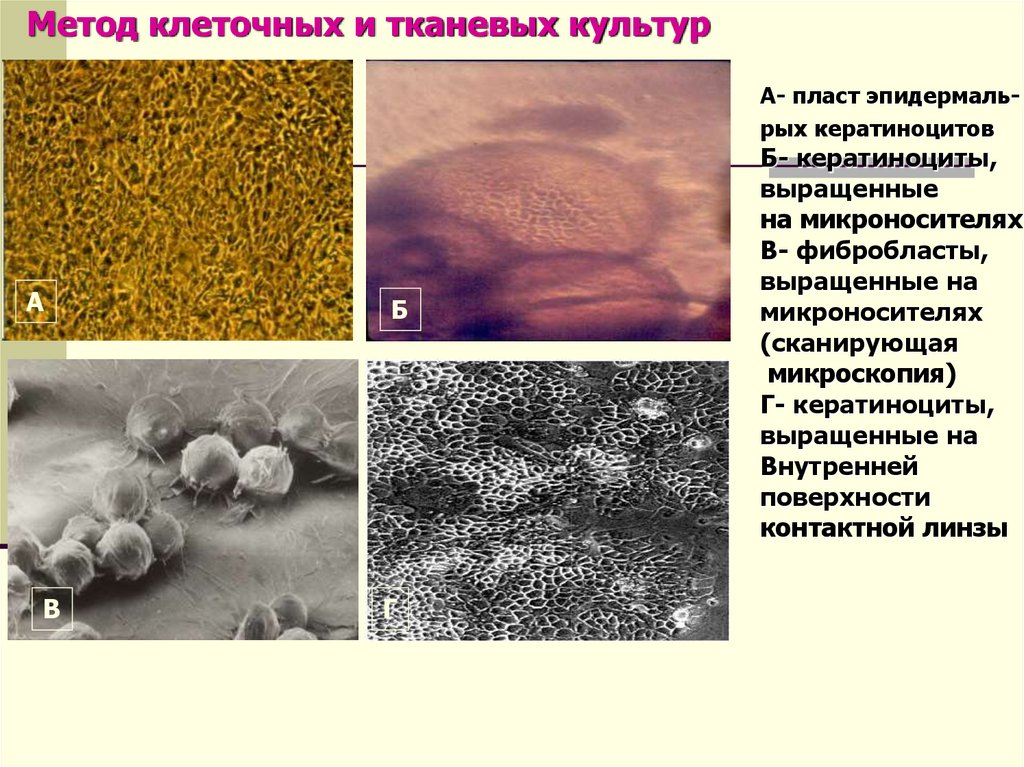
Метод клеточных и тканевых культурА- пласт эпидермальрых кератиноцитов
А
Б
В
Г
Б- кератиноциты,
выращенные
на микроносителях
В- фибробласты,
выращенные на
микроносителях
(сканирующая
микроскопия)
Г- кератиноциты,
выращенные на
Внутренней
поверхности
контактной линзы
18.
Использование методов молекулярнойбиологии стало основой для создания нового
раздела в патологии – молекулярной
патологии.
Клетка – основная структурная единица при
изучении патологических процессов (Струков
А.И., 1976).
Познание патологии клетки – структурная
основа патологии человека.
19.
Повреждение клеток и тканейНормальные клетки находятся в состоянии
гомеостаза или равновесия с окружающей
средой.
Повреждение клетки характеризуется
комплексом биохимических и морфологических изменений, нарушающих состояние
гомеостаза и развивающихся под действием
повреждающих факторов.
20.
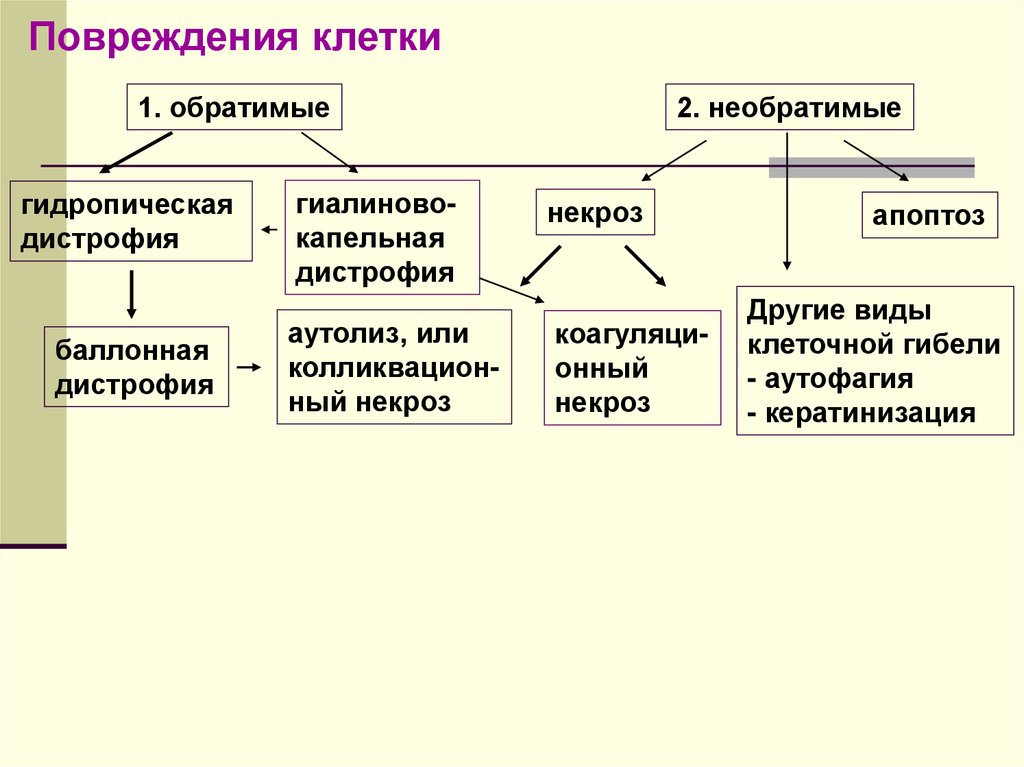
Повреждения клетки1. обратимые
гидропическая
дистрофия
баллонная
дистрофия
гиалиновокапельная
дистрофия
аутолиз, или
колликвационный некроз
2. необратимые
некроз
коагуляционный
некроз
апоптоз
Другие виды
клеточной гибели
- аутофагия
- кератинизация
21.
Причины повреждения клетокэкзогенные – физические (механическая
травма, ↑ или↓ t°, радиация, электрический
ток), химические (алкоголь, курение, высокие
дозы лекарств, яды) и биологические факторы
(вирусы, бактерии, грибы)
эндогенные – различные метаболиты, в том
числе свободные радикалы, гормоны,
цитокины, иммунные комплексы, генетические
дефекты
22.
Наиболее чувствительные мишени привоздействии повреждающих факторов
клеточные мембраны, обеспечивающие
сохранение ионного и осмотического
гомеостаза клетки
митохондрии, обеспечивающие
окислительное фосфорилирование
и образование АТФ
процессы синтеза ферментов и структурных
белков
генетический аппарат клетки
23.
Реакция клеток на повреждение зависит от- типа, продолжительности и тяжести
повреждения
- особенностей самой клетки (ткани) – ее
метаболических потребностей, характера
питания и т.д. Этими же факторами определяется время появления морфологических
изменений (от нескольких минут при набухании
клетки до нескольких часов при развитии
некроза)
24.
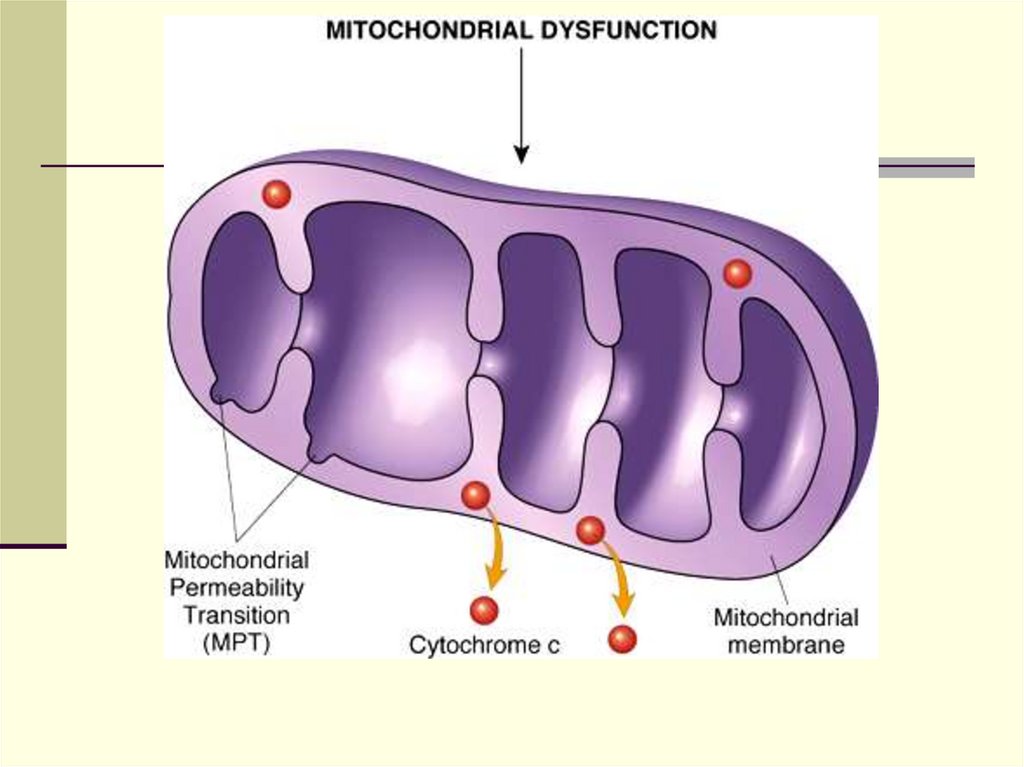
Механизмы развития повреждения исмерти клетки
образование свободных радикалов →
перекисное окисление липидов →
повреждение мембран
утрата плазматической мембраной
избирательной проницаемости – постоянный
признак всех видов повреждения (↓ или
прекращение синтеза АТФ → проникновение
ионов кальция в клетку → активация
фосфолипаз → повреждение мембраны)
25.
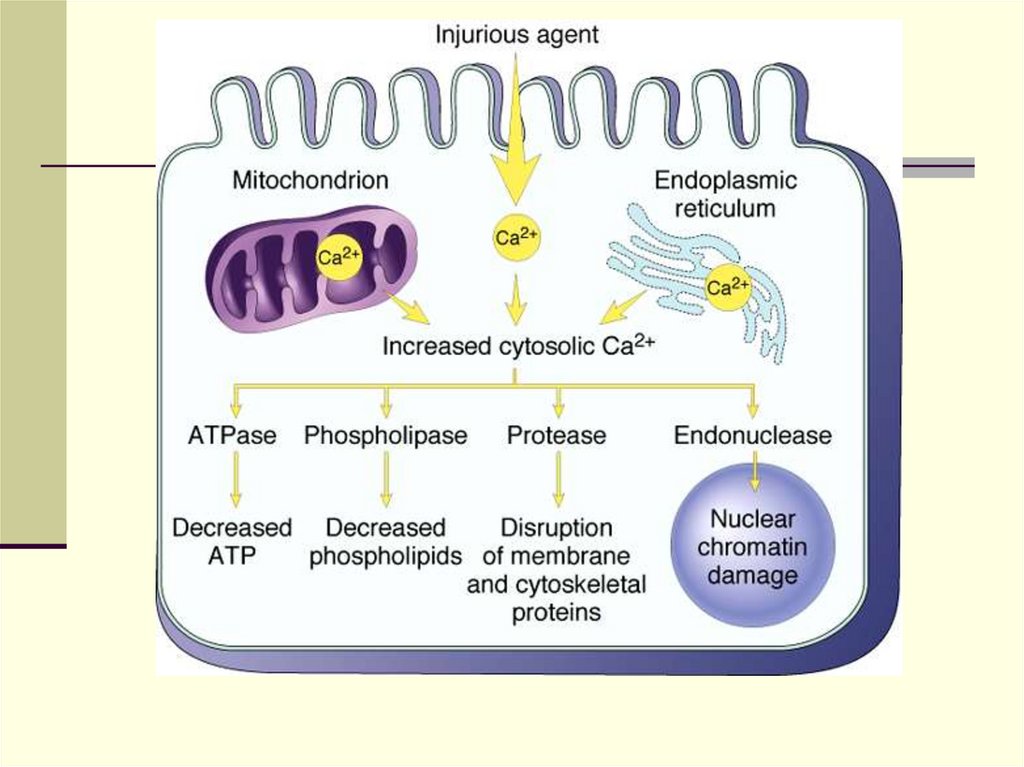
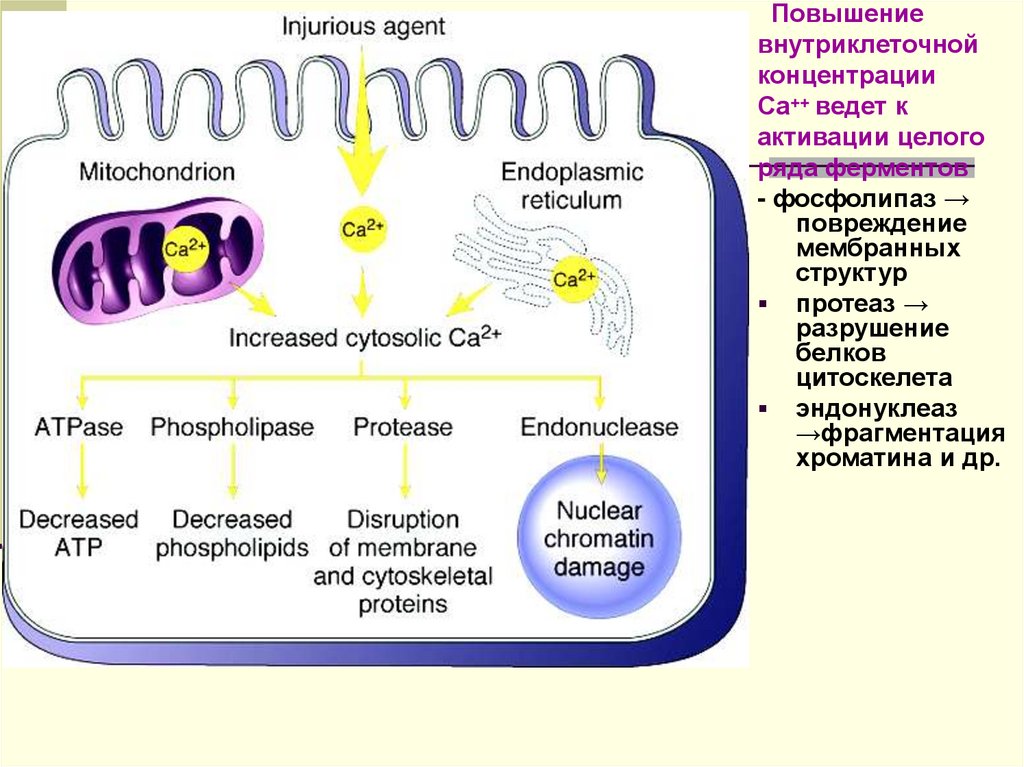
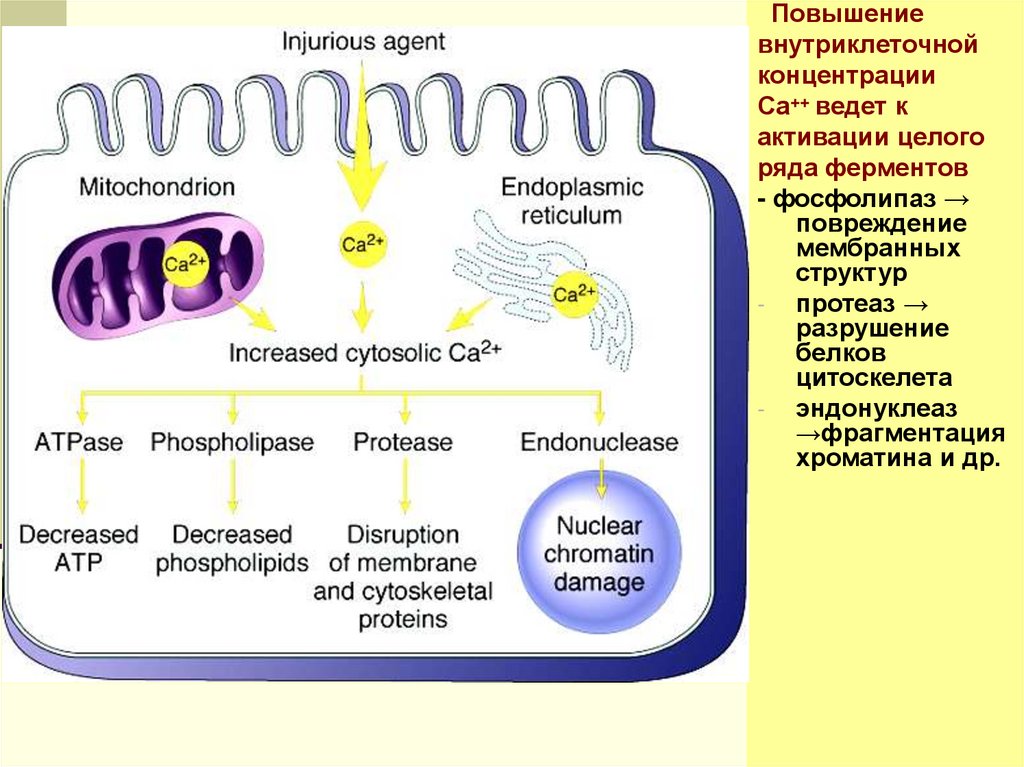
(механизмы развития повреждения и смерти клетки)нарушение гомеостаза кальция → ↑ концентрации Са2+
в цитоплазме → активация ферментов, повреждающих
клетку: фосфолипаз (повреждение клеточных
мембран), протеаз (разрушение мембран и
цитоскелета), активация АТФаз (истощение запасов
АТФ) и эндонуклеаз (фрагментация хроматина)
снижение синтеза АТФ в митохондриях (→ нарушение
процессов синтеза белка, мембранного транспорта,
липогенеза и др. обменных процессов)
26.
Универсальные механизмы повреждения клетки27.
28.
29.
Независимо от причины на уровнеклеточных структур различные виды
повреждений укладываются в 3 возможных
инициальных механизма:
1. Гипоксия
2. Свободно радикальный механизм
3. Токсическое повреждение
Каждый из этих механизмов имеет свои
особенности
30.
Гипоксия – патологический процесс,развивающийся вследствие недостаточности
и/или неэффективности биологического
окисления и приводящий к снижению или
прекращению энергетического обеспечения
жизненно важных процессов.
31.
Основные причины развития гипоксииишемия (←уменьшение кровоснабжения ←
спазм, склероз, тромбоз сосудов)
неадекватная оксигенация крови (болезни
сердца, легких)
снижение способности крови к
транспортировке О2 (анемия, отравление)
32.
Изменения метаболизма при гипоксиидефицит макроэргов – прогрессивное ↓ содержания
АТФ и креатинфосфата при одновремен-ном
увеличении продуктов его распада - АДФ, АМФ и
неорганического фосфата
преобладание гликолиза → падение содержания
гликогена в клетках и ↑ концентрации лактата и
пирувата → развитие внутриклеточного
метаболического ацидоза → активация лизосо-мальных
протеаз, ↑ распада и ↓ синтеза белков
снижение выработки АТФ → нарушение работы
различных (K+/Na+ , Ca++-, Mg++-) АТФаз → нарушение
ионного гомеостаза клетки - ↑ внутриклеточного Ca + + ,
Na + и внеклеточного K+.
33.
Повышениевнутриклеточной
концентрации
Са++ ведет к
активации целого
ряда ферментов
- фосфолипаз →
повреждение
мембранных
структур
протеаз →
разрушение
белков
цитоскелета
эндонуклеаз
→фрагментация
хроматина и др.
34.
Дополнительные факторы гипоксическогоповреждения клетки
↓ рН
накопление молочной кислоты в цитоплазме
(компенсаторно в условиях гипоксии клетка переходит
на анаэробный гликолиз) → ↑ осмотического давления
в клетке и увеличение ее отека (набухания)
накопление воды в цитоплазме, митохондриях,
цистернах гранулярной ЭПС → отщепление от ее
мембран рибосом → снижение синтеза белка
появление в цитоплазме клеток и внеклеточном
пространстве «миелиновых фигур», формирующихся
из мембран органелл
35.
Морфологические проявления гипоксическогоповреждения клетки (ткани)
нарушения микроциркуляции – стаз и сладж крови, ↑
сосудистой проницаемости, паравазальный отек,
микрокровоизлияния, мукоидное и фибриноидное
набухание соединительной ткани → фиброз стромы
при хр. гипоксии
обратимое набухание паренхиматозных клеток (ранний
признак)
при сохраняющейся гипоксии прогрессирование в
необратимую гидропическую, а позднее – баллонную
дистрофию, являющуюся, по сути, колликвационным
некрозом
36.
Гидропическая дистрофия.В результате инвагинации
цитоплазматической мембраны образуются вакуоли,
заполненные водой («гипоксические вакуоли» — ГВ),
Вода также накапливается в
расширенных цистернах
гранулярной эндоплазматической сети (ГрЭПС) и
набухших митохондриях (М)
37.
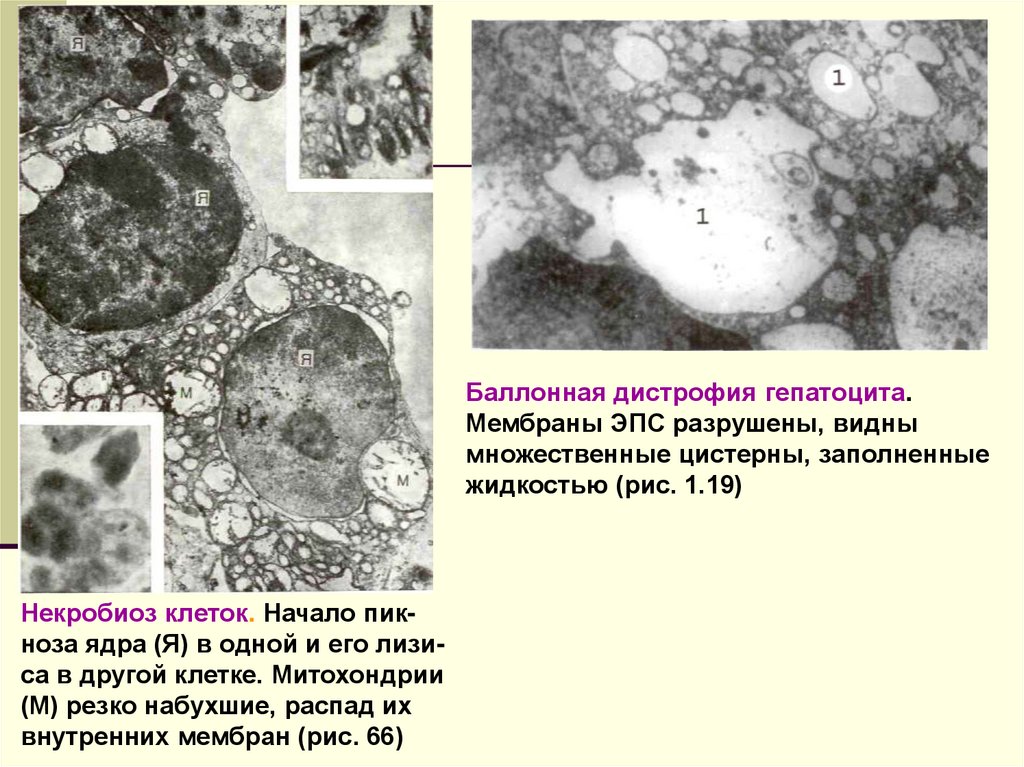
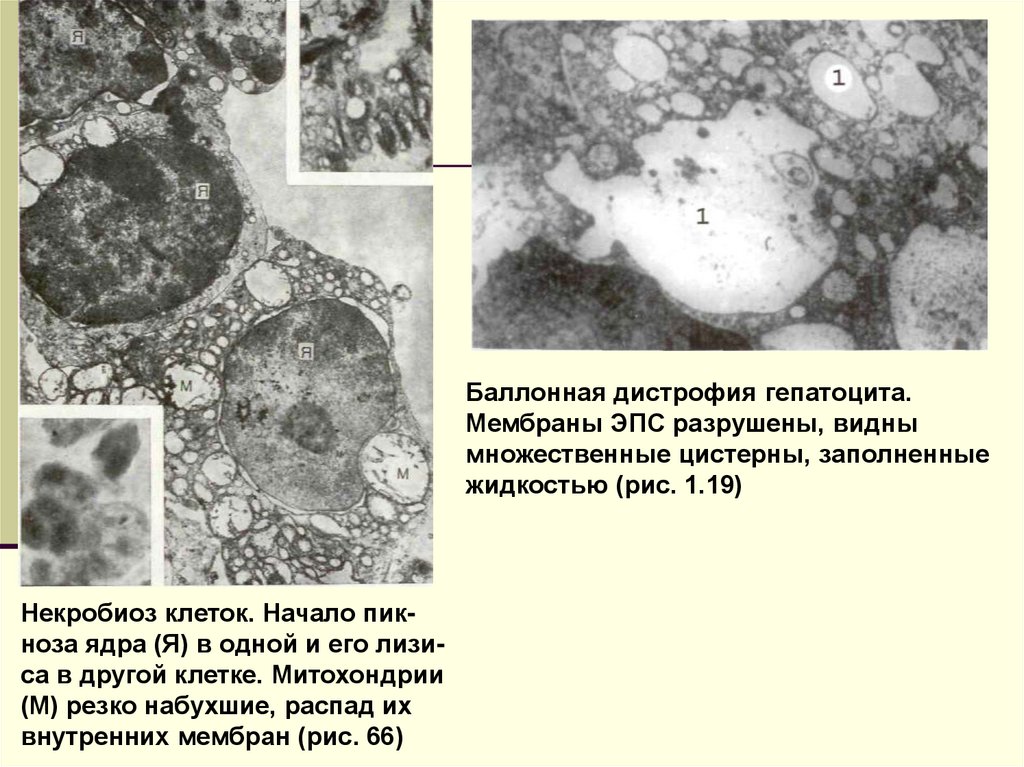
Баллонная дистрофия гепатоцита.Мембраны ЭПС разрушены, видны
множественные цистерны, заполненные
жидкостью (рис. 1.19)
Некробиоз клеток. Начало пикноза ядра (Я) в одной и его лизиса в другой клетке. Митохондрии
(М) резко набухшие, распад их
внутренних мембран (рис. 66)
38.
Патогенез набухания клеток связан смодификацией структуры и увеличением
проницаемости как клеточной, так и
внутренней мембраны митохондрий
ингибированием дыхательной цепи
митохондрий
снижением, а позднее прекращением синтеза
АТФ
ингибированием различных ферментов клетки
39.
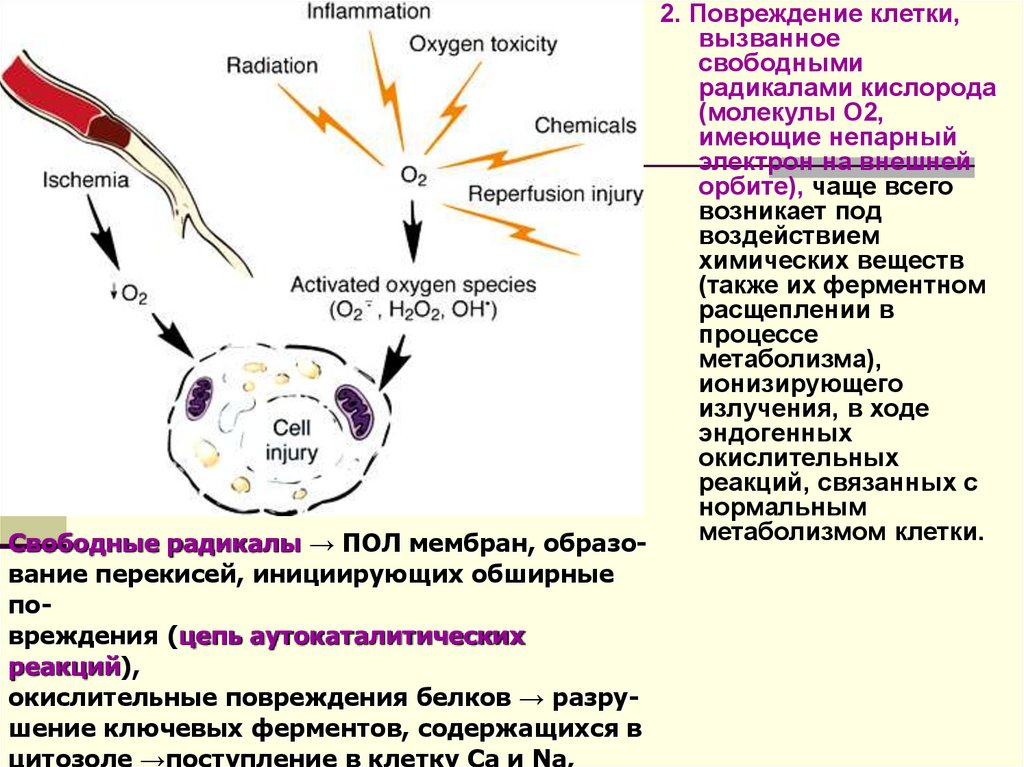
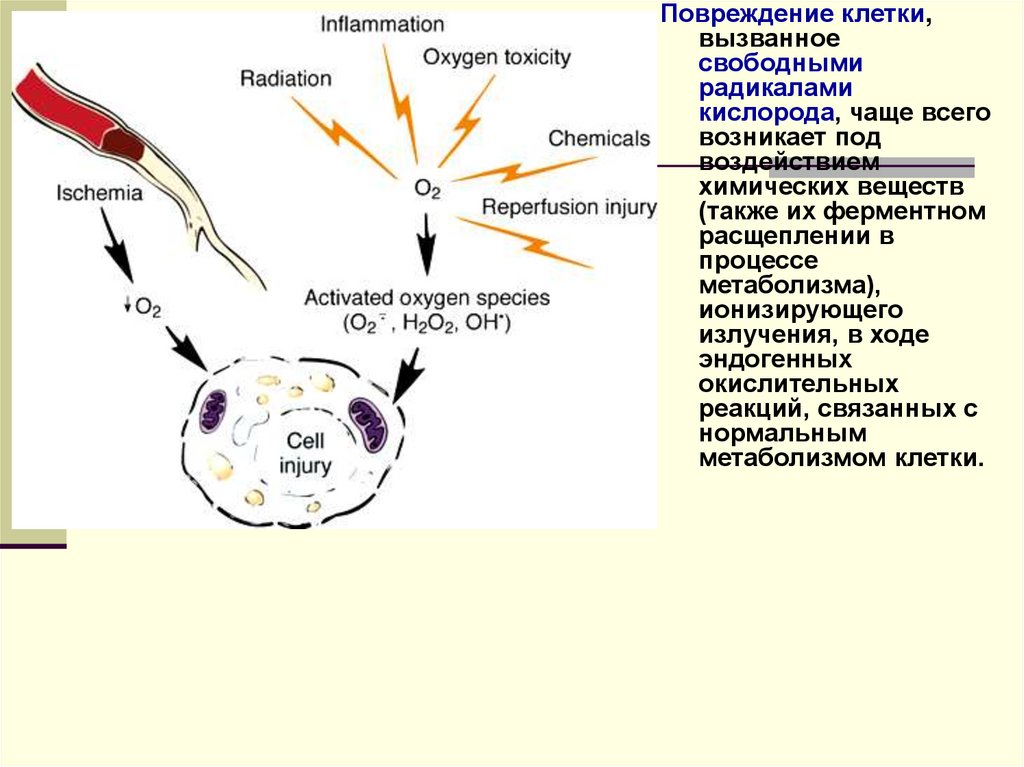
2. Повреждение клетки,вызванное
свободными
радикалами кислорода
(молекулы О2,
имеющие непарный
электрон на внешней
орбите), чаще всего
возникает под
воздействием
химических веществ
(также их ферментном
расщеплении в
процессе
метаболизма),
ионизирующего
излучения, в ходе
эндогенных
окислительных
реакций, связанных с
нормальным
метаболизмом клетки.
Свободные радикалы → ПОЛ мембран, образование перекисей, инициирующих обширные
повреждения (цепь аутокаталитических
реакций),
окислительные повреждения белков → разрушение ключевых ферментов, содержащихся в
цитозоле →поступление в клетку Са и Na,
40.
В норме под воздействием цитохромоксидазыО2 превращается в воду, но при избытке
промежуточных соединений О2 (супероксид,
перекись водорода, гипероксильные ионы)
развивается цепь аутокаталитических
реакций, ведущих к обширному повреждению
клетки (перекисное окисление липидов
мембран клетки, связывание лабильных
аминокислот, фрагментация полипептидных
цепей, повреждение ДНК).
41.
3. Особенности инициальных механизмов поврежденияклетки при действии химических веществ
определяются
тропностью к определенным видам клеток и
внутриклеточных органелл, участвующих в
метаболизме этих веществ (например, хлорид ртути →
больше страдают клетки желудочно-кишечного тракта и
почек)
характером образующихся токсических метаболитов
(при изначально биологически неактивных токсинах)
Наиболее важным и при воздействии химических
веществ оказывается образование свободных
радикалов О2 и последующее перекисное окисление
липидов
42.
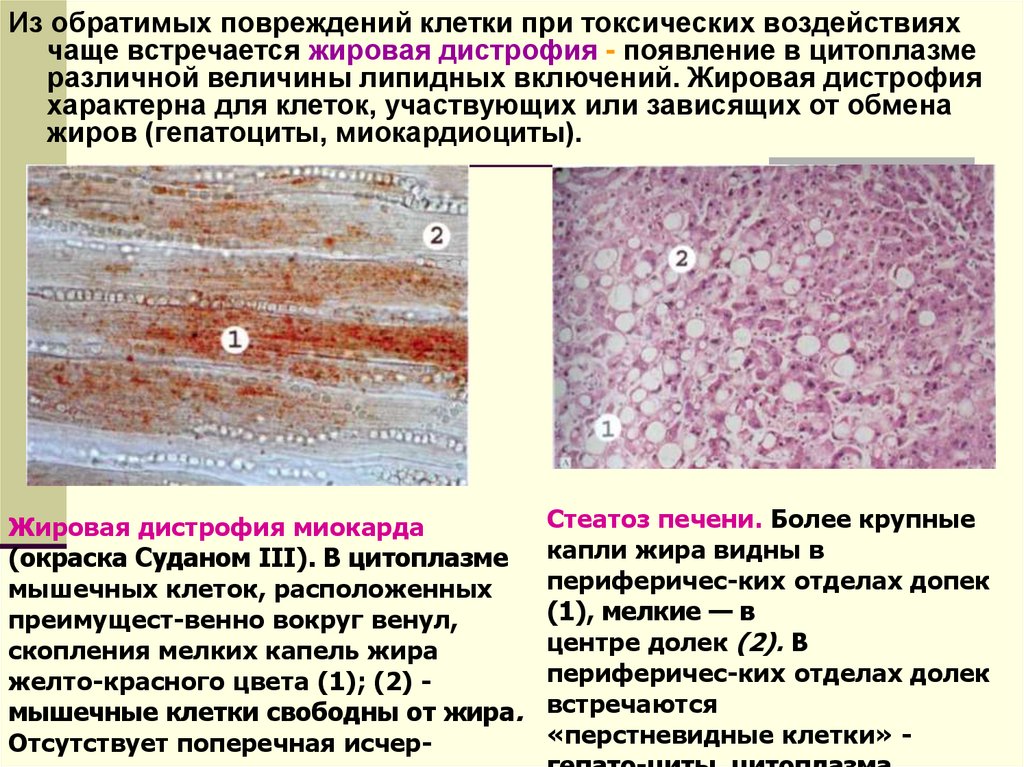
Из обратимых повреждений клетки при токсических воздействияхчаще встречается жировая дистрофия - появление в цитоплазме
различной величины липидных включений. Жировая дистрофия
характерна для клеток, участвующих или зависящих от обмена
жиров (гепатоциты, миокардиоциты).
Жировая дистрофия миокарда
(окраска Суданом III). В цитоплазме
мышечных клеток, расположенных
преимущест-венно вокруг венул,
скопления мелких капель жира
желто-красного цвета (1); (2) мышечные клетки свободны от жира.
Отсутствует поперечная исчер-
Стеатоз печени. Более крупные
капли жира видны в
периферичес-ких отделах допек
(1), мелкие — в
центре долек (2). В
периферичес-ких отделах долек
встречаются
«перстневидные клетки» -
43.
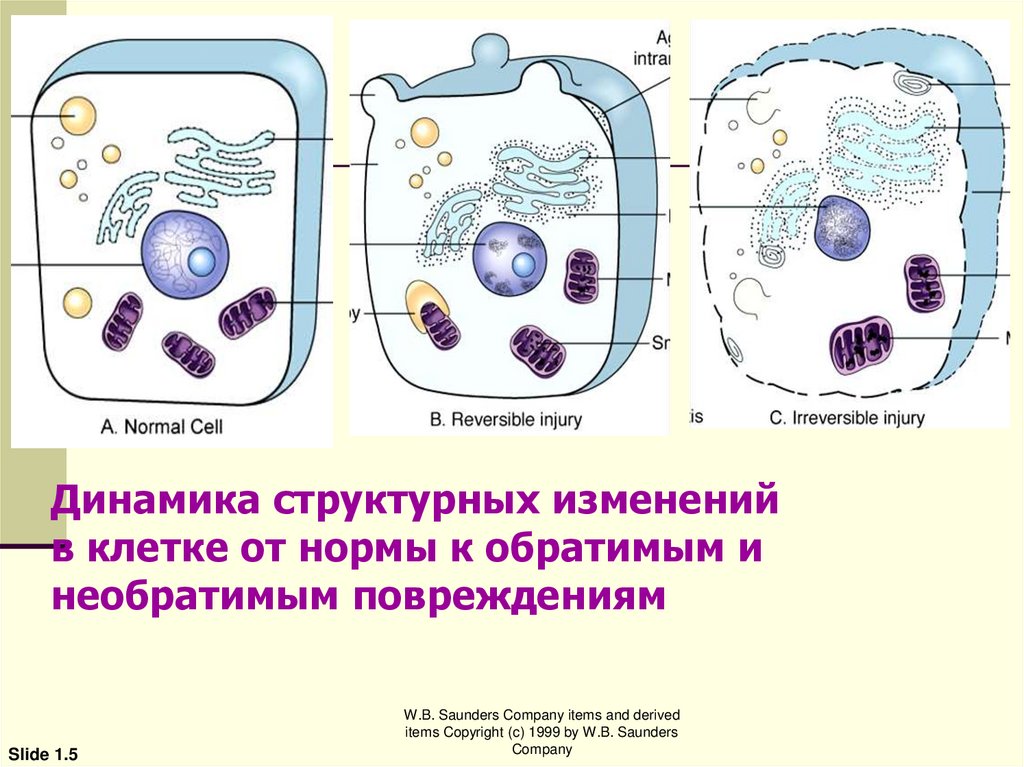
Динамика структурных измененийв клетке от нормы к обратимым и
необратимым повреждениям
Slide 1.5
W.B. Saunders Company items and derived
items Copyright (c) 1999 by W.B. Saunders
Company
44.
Обратимые повреждения клеткиДистрофия – нарушение обмена веществ в
клетках и тканях (dys – отклонение; trophe –
питание)
Трофика – совокупность механизмов,
определяющих метаболизм (обмен
веществ) и структурную организацию
клетки (ткани), необходимых для
выполнения специализированной функции
45.
Механизмы осуществления трофики клетки (ткани)клеточные
- структурная организация
клетки (ферментные системы,
м/х);
- ауторегуляция, обусловленная
генетическим кодом
внеклеточные
- транспортные системы
организма (кровь, лимфа);
- интегративные
системы организма
(нервная, эндокринная,
система соединительной ткани)
46.
Патогенетические механизмы нарушения трофикиклетки (или развития дистрофии)
клеточные
- энергодефицит вследствие
поражения митохондрий →
внутриклеточная гипоксия
→ нарушение работы
ферментов
- наследственные или
приобретенные
ферментопатии
внеклеточные
- нарушения микроциркуляции
→ гипоксия (тот же механизм
нарушения работы митохондрий,
ферментов) → дисциркуляторная
дистрофия
- расстройства нейро-эндокринной и гуморальной регуляции
→ нервные или эндокринные
дистрофии
Накопление в клетке и/или межклеточном веществе различных
продуктов обмена → нарушение структуры и функции клетки
47.
Механизмы формирования структурных проявленийдистрофии
1. Скопление естественных эндогенных метаболитов,
которые образуются и в норме, повышена скорость
их образования или снижена скорость их удаления
из клетки
2. Накопление эндогенных веществ, которые не могут
метаболизироваться в клетке (генетические дефекты
ферментов → болезни накопления, или
тезаурисмозы
3. Накопление экзогенных веществ, которые клетка не
может ни разрушить с помощью ферментов, ни
транспортировать во внеклеточное пространство
(угольная пыль)
Какие вещества могут накапливаться в клетке?
вода, жиры, белки, углеводы, пигменты, аномальные
вещества – продукты нарушенного метаболизма
(липофусцин, экзогенные вещества)
48.
Морфогенетические механизмы развития дистрофий:Инфильтрация - избыточное проникновение веществ
или продуктов обмена в клетки, стенки сосудов,
межклеточное вещество и/или нарушение включения
их в метаболизм с последующим накоплением
(инфильтрация белком эпителия проксимальных
канальцев почек при увеличении его количества в
первичной моче или метастатическое
обызвествление, обусловленное гиперкальциемией).
Продукты обмена могут не метаболизироваться, а
накапливаться в связи с генетическим дефектом
фермента (болезни накопления, или тезаурисмозы).
Могут аккумулироваться экзогенные вещества,
которые клетка не может ни разрушить с помощью
ферментов, ни транспортировать в другое место
(частицы угля).
49.
Морфогенетические механизмы развития дистрофий(продолжение):
Декомпозиция (фанероз) - распад ультраструктур
клеток и межклеточного вещества или сложных в
химическом отношении веществ, ведущий к
нарушению метаболизма и накоплению продуктов
нарушенного обмена в клетке. Распад полисахариднобелковых комплексов лежит в основе фибриноидных
изменений соединительной ткани при ревматических
болезнях.
Трансформация - переход одного вещества в другое
(трансформация углеводов в жиры при сахарном
диабете).
Извращённый синтез - образование в клетках или в
тканях веществ, не встречающихся в норме (синтез
аномального белка амилоида и его отложение в
межклеточном веществе).
Механизмы развития дистрофий могут сочетаться.
50.
Белковые дистрофии (диспротеинозы).Механизм развития белковой дистрофии -
повреждающий стимул → «стрессовые протеины» или
белки-чапероны (chaperones). В норме чапероны
обеспечивают восстановление повреждённых,
частично денатурированных белков. Если этого не
происходит и возникает опасность дальнейшего
повреждения клетки изменёнными белками, то
включается убиквитиновый механизм - необратимо
повреждённые белки присоединяются к другому
белку убиквитину, который обеспечивает их
расщепление нелизосомальными протеиназами до
нетоксичных фрагментов.
Фрагменты белков накапливаются, в цитоплазме
появляются эозинофильные гиалиноподобные
белковые глыбки и капли, сливающиеся между собой
и заполняющие тело клетки (гиалиново-капельная
дистрофия).
51.
Белковая дистрофия часто встречается в почках,реже в печени и очень редко в миокарде.
В почках при повреждении гломерулярного фильтра
эозинофильные капли (гиалиново-капельная
дистрофия) встречаются в цитоплазме эпителия
проксимальных канальцев.
Примерами избыточного накопления белка являются
также тельца Русселя — продукты избыточного
синтеза белка в плазматических клетах и тельца
Маллори — эозинофильные включения в клетках
печени при алкогольной болезни.
Исход белковой дистрофии — неблагоприятный.
Гиалиново-капельная дистрофия завершается
фокальным, а затем тотальным коагуляционным
некрозом клетки, функция клетки и органа снижается.
52.
Гиалиноз - внеклеточное (в стенках сосудов имежклеточном веществе) образование гиалина.
Внеклеточный гиалин — сложное вещество, в состав
которого могут входить фибрин, различные белки
плазмы, иммуноглобулины, липиды.
К развитию гиалиноза ведёт деструкция волокнистых
структур и повышение тканево-сосудистой
проницаемости в связи с дисциркуляторными,
метаболическими и иммунопатологическими
процессами. Гиалинозу могут предшествовать
плазматическое пропитывание, фибриноидное
набухание, воспаление, некроз, склероз.
53.
Особенности сосудисто-стромальной дистрофииМукоидное набухание — увеличение количества и
перераспределение мукополисахаридов,
преимущественно гликозаминогликанов (за счёт
отщепления их от белка), в основном веществе
соединительной ткани. Накопление
гликозаминогликанов всегда начинается с повреждения
сосудов микроциркуляторного русла, что ведёт к
развитию тканевой гипоксии, активации гиалуронидазы
и ослаблению связи между гликозаминогликанами и
белком.
Гликозаминогликаны обладают выраженными
гидрофильными свойствами, что на фоне повышенной
сосудисто-тканевой проницаемости ведёт к
выраженной гидратации (набуханию) основного
вещества соединительной ткани. Одновременно
увеличивается концентрация протеогликанов и в
меньшей степени гликопротеидов.
54.
Для выявления гликозаминогликанов используются специальныеокраски (альциановый синий, коллоидное железо). При окраске
гематоксилином и эозином они имеют слабо базофильную
окраску, так как при ослабевании связи с белком высвобождаются
их кислотные радикалы. Чаще всего в практике используют
метахроматические катионные красители, которые окрашивают
гликозаминогликаны в цвет, отличный от собственного цвета
красителя. Толуидиновый синий придаёт гликозаминогликанам
(гиалуроновая кислота, хондроитинсульфаты) сиреневый или
пурпурный цвет в связи с закислением среды.
Микроскопически коллагеновые волокна обычно сохраняют
пучковое строение, но набухают и разволокняются. Набухание и
увеличение в объёме основного вещества приводит к тому, что
клетки соединительной ткани удаляются друг от друга.
Макроскопически органы практически не изменены.
Мукоидное набухание развивается чаще всего в стенках артерий,
сердечных клапанах, эндо- и эпикарде, в капсулах суставов при
инфекционно-аллергических заболеваниях, ревматических
заболеваниях, гипертонической болезни.
Исход может быть двояким. Мукоидное набухание обратимо, при
прекращении воздействия патогенного фактора происходит
полное восстановление структуры и функции. Если воздействие
патогенного фактора продолжается, мукоидное набухание может
перейти в фибриноидное набухание.
55.
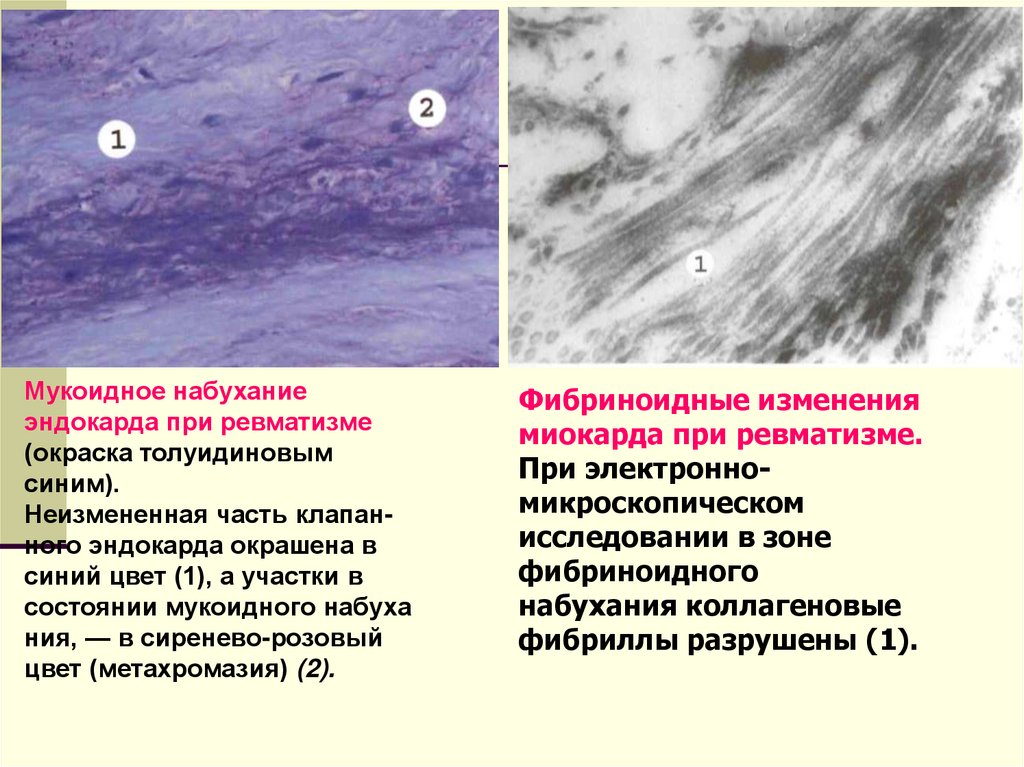
Мукоидное набуханиеэндокарда при ревматизме
(окраска толуидиновым
синим).
Неизмененная часть клапанного эндокарда окрашена в
синий цвет (1), а участки в
состоянии мукоидного набуха
ния, — в сиренево-розовый
цвет (метахромазия) (2).
Фибриноидные изменения
миокарда при ревматизме.
При электронномикроскопическом
исследовании в зоне
фибриноидного
набухания коллагеновые
фибриллы разрушены (1).
56.
Фибриноидное набухание — глубокая и необратимаядезорганизация СТ - распад белка (коллагена, фибронектина,
ламинина) и деполимеризация гликозаминогликанов →
деструкция основного вещества и волокон → резкое повышение
сосудистой проницаемости и образованием фибриноида.
Фибриноид — сложное вещество, образованное за счёт белков и
полисахаридов, распадающихся коллагеновых волокон, а также
плазменных белков крови и нуклеопротеидов разрушенных
клеток соединительной ткани. Обязательным компонентом
фибриноида является фибрин.
Микроскопически пучки коллагеновых волокон становятся
гомогенными, эозинофильными (вследствие блокирования
кислотных радикалов гликозаминогликанов плазменными
белками и смещения pH среды в щелочную сторону), резко ШИКпозитивными, что свидетельствует о значительном увеличении в
них количества гликопротеидов. Окраска на фибрин всегда
положительная, но интенсивность её колеблется. Метахромазия
при окрашивании толуидиновым синим отсутствует. Это связано
с практически полной деструкцией гликозаминогликанов.
Макроскопически органы и ткани, в которых развивается
фибриноидное набухание, мало изменены.
57.
Фибриноидное набухание может носить системный(распространённый) или локальный (местный)
характер.
Системное поражение - при инфекционноаллергических заболеваниях (фибриноид сосудов при
туберкулёзе с гиперергическими реакциями);
аллергических и аутоиммунных болезнях
(ревматические болезни, гломерулонефрит);
ангионевротических реакциях (фибриноид артериол
при гипертонической болезни и артериальных
гипертензиях).
При гипертонической болезни - гиалиноз мелких
артериол и артерий в связи с плазматическим
пропитыванием и фибриноидным набуханием
сосудистой стенки при длительном спазме,
сопровождающимся гипоксическим повреждением
структур сосудистой стенки.
Локально фибриноид выявляется при хроническом
воспалении. Например, в дне хронической язвы
желудка, трофических язв кожи.
58.
В исходе фибриноидного набухания может развиватьсяфибриноидный некроз, характеризующийся полной
деструкцией СТ. Вокруг очагов некроза обычно
выражена реакция макрофагов. В дальнейшем
происходит замещение очага деструкции рубцовой СТ
(склероз) или гиалиноз.
Фибриноидное набухание ведёт к нарушению и
прекращению функции органа (острая почечная
недостаточность при злокачественной гипертонии,
которая характеризуется фибриноидными изменениями
и некрозом артериол и капилляров клубочков).
Развивающиеся в исходе фибриноидного некроза
склероз или гиалиноз ведут к нарушению функции
клапанов сердца (формированию пороков сердца),
неподвижности суставов, сужению просвета и
уменьшению эластичности стенок сосудов.
При фибриноидном набухании и гиалинозе функция
органа снижается или может прекращаться. Гиалиноз
сосудов приводит к сужению просвета, гипоксии,
атрофии и склерозу органов. Гиалиноз клапанов лежит
в основе ревматического порока сердца.
59.
Необратимые повреждения клеткиНекроз (nekros - мертвый) – гибель клеток,
тканей или целых органов в живом
организме, механизмы которой связаны с
денатурацией белка и активацией
гидролитических ферментов, ведущей к
аутолизу.
60.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ НЕКРОЗАВыделяют пять основных этиологических
факторов некроза:
травматический;
токсический;
трофоневротический;
аллергический;
сосудистый.
По механизму действия этиологического
фактора некроз может быть прямым
(травматическим, токсическим) и непрямым
(трофоневротическим, аллергическим и
сосудистым).
61.
Травматический некроз - результат прямогодействия на ткань физических
(механических, температурных,
вибрационных, радиационных), химических
(кислот, щелочей) факторов.
Токсический некроз развивается при
действии на ткани токсических факторов
бактериальной и другой природы.
Трофоневротический некроз развивается при
нарушении циркуляции и иннервации тканей
при заболеваниях центральной и
периферической нервной системы.
Примером трофоневротического некроза
могут служить пролежни.
62.
Аллергический некроз - результат иммунногоцитолиза тканей в ходе реакций
гиперчувствительности немедленного и
замедленного типа. Классическим примером
аллергического некроза при реакциях немедленного
типа с участием иммунных комплексов, содержащих
комплемент, может служить фибриноидный некроз
при феномене Артюса. Иммунный цитолиз с
участием T-лимфоцитов киллеров и макрофагов
приводит к развитию некроза ткани печени при
хроническом активном гепатите.
63.
Сосудистый некроз связан с абсолютной илиотносительной недостаточностью
циркуляции в артериях, венах и
лимфатических сосудах. Наиболее часто он
обусловлен нарушением кровообращения в
артериях в связи с их тромбозом, эмболией,
длительным спазмом, а также с
функциональным перенапряжением органа в
условиях гипоксии. Недостаточная
циркуляция крови вызывает ишемию тканей,
гипоксию и развитие ишемического некроза.
64.
Помимо особенностей этиологии механизмы некрозаво многом зависят от структурно-функционального
своеобразия клеток, тканей и органов.
Конечный результат всех механизмов некроза —
возникновение внутриклеточного хаоса. Вместе с тем
можно выделить пять наиболее значимых
патогенетических механизмов некроза.
65.
5 наиболее значимых патогенетических механизмовнекроза:
Связывание клеточных белков с убиквитином
Убиквитин — белок, состоящий из 76 аминокислотных
оснований, широко распространён во всех клетках эукариотов,
в присутствии АТФ формирует ковалентные связи с
лизиновыми основаниями других белков.
Синтез убиквитина инициируется различными
видами повреждений. Связываясь с белками,
убиквитин уменьшает длительность их жизни путём
их частичной денатурации. Так, при некрозе клеток
центральной нервной системы при болезнях
Альцгеймера, Паркинсона, а также в гепатоцитах при
алкогольном поражении печени (тельца Мэллори)
обнаруживаются цитоплазматические тельца,
построенные из комплекса белков с убиквитином;
дефицит АТФ;
генерация активных форм кислорода;
нарушение кальциевого гомеостаза;
потеря селективной проницаемости клеточными
мембранами
66.
Некротический процесс структурно(морфогенетически) проходит через стадии
паранекроза, некробиоза, смерти клетки, аутолиза.
Паранекроз - изменения подобные некротическим, но
обратимые (?).
Некробиоз - необратимые дистрофические
изменения, характеризующиеся преобладанием
катаболических реакций над анаболическими.
Смерть клетки - время установить трудно.
Аутолиз - разложение мёртвого субстрата под
действием гидролитических ферментов погибших
клеток и клеток воспалительного инфильтрата. Для
работы гидролитических ферментов необходимы
кислые значения pH.
При чрезмерно высоком pH преобладает
денатурация белков и развивается коагуляционный
некроз.
67.
Каким клиническим признакам соответствуетнеобратимое повреждение клеток?
Необратимое повреждение клеток приводит к потере
выполняемой ими функции. Например:
повреждение клеток миокарда — прекращение сердечных
сокращений.
Повреждение мотонейронов — паралич скелетной мускулатуры.
Повреждение клеток островков Лангерганса— сахарный диабет с
повышением концентрации глюкозы в крови.
Существуют ли лабораторные признаки повреждения
клеток, используемые в клинической практике?
Тяжелое повреждение клеток обычно сопровождается
поступлением в кровь их цитоплазматических ферментов,
например:
Креатинкиназы (креатинфосфокиназы) — при повреждение
сердечной мышцы или скелетной мускулатуры.
Аспартатаминотрансфераза и аланинаминотрансфераза
высвобождаются из поврежденных клеток печени.
Лактатдегидрогеназа выбрасывается из поврежденных
эритроцитов и многих других клеток
68.
Ранняя диагностика некрозаопределение этих ферментов биохимическими
методами
электрофизиологические методы
специальные морфологические методы
(макроскопическая диагностика с теллуритом
калия или солями тетразолия, ШИК-реакция),
светооптически изменения становятся
видимыми на стадии аутолиза.
69.
Необратимые изменения клеткиморфологически ассоциируются с
разрушением ее мембран, обусловленным
как действием фосфолипаз и уменьшением
фосфолипидов в клеточной мембране, так и
активацией протеаз, повреждающих
цитоскелет, в норме выполняющий роль
якоря между плазматической мембраной и
внутренним содержимым клетки.
70.
Макроскопические проявлениянекроза зависят от своеобразия
органа, в котором он возникает, и
характера повреждающего фактора.
Микроскопические проявления
выражаются в изменении ядер
(кариопикноз, кариорексис,
кариолизис) и цитоплазмы (разрыв
цитоплазматической мембраны,
коагуляция и лизис цитоплазмы).
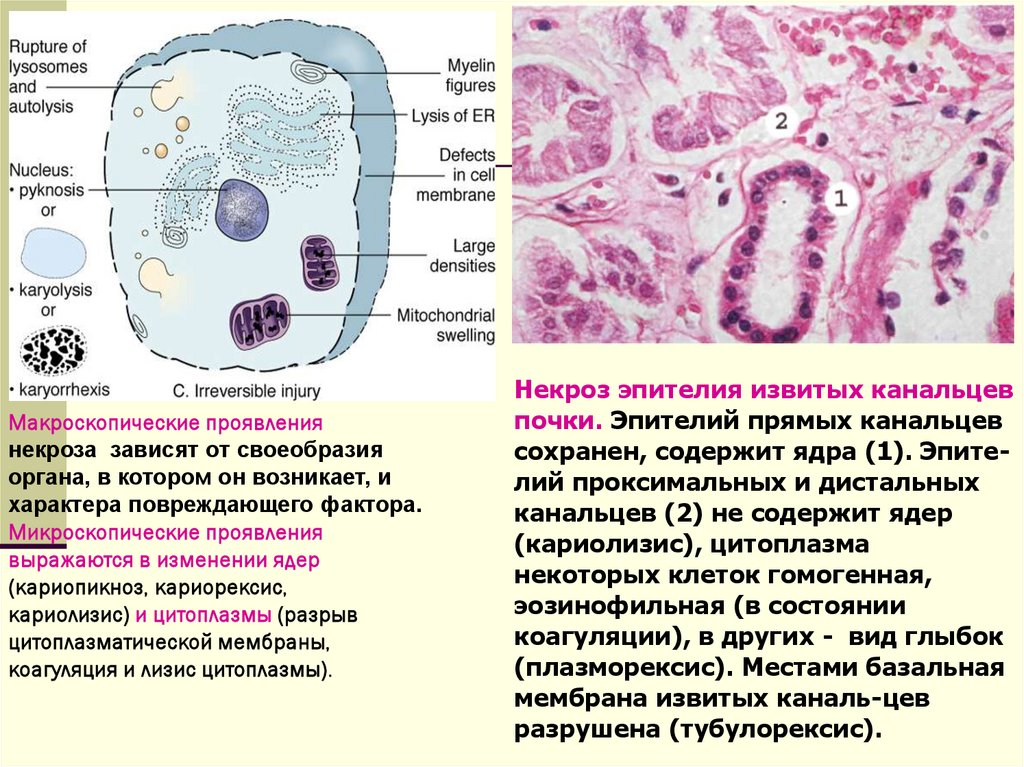
Некроз эпителия извитых канальцев
почки. Эпителий прямых канальцев
сохранен, содержит ядра (1). Эпителий проксимальных и дистальных
канальцев (2) не содержит ядер
(кариолизис), цитоплазма
некоторых клеток гомогенная,
эозинофильная (в состоянии
коагуляции), в других - вид глыбок
(плазморексис). Местами базальная
мембрана извитых каналь-цев
разрушена (тубулорексис).
71.
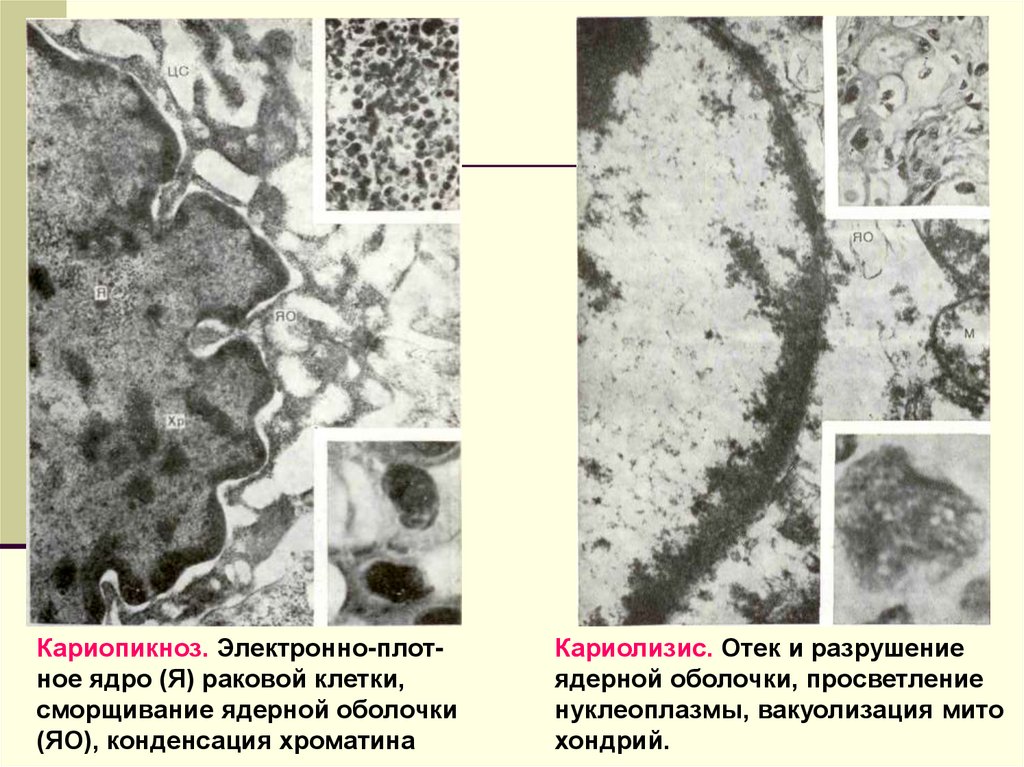
Кариопикноз. Электронно-плотное ядро (Я) раковой клетки,сморщивание ядерной оболочки
(ЯО), конденсация хроматина
Кариолизис. Отек и разрушение
ядерной оболочки, просветление
нуклеоплазмы, вакуолизация мито
хондрий.
72.
Изменения цитоплазмыплазмокоагуляция - денатурация и коагуляция
белков цитоплазмы. Светооптически цитоплазма
становится гомогенной, нарастает ее ацидофилия
(розовый цвет при окраске эозином) вследствие
коагуляции цитоплазматических белков и утраты
рибосом.
плазморексис - распад цитоплазмы на глыбки
плазмолизис - расплавление цитоплазмы вследствие
воздействия ферментов лизосом (колликвация,
аутолиз).
73.
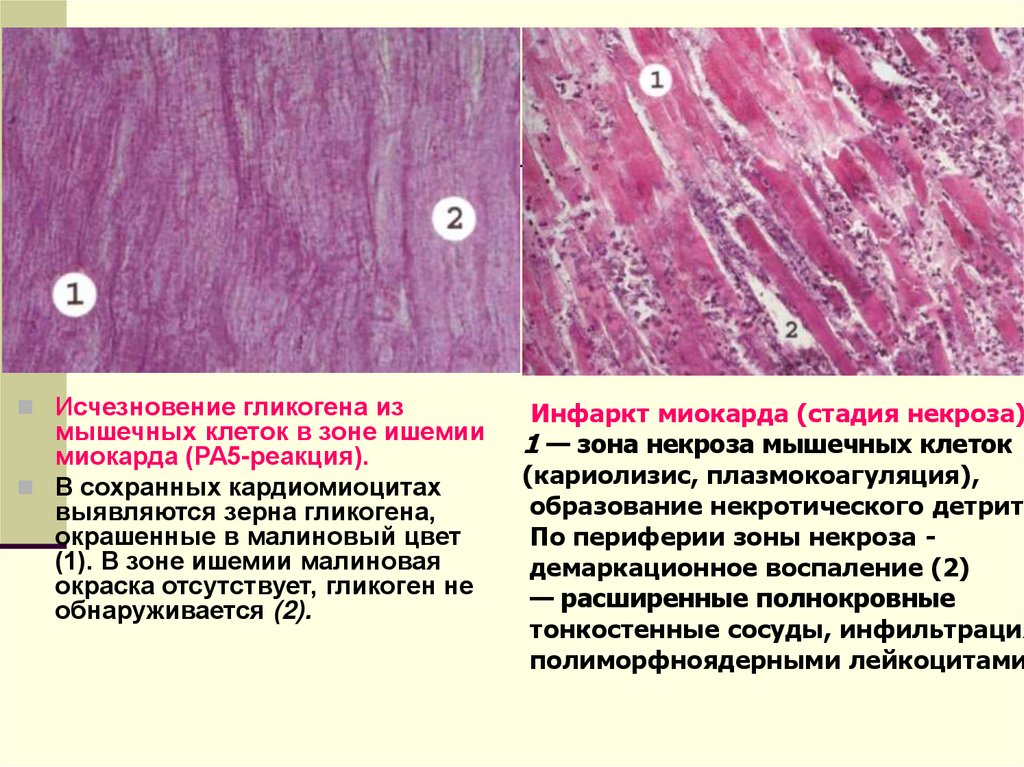
Исчезновение гликогена измышечных клеток в зоне ишемии
миокарда (РА5-реакция).
В сохранных кардиомиоцитах
выявляются зерна гликогена,
окрашенные в малиновый цвет
(1). В зоне ишемии малиновая
окраска отсутствует, гликоген не
обнаруживается (2).
Инфаркт миокарда (стадия некроза)
1 — зона некроза мышечных клеток
(кариолизис, плазмокоагуляция),
образование некротического детрита
По периферии зоны некроза демаркационное воспаление (2)
— расширенные полнокровные
тонкостенные сосуды, инфильтрация
полиморфноядерными лейкоцитами
74.
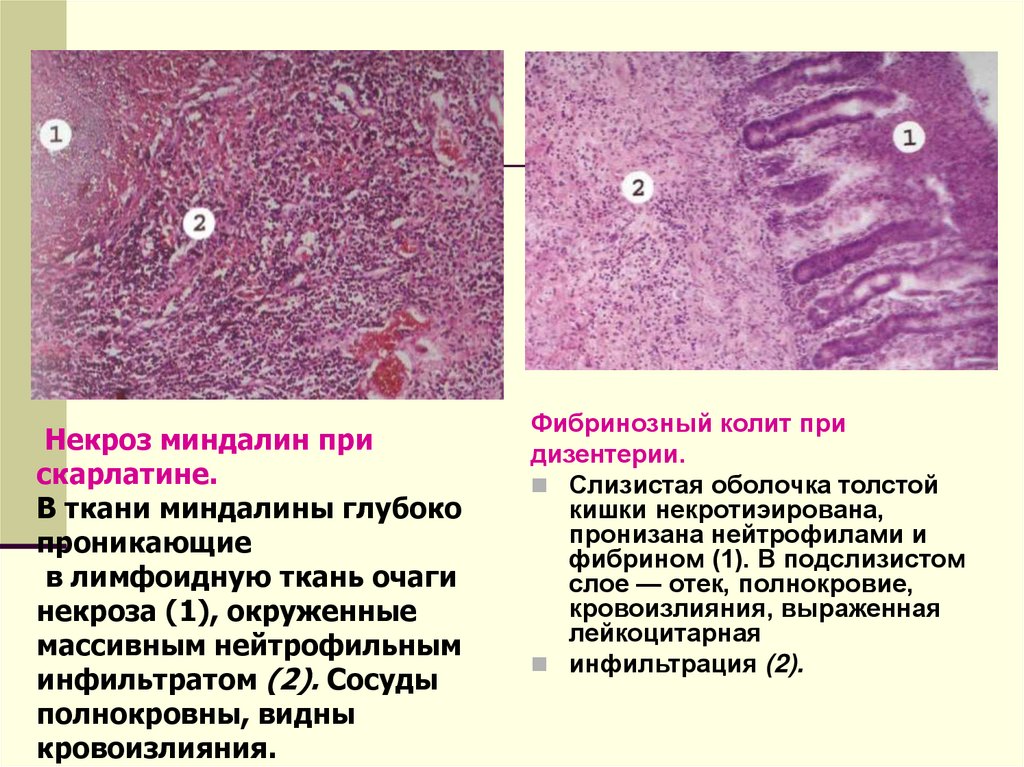
Некроз миндалин прискарлатине.
В ткани миндалины глубоко
проникающие
в лимфоидную ткань очаги
некроза (1), окруженные
массивным нейтрофильным
инфильтратом (2). Сосуды
полнокровны, видны
кровоизлияния.
Фибринозный колит при
дизентерии.
Слизистая оболочка толстой
кишки некротиэирована,
пронизана нейтрофилами и
фибрином (1). В подслизистом
слое — отек, полнокровие,
кровоизлияния, выраженная
лейкоцитарная
инфильтрация (2).
75.
Изменения межклеточного веществавыражаются в деструкции коллагеновых,
эластических и ретикулярных волокон,
превращающихся в плотные гомогенные
розовые массы, которые могут
пропитываться фибрином с развитием
фибриноидного некроза.
76.
Коагуляционный некроз – погибшие клеткисохраняют свои очертания в течение
нескольких дней. Коагуляция
цитоплазматических белков придает им
устойчивость к действию лизосомальных
ферментов.
Коагуляционный некроз характерен для органов
богатых белками и бедных жидкостью
(миокард, почки, надпочечники, селезенка,
печень)
77.
Инфаркт - сосудистый (ишемический) некрозвнутренних органов (самый частый вид
некроза).
Казеозный (творожистый) некроз развивается при
туберкулезе, сифилисе, лепре, грануломатозе,
выявляется как сухой, крошащийся
ограниченный участок ткани беловато-желтого
цвета, гибнет и паренхима, и строма.
78.
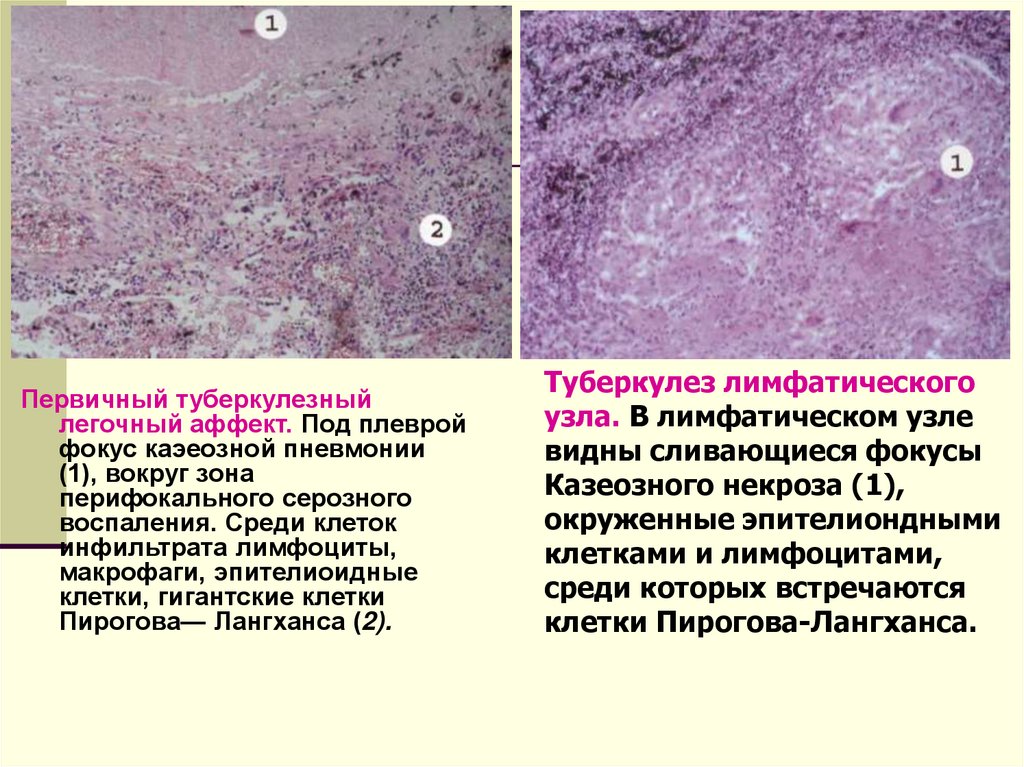
Первичный туберкулезныйлегочный аффект. Под плеврой
фокус каэеозной пневмонии
(1), вокруг зона
перифокального серозного
воспаления. Среди клеток
инфильтрата лимфоциты,
макрофаги, эпителиоидные
клетки, гигантские клетки
Пирогова— Лангханса (2).
Туберкулез лимфатического
узла. В лимфатическом узле
видны сливающиеся фокусы
Казеозного некроза (1),
окруженные эпителиондными
клетками и лимфоцитами,
среди которых встречаются
клетки Пирогова-Лангханса.
79.
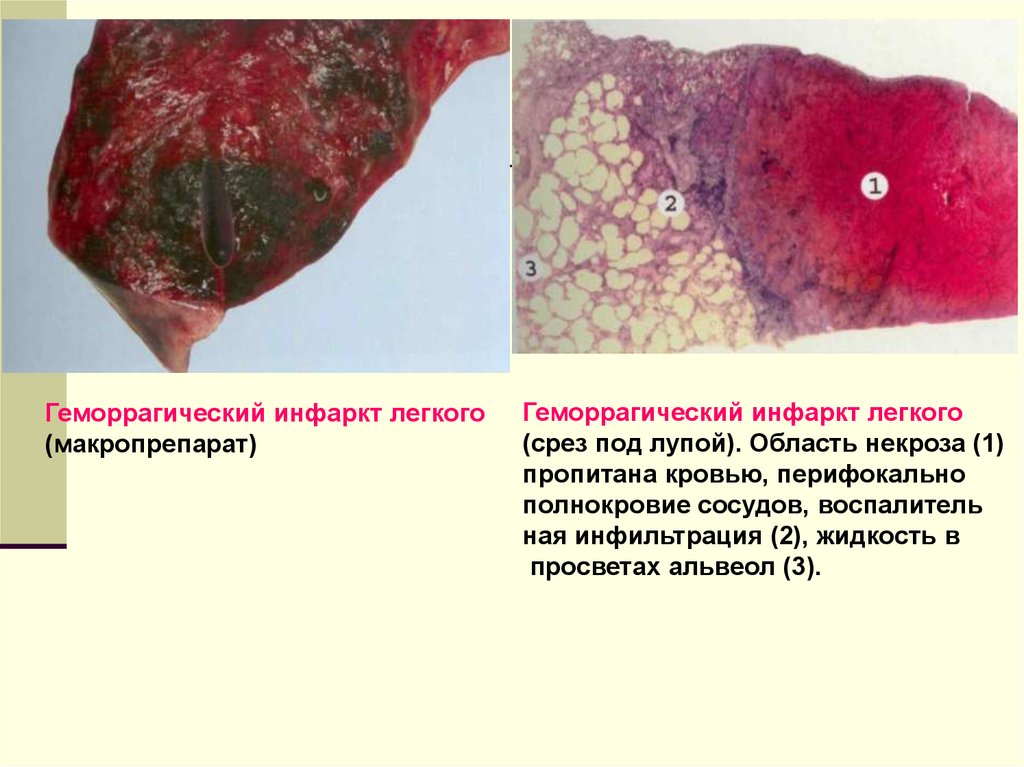
Геморрагический инфаркт легкого(макропрепарат)
Геморрагический инфаркт легкого
(срез под лупой). Область некроза (1)
пропитана кровью, перифокально
полнокровие сосудов, воспалитель
ная инфильтрация (2), жидкость в
просветах альвеол (3).
80.
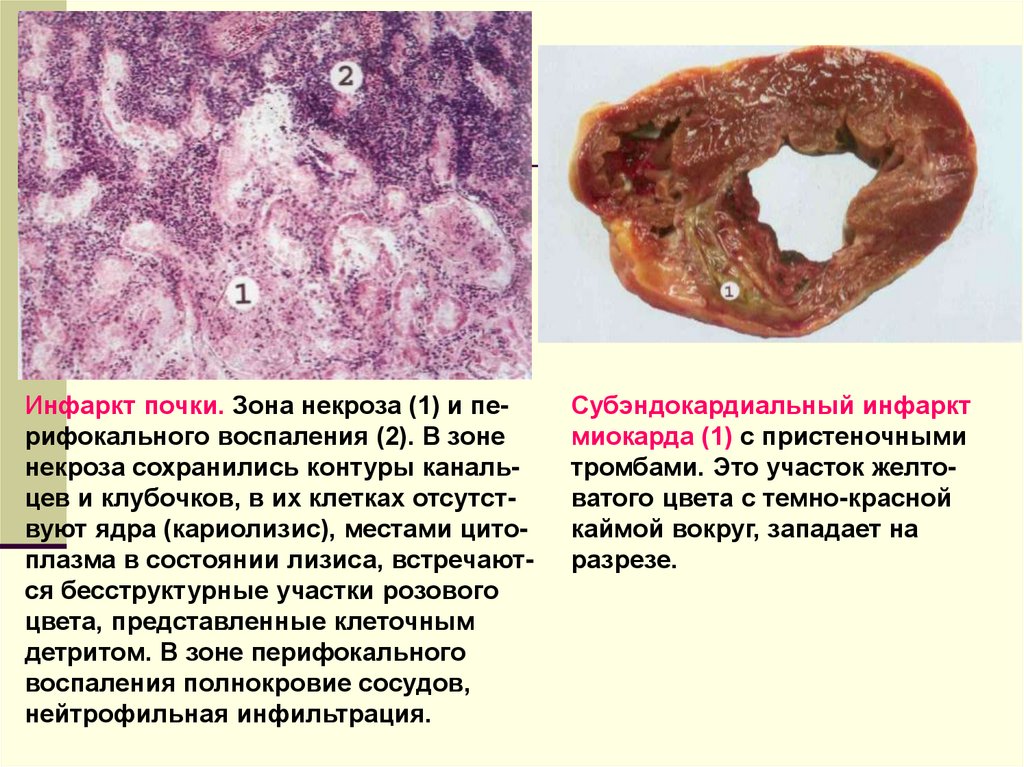
Инфаркт почки. Зона некроза (1) и перифокального воспаления (2). В зоненекроза сохранились контуры канальцев и клубочков, в их клетках отсутствуют ядра (кариолизис), местами цитоплазма в состоянии лизиса, встречаются бесструктурные участки розового
цвета, представленные клеточным
детритом. В зоне перифокального
воспаления полнокровие сосудов,
нейтрофильная инфильтрация.
Субэндокардиальный инфаркт
миокарда (1) с пристеночными
тромбами. Это участок желтоватого цвета с темно-красной
каймой вокруг, западает на
разрезе.
81.
Жировой (ферментный) некроз (стеатонекроз) – чащеразвивается при остром панкреатите и повреждениях
поджелудочной железы, обусловлен действием
панкреатической липазы на триглицериды жировых клеток с
расщеплением их на глицерин и жирные кислоты,
образующие при взаимодействии с кальцием плазмы мыла.
Выявляется в ткани железы в виде белых непрозрачных
бляшек. Неферментный жировой некроз (подкожная жировая
ткань, молочная железа, брюшная полость) чаще обусловлен
травмой.
82.
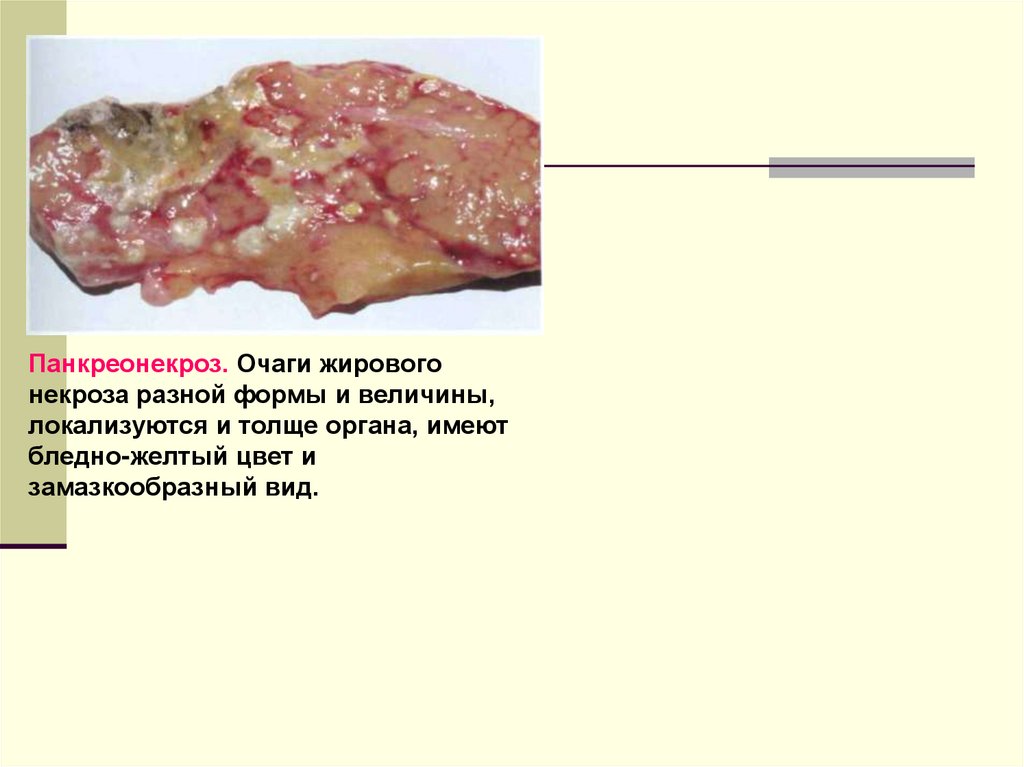
Панкреонекроз. Очаги жировогонекроза разной формы и величины,
локализуются и толще органа, имеют
бледно-желтый цвет и
замазкообразный вид.
83.
Гангрена — некроз черного или темнокоричневого цвета, развивающийся в тканях,прямо или через анатомические отверстия
соприкасающиеся с внешней средой.
Различают сухую, влажную гангрену и
пролежень.
84.
Сухая гангрена - некроз тканей, соприкасающихся свнешней средой, протекающий без участия
микроорганизмов, имеет выраженную зону
демаркационного воспаления, часто возникает в
нижних конечностях (поражение сосудов, ожог,
отморожение).
Влажная гангрена развивается при наслоении на
некротические изменения ткани бактериальной
инфекции, обусловливающей вторичную
колликвацию – лизис погибших клеток
бактериальными ферментами (может встречаться в
конечностях, но чаще во внутренних органах –
кишечник, легкие, мягкие ткани). Участок некроза
вследствие присоединившегося воспаления отекает,
демаркационная зона не определяется.
85.
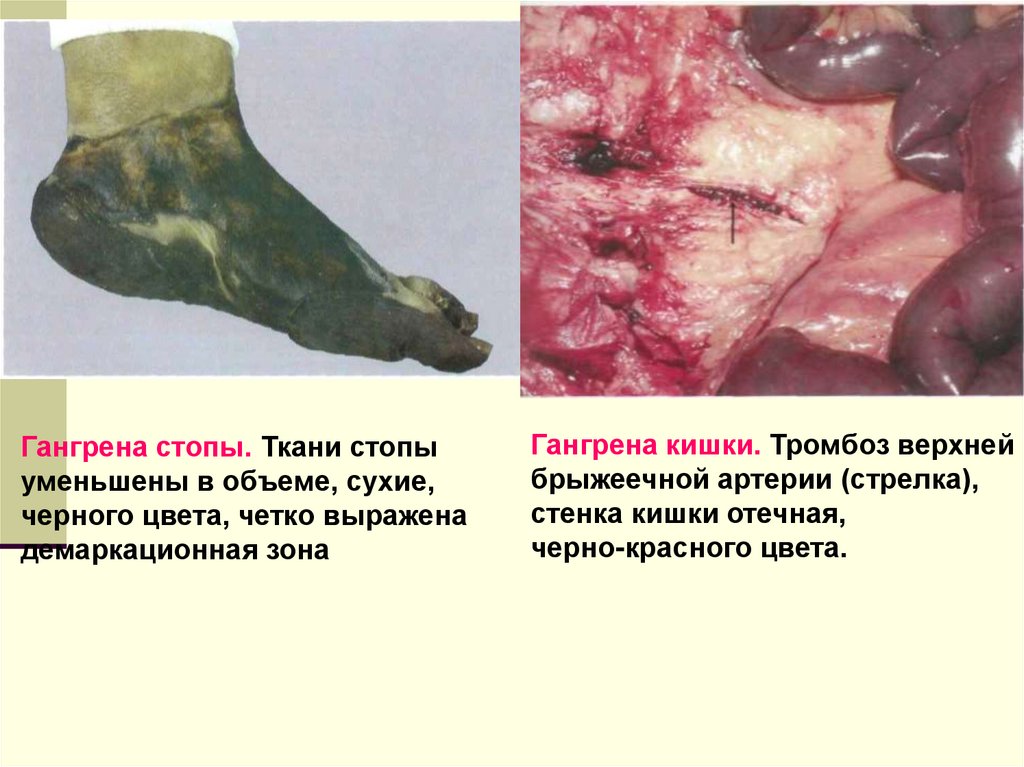
Гангрена стопы. Ткани стопыуменьшены в объеме, сухие,
черного цвета, четко выражена
демаркационная зона
Гангрена кишки. Тромбоз верхней
брыжеечной артерии (стрелка),
стенка кишки отечная,
черно-красного цвета.
86.
Пролежень (разновидность гангрены) –омертвление поверхностных участков тела
(кожи, мягких тканей), возникает вследствие
трофоневротических нарушений на участках,
подвергающихся сдавлению.
87.
Газовая гангрена развивается приинфицировании погибших тканей анаэробной
флорой (группа клостридий).
Характеризуется обширным некрозом ткани и
образованием газов в результате
ферментативной деятельности бактерий.
88.
Колликвационный (влажный) некрозхарактеризуется расплавлением
некротизированной ткани в результате
аутолиза, характерен для тканей бедных
белками и богатых жидкостью (ишемический
инфаркт головного мозга является примером
колликвационного некроза)
89.
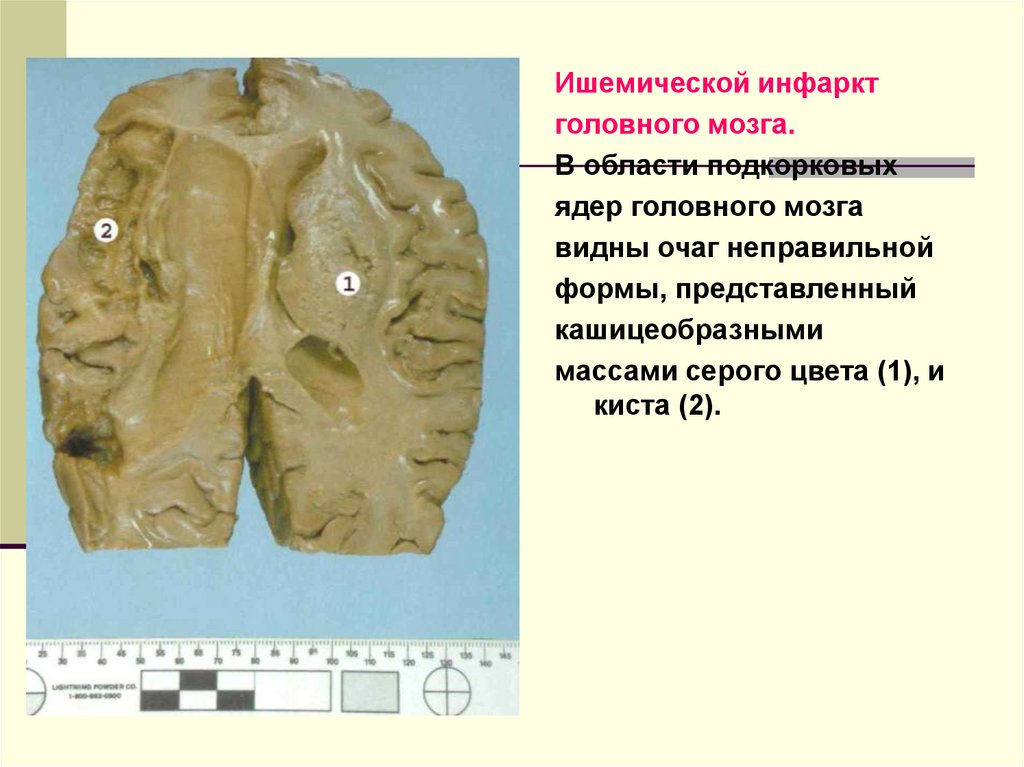
Ишемической инфарктголовного мозга.
В области подкорковых
ядер головного мозга
видны очаг неправильной
формы, представленный
кашицеобразными
массами серого цвета (1), и
киста (2).
90.
Исходы некроза зависят от условий еговозникновения и развития.
Мелкоочаговые некрозы при благоприятных
условиях подвергаются расплавлению,
резорбции и замещаются исходной тканью
(печень, костный мозг, СО желудочнокишечного тракта, поверхностные некрозы
кожи – клеточный тип регенерации).
91.
В органах и тканях, где регенерация протекаетпо внутриклеточному механизму (сердце,
головной мозг) на месте некроза разрастается
соединительная ткань с образованием рубца,
полости или кисты (головной мозг).
Если полного рассасывания некротизированной ткани не происходит, то она может
подвергнуться инкапсуляции (отграничению)
или отделиться от живой ткани
(секвестрация).
92.
При некрозах в соединительной ткани могутнаблюдаться различные процессы –
гиалиноз, отложение солей кальция,
оссификация.
93.
При некрозе во внутренних органах, имеющихестественные выводные протоки, остатки
мертвой ткани могут выходить наружу, а на
их месте образуются полости. Возможно
образование искусственных протоков –
свищей.
Подвергшиеся некрозу наружные части тела
могут отторгаться (мутиляция).
94.
Апоптоз – («запрограммированная гибельклетки») генетически опосредованная гибель
клетки. Как физиологический
процесс апоптоз обеспечивает регуляцию
численности клеточных популяций в
эмбриогенезе и постнатальном морфогенезе,
участвует в сохранении тканевого
гомеостаза, защите организма от
мутировавших или поврежденных клеток.
95.
Вместе с тем, апоптоз может быть патогенетическимфактором различных заболеваний:
сердечно-сосудистой системы (дилатационная и
ишемическая кардиомиопатия, аритмии различного
генеза)
слизистых оболочек (атрофический гастрит,
целиакия, или глютеновая энтеропатия)
нейродегенеративных и аутоиммунных заболеваний
(при последних имеет место подавление апоптоза
аутореактивных иммунных клеток (Т-супрессоров) и
усиленная программированная гибель клеток в
тканях-мишенях.
96.
Апоптоз осуществляется посредствомгенетической программы при запуске каскада
реакций с участием специализированных
внутриклеточных протеаз из семейства
каспаз и морфологически выражается в
фрагментации клеток на окруженные
мембраной тельца, подвергающиеся
фагоцитозу без сопутствующих
воспалительной реакции и склероза.
97.
98.
Как процесс, осуществляемый посредствомгенетической программы, апоптоз нуждается
в синтезе РНК, белка и характеризуется
специфическими структурнофункциональными изменениями
митохондрий.
Процесс контролируется экспрессией ряда
генов, связанных с клеточной
пролиферацией и канцерогенезом.
99.
Для поддержания тканевого гомеостазанеобходим определенный баланс между
активностью механизмов, контролирующих
клеточный цикл, или
процессы пролиферации, и механизмами
блокады-индукции апоптоза. Регуляция этих
процессов имеет общие молекулярногенетические механизмы.
100.
Апоптоз может быть вызван различнымистимулами. Эндогенные стимулы апоптоза
действуют через рецепторы, не оказывая
выраженного токсического или
деструктивного эффекта, так как
регулируются системой клеточного
(тканевого) гомеостаза, или эволюционно
детерминированной нормы.
101.
Стимуляторы (промоторы) апоптоза:Эндогенные:
- дефицит ФР, повреждения ДНК, нарушения цитоскелета, потеря
контакта клеток друг с другом и матриксом, нарушение осмотического
равновесия
- ↑ продукции ФНОα, ↑ внутриклеточного Са++, ↑ оксида азота (NO) и
его синтетазы (NOS III), цитокины воспаления, гормоны
- активация Bad, bax, bik, bid, Bclxs, р38, ангиотензин II
- ↑ пролиферации
Экзогенные:
- ионизирующее и др. виды излучения, ↑ tº, ишемия, гипоксия,
вирусные и бактериальные инфекции, гормоны, лечебные факторы,
активация свободно-радикального окисления (окислительный
стресс)
Ингибиторы (протекторы) апоптоза:
-активация Bcl 2, Bcl XL, репрессия или мутация р53,
некоторые лекарственные препараты (ингибиторы АПФ,
антиоксиданты)
102.
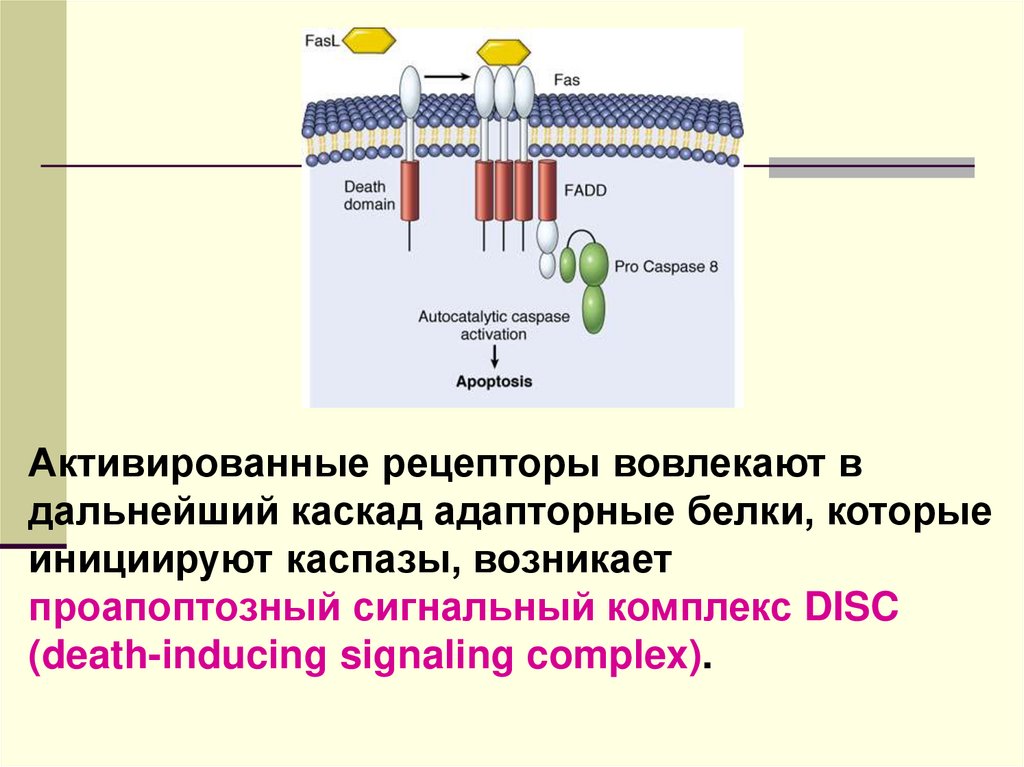
Экстрацеллюлярный запуск апоптоза осуществляетсяпосредством взаимодействия специфических
лигандов с соответствующими рецепторами с
последующей передачей сигналов из микроокружения
в интрацеллюлярное пространство и активацией
программы апоптоза. К таким лигандам («лигандам
смерти») относятся ФНОα (TNFα), лимфотоксин, Fasлиганд, Apo3-лиганд и TRAIL (related apoptosisinducing ligand), а к рецепторам – рецептор ФНО1, Fasрецепторы, рецепторы смерти DR 3,4, 5 и 6 (death
receptor).
103.
Активированные рецепторы вовлекают вдальнейший каскад адапторные белки, которые
инициируют каспазы, возникает
проапоптозный сигнальный комплекс DISC
(death-inducing signaling complex).
104.
Интрацеллюлярный путь апоптоза активируется черезспецифические процессоры, находящиеся в
клеточном ядре или цитоплазме. Этот путь приводит к
формированию другого комплекса называемого
апоптосомой.
Внутренний и внешний пути активации апоптоза на
определенном этапе перекрещиваются и сливаются в
единую последовательность при активации
эффекторного звена, представленного
специфическими протеолитическими ферментами –
каспазами.
105.
Запуск апоптоза возможен через:1. активацию Fas-рецепторов и каскада каспаз –
протеинов, расположенных в ядре,
цитоплазме, на различных мембранах и
образующих наборы регуляторных пептидов,
которые сами по себе или через каскад новых
реакций осуществляют аутокринную
регуляцию апоптоза, контролируя баланс
многочисленных проапоптотических и
антиапоптотических протеинов;
106.
2.митохондриальные активаторы апоптоза –гены семейства Bcl2 (Bax, Bad, Bid и др.):
1). выход в цитоплазму цитохрома С → Apaf-1
→ каскад каспаз → апоптоз;
2). выход в цитоплазму AIF → ядро→ апоптоз.
107.
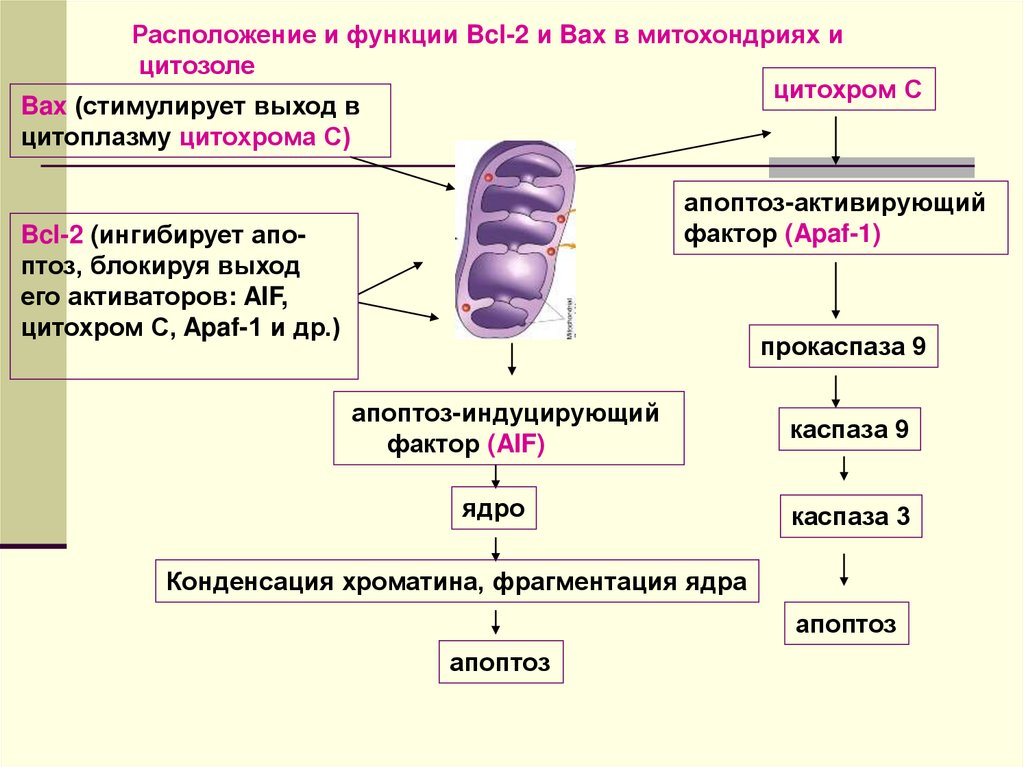
Расположение и функции Bcl-2 и Bax в митохондриях ицитозоле
цитохром С
Bax (стимулирует выход в
цитоплазму цитохрома С)
апоптоз-активирующий
фактор (Apaf-1)
Bcl-2 (ингибирует апоптоз, блокируя выход
его активаторов: AIF,
цитохром С, Apaf-1 и др.)
прокаспаза 9
апоптоз-индуцирующий
фактор (AIF)
каспаза 9
ядро
каспаза 3
Конденсация хроматина, фрагментация ядра
апоптоз
апоптоз
108.
Морфологические особенности апоптоза(фазы - начальная, эффекторная, деградации; продолжительность 12-24 часа)
конденсация и маргинальное расположение
хроматина (конец 1-го часа), фрагментация ядра
клетка округляется, утрачивает микроворсинки и
контакты с соседними клетками, отделяется от них, на
поверхности появляются пузырьковидные выросты
(через 4-5 часов), затем клетка уплотняется или
сморщивается
распад клеток на апоптозные тельца или они
спадаются в единичные округлые плотные
апоптозные тельца
фагоцитоз апоптозных тел
109.
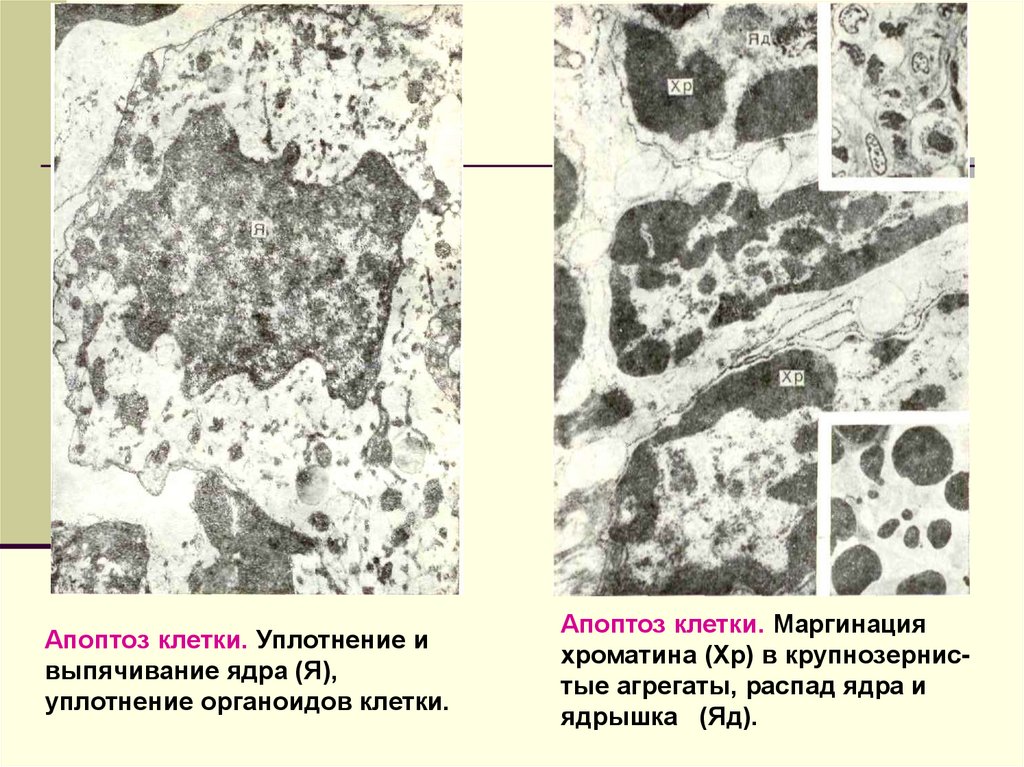
Апоптоз клетки. Уплотнение ивыпячивание ядра (Я),
уплотнение органоидов клетки.
Апоптоз клетки. Маргинация
хроматина (Хр) в крупнозернистые агрегаты, распад ядра и
ядрышка (Яд).
110.
Существуют гены (или комплекс молекулярных путей),интегрирующие правильное восприятие клеткой
средовых сигналов и соответствующих ответов на
них. Мутации на этих путях могут вести к опухолевой
прогрессии.
Такую интегральную функцию выполняет ген р53,
который называют «хранителем генома» (guardian of
the genom), а также супрессором опухолей, так как он
осуществляет остановку клеточного цикла при
неблагоприятных для клетки условиях.
111.
В норме при получении информации о поврежденииДНК р53 запускает каскад реакций,
приводящих к остановке клеточного цикла.
Р53 → активация транскрипции р21 → блокада
Cdk (циклинзависимая киназа)→ ингибирование
фосфорилирования Pb → остановка прогрессии
клеточного цикла.
После завершения репарации ДНК блок
снимается и клеточное деление может продолжаться.
112.
При мутации р53 этот блок не развиваетсяи клетки с поврежденной и нерепарированной ДНК входят в клеточный цикл. Мутации
р53 дестабилизируют геном клетки.
В норме, если повреждение ДНК необратимо, белок р53 направляет клетку на путь
апоптоза. Основные механизмы проапоптотической функции р53 заключаются в активации гена р21, проапоптозных белков Bax и
Fas и подавлении экспрессии ингибитора
апоптоза Bcl2.
113.
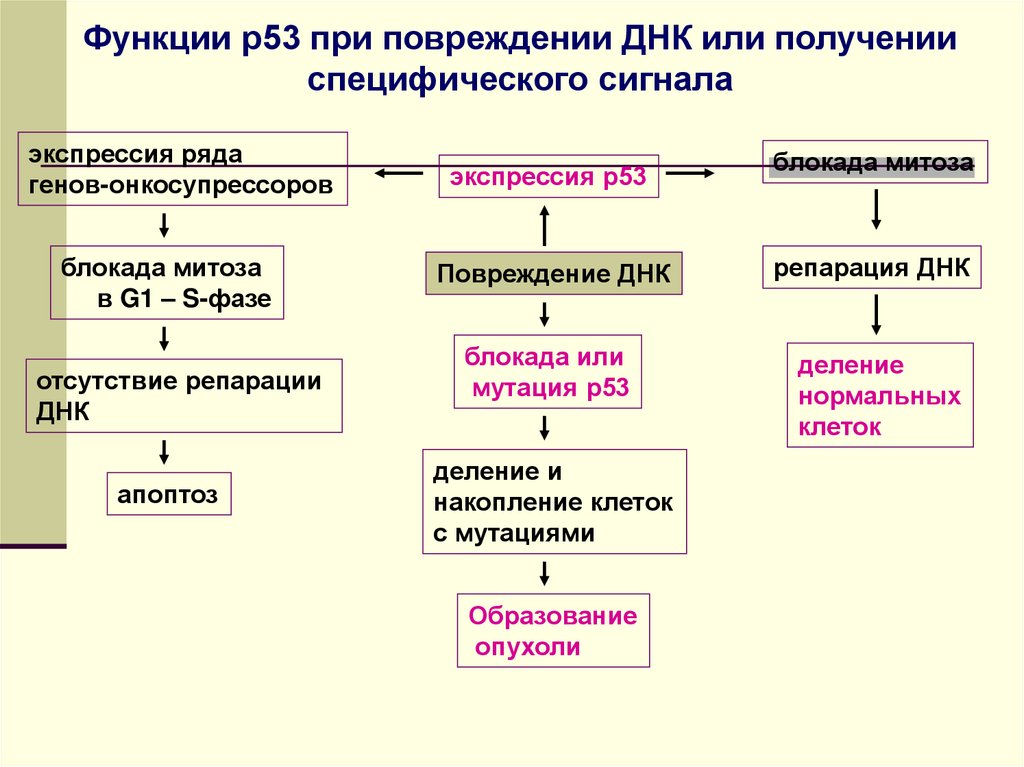
Функции р53 при повреждении ДНК или полученииспецифического сигнала
экспрессия ряда
генов-онкосупрессоров
экспрессия р53
блокада митоза
в G1 – S-фазе
Повреждение ДНК
репарация ДНК
блокада или
мутация р53
деление
нормальных
клеток
отсутствие репарации
ДНК
апоптоз
деление и
накопление клеток
с мутациями
Образование
опухоли
блокада митоза
114.
Дополнительно. Типы клеточной гибелиВ 90-е годы был создан номенклатурный комитет по клеточной гибели – the
Nomenclature Committee on cell death – NCCD. В основу классификации
могут быть положены морфологические проявления гибели клетки
- некроз
- апоптоз
- аутофагия
- кератинизация, или ороговение
Биохимические критерии
- без или с участием нуклеаз или определенных классов протеаз – каспаз,
кальпаинов, катепсинов, трансглутаминазы
Функциональные критерии
- запрограммированная или спонтанная, физиологическая или
патологическая
Иммунный аспект
- иммуногенная, неиммуногенная гибель
115.
Процесс, в который вовлекаются погибающие клетки,является обратимым до тех пор пока они не входят в
первую необратимую фазу или не минуют точку
необратимости- это
-массированная активация каспаз
Утрата митохондриального трансмембранного
потенциала
Полная проницаемость внешней мембраны митохондрий
или экспонирование на внешней стороне цитолеммы
фосфатидилсериновых остатков (PS), имитирующих
сигнал «съешь меня» для расположенных рядом
нормальных клеток или макрофагов.
Вместе с тем, даже наличие этих критериев может не
сопровождаться гибелью клетки.
116.
Атипичные формы клеточной гибели117.
Slide 1.15W.B. Saunders Company items and derived
items Copyright (c) 1999 by W.B. Saunders
Company
118.
119.
120.
Каковы гистологические признаки некроза?Микроскопические признаки некроза
выявляются лишь на поздних стадиях
процесса на фоне аутолиза.
Ранние стадии некроза можно
диагностировать лишь с использованием
специальных методов, таких как
поляризационная и люминесцентная
микроскопия с окраской акридиновым
оранжевым, гистохимических методик (ШИКреакция) и электронной микроскопии.
Ультраструктурные признаки некроза
отражают изменения в ядре, митохондриях,
полисомах, лизосомах, цитоплазматической
сети клетки.
121.
Каковы гистологические признаки некроза?Ядро - агрегация хроматина, фрагментация фибрилл,
полное разрушение.
Митохондрии - набухание, уменьшение плотности
матрикса, образование в нём агрегатов неправильной
формы, отложение солей кальция.
Цитоплазматическая сеть - набухание, фрагментация и
распад мембранных структур.
Полисомы и рибосомы - распад полисом, отделение
рибосом от поверхности цистерн эндоплазматической
системы, уменьшение размеров и количества
рибосом.
Лизосомы - агрегация гранул матрикса и его
просветление, разрыв мембран.
Цитоплазматический матрикс - исчезновение гранул
гликогена, снижение активности ферментов.
При местных изменениях говорят о фокальном
коагуляционном или колликвационном некрозах.
122.
Каковы гистологические признаки некроза?Микроскопические признаки некроза
Изменения ядер связаны с активацией
гидролаз (рибонуклеаз и дезоксирибонуклез).
Ядра последовательно подвергаются
сморщиванию (кариопикноз), распаду на
глыбки (кариорексис) и лизируются
(кариолизис).
В цитоплазме происходит денатурация и
коагуляция белков, сменяемая обычно
колликвацией. Коагуляция цитоплазмы,
сменяется распадом её на глыбки
(плазморексис) и лизисом органелл
(плазмолизис). При местных изменениях
говорят о фокальном коагуляционном или
колликвационном некрозах.
123.
Особенности отдельных видов повреждения1. гипоксическое или ишемическое повреждение
- ↓ окислительного фосфорилирования в м/х,
↓ или прекращение образования АТФ, переход на
аэробный гликолиз, образование АТФ из гликогена и
креатинфосфата → накопление в клетке молочной
кислоты и неорганического фосфата → ↓
внутриклеточного рН, конденсация ядерного
хроматина
- ↓ АТФ → строе набухание (отек) клетки
- ↓ работы АТФаз → поступление в клетку Na и Ca
- Набухание м/х, вакуолизация ЭПС → ↓ синтеза белка и
ферментов, отек лизосом → выход в цитоплазму
гидролаз → расщепление компонентов клетки
124.
2.повреждение клетки, вызванное вободнымирадикалами (молекулы кислорода, имеющие
непарный электрон на внешней орбите) →
перекисное окисление липидов мембран, образование
перекисей, инициирующих обширные повреждения
(цепь аутокаталитических реакций), окислительные
повреждения белков → разрушение ключевых
ферментов, содержащихся в цитозоле →поступление
в клетку Са и Na, повреждения ДНК
125.
3. Токсические поврежденияБольшинство химических соединений биологи-чески
неактивно и вначале превращается в токсичные
метаболиты, которые действуют на клетки мишени →
повреждения мембран клеток путем прямого ковалентного
связывания с мембранными белками и липидами; наиболее важно
образование свободных радикалов и последующее перекисное
окисление липидов мембран.
Часть водорастворимых соединений может действовать
непосредственно связываясь с некоторыми молекулами или
органеллами, например, хлорид ртути → связывание
сульфгидрильных групп клеточной мембраны → ↑ ее
проницаемости и торможение АТФаза-зависимого транспорта.
Цианиды действуют непосредственно на ферменты м/х
126.
Механизмы развития повреждения и смертиклетки
гипоксия ткани → активация фосфолипаз,
образование свободных радикалов → перекисное
окисление липидов → повреждение мембран → ↑
интрацеллюлярной концентрации кальция
утрата плазматической мембраной избирательной
проницаемости – постоянный признак всех видов
повреждения (↓ или прекращение синтеза АТФ →
проникновение ионов кальция в клетку → активация
фосфолипаз → повреждение мембраны
прямое повреждающее действие токсинов, ряда
физических и химических агентов → перекисное
окисление липидов → повреждение мембран)
127.
(механизмы развития повреждения и смерти клетки)нарушение гомеостаза кальция → ↑ концентрации Са2+
в цитоплазме → активация ферментов, повреждающих
клетку: фосфоли-паз (повреждение клеточных
мембран), протеаз (разрушение мембран и
цитоскелета), активация АТФаз (истощение запасов
АТФ) и эндонуклеаз (фрагментация хроматина)
снижение синтеза АТФ в митохондриях (→ нарушение
процессов синтеза белка, мембранного транспорта,
липогенеза и др. обменных процессов)
128.
Различают три основных видаповреждения клетки
гипоксическое
2. обусловленное свободными радикалами
3. токсическое
1.
129.
Гипоксия – патологический процесс, разви-вающийсявследствие недостаточности и/или неэффективности
биологического окисления и приводящий к снижению
или прекращению энергетического обеспече-ния
жизненно важных процессов.
Основные причины развития гипоксии
- ишемия (←уменьшение кровоснабжения ← спазм,
склероз, тромбоз сосудов)
- неадекватная оксигенация крови (болезни сердца,
легких)
- снижение способности крови к транспортировке О2
(анемия, отравление)
130.
Изменения метаболизма при гипоксии- дефицит макроэргов – прогрессивное ↓ содер-жания
АТФ и креатинфосфата при одновремен-ном
увеличении продуктов его распада - АДФ, АМФ и
неорганического фосфата
- преобладание гликолиза → падение содержа-ния
гликогена в клетках и ↑ концентрации лак-тата и
пирувата → развитие внутриклеточного
метаболического ацидоза →
- активация лизосомальных протеаз, ↑ распада и ↓
синтеза белков
- снижение выработки АТФ → нарушение работы
различных (K+/Na+ , Ca++-, Mg++-) АТФаз → нару-шение
ионного гомеостаза клетки - ↑ внутрикле-точного Ca + + ,
Na + и внеклеточного K+.
131.
Повышениевнутриклеточной
концентрации
Са++ ведет к
активации целого
ряда ферментов
- фосфолипаз →
повреждение
мембранных
структур
- протеаз →
разрушение
белков
цитоскелета
- эндонуклеаз
→фрагментация
хроматина и др.
132.
Дополнительные факторы гипоксическогоповреждения клетки
- ↓ рН
- накопление молочной кислоты в цитоплазме
(компенсаторно в условиях гипоксии клетка переходит
на анаэробный гликолиз) → ↑ осмо-тического
давления в клетке и увеличение ее отека (набухания)
- накопление воды в цитоплазме, митохондри-ях,
цистернах гранулярной ЭПС → отщепление от ее
мембран рибосом → снижение синтеза белка
- появление в цитоплазме клеток и внеклеточ-ном
пространстве «миелиновых фигур», фор-мирующихся
из мембран органелл
133.
Морфологические проявления гипоксическогоповреждения клетки (ткани)
- нарушения микроциркуляции – стаз и сладж крови, ↑
сосудистой проницаемости, парава-зальный отек,
микрокровоизлияния, мукоид-ное и фибриноидное
набухание соединитель-ной ткани → фиброз стромы
при хр. гипоксии
- обратимое набухание паренхиматозных клеток (ранний
признак)
- при сохраняющейся гипоксии прогрессирова-ние в
необратимую гидропическую, а позднее – баллонную
дистрофию, являющуюся по сути колликвационным
некрозом
134.
Баллонная дистрофия гепатоцита.Мембраны ЭПС разрушены, видны
множественные цистерны, заполненные
жидкостью (рис. 1.19)
Некробиоз клеток. Начало пикноза ядра (Я) в одной и его лизиса в другой клетке. Митохондрии
(М) резко набухшие, распад их
внутренних мембран (рис. 66)
135.
Патогенез набухания клеток связан с модификацией структуры и увеличением проницаемости как клеточной, так и внутренней мембранымитохондрий
- ингибированием дыхательной цепи
митохондрий
- снижением, а позднее прекращени-ем
синтеза АТФ
- ингибированием различных ферментов
клетки
136.
Повреждение клетки,вызванное
свободными
радикалами
кислорода, чаще всего
возникает под
воздействием
химических веществ
(также их ферментном
расщеплении в
процессе
метаболизма),
ионизирующего
излучения, в ходе
эндогенных
окислительных
реакций, связанных с
нормальным
метаболизмом клетки.
137.
В норме под воздействием цитохром-оксидазыО2 превращается в воду, но при избытке
промежуточных соедине-ний О2 (супероксид,
перекись водорода, гипероксильные ионы)
развивается цепь аутокаталитических
реакций, ведущих к обширному повреждению
клетки (перекисное окисление липидов
мембран клетки, связывание лабильных
аминокислот, фрагментация полипептидных
цепей, повреждение ДНК).
138.
Особенности инициальных механизмов поврежденияклетки при действии химических веществ
определяются
- тропностью к определенным видам клеток и
внутриклеточных органелл, участвующих в
метаболизме этих веществ (например, хлорид ртути →
больше страдают клетки желудочно-кишечного тракта и
почек)
- характером образующихся токсических мета-болитов
(при изначально биологически неак-тивных токсинах)
Наиболее важным и при воздействии химичес-ких
веществ оказывается образование свобод-ных
радикалов О2 и последующее перекисное окисление
липидов
139.
Из обратимых повреждений клетки притоксических воздействиях чаще
встречается жировая дистрофия появление в цитоплазме различной
величины липидных включений.
Жировая дистрофия характерна для
клеток, участвующих или зависящих от
обмена жиров (гепатоциты,
миокардиоциты).