





















































 Медицина
МедицинаПохожие презентации:










Гемопоэз у детей
1.
Гемопоэз у детейАНЕМИИ
ЧГМА, ПДБ
Доц. Максимова О.Г.
2.
Апластические анемии (АА)• Группа наследственных и
приобретенных заболеваний крови,
обусловленных дефектами стволовой
клетки, приводящих к уменьшению или
отсутствию продукции ГК и жировому
замещению КМ
• Врождённые
• Приобретённые
3.
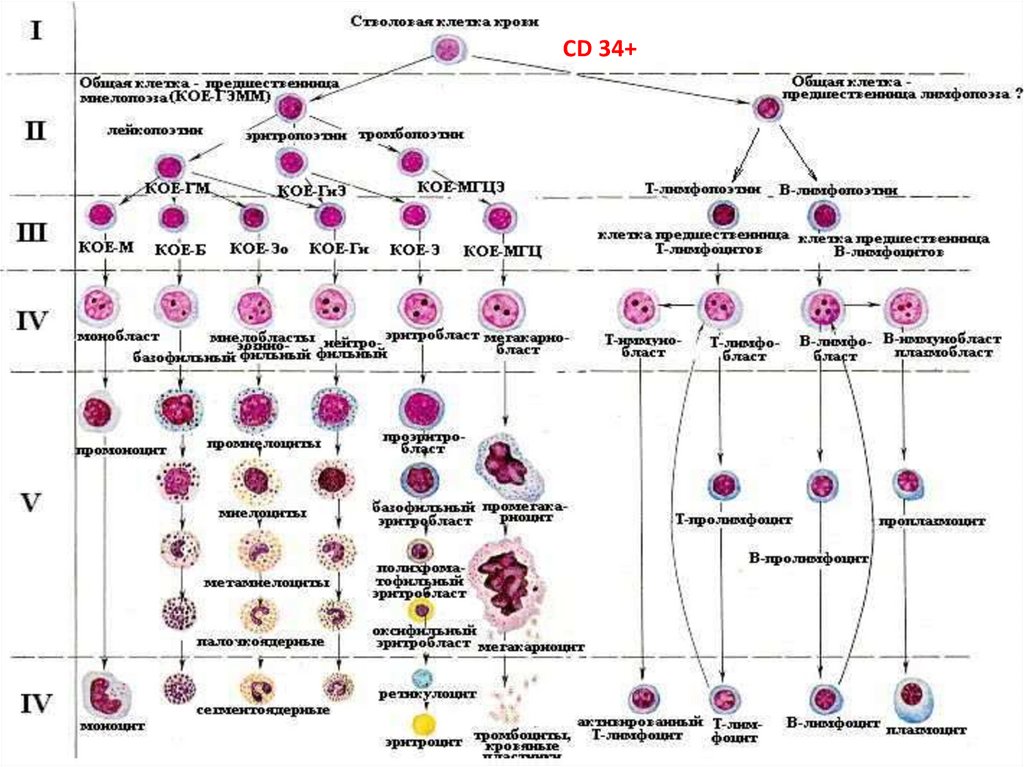
CD 34+4.
Приобретённые: острые, хроническиеЭрлих ,1888 г
Вирус ассоциированные: вирусы гепатита
А (флавовирусы), Эбштейн-Бар,
ветряной оспы, парвовирусом 19, и др.
Вызванные лекарственными и
химическими веществами
Идиопатические Идиопатические
5.
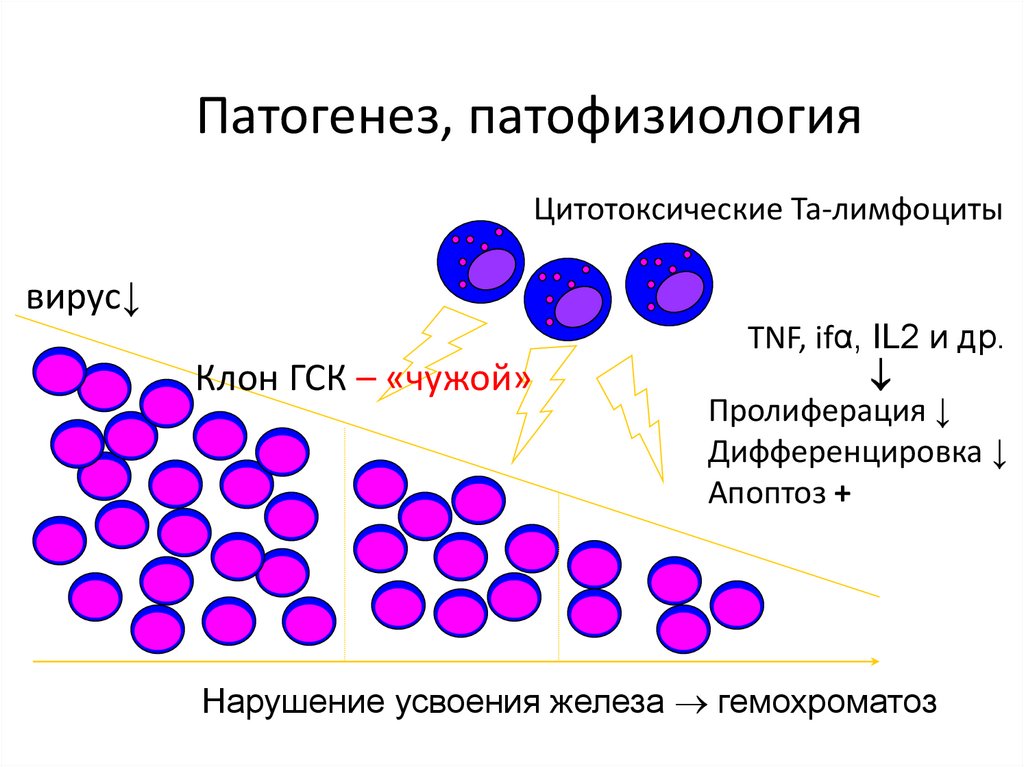
Патогенез, патофизиологияЦитотоксические Та-лимфоциты
вирус↓
Клон ГСК – «чужой»
TNF, ifα, IL2 и др.
Пролиферация ↓
Дифференцировка ↓
Апоптоз +
Нарушение усвоения железа гемохроматоз
6.
Клинические синдромыАнемический
Геморрагический
(тромбоцитопения)
ПИДС ( нейтропения):
бессимптомная лихорадка,
рецидивирующие
бактериальные инфекции
7.
Лабораторная диагностикаГемограмма:
абсолютная
нейтропения (н<500/мкл – до 1года,
н 1000/мкл
–
после
1
года),
тромбоцитопения (< 150000/мкл),
ретикулоцитопения (< 0,5%)
Анемия нормохромная,
макроцитарная
Начало: одноцитопения
или
нормо-
или
двухростковая
8.
Диагностика в специализированном онкогематологическом отделении• Госпитализация в онко-гематологический
стационар
• Исследование пунктата костного мозга,
полученного из 4 точек
• Миелограмма: обеднение клеточными
элементами, замещение красного костного
мозга жировой тканью
• Трепанобиопсия
9.
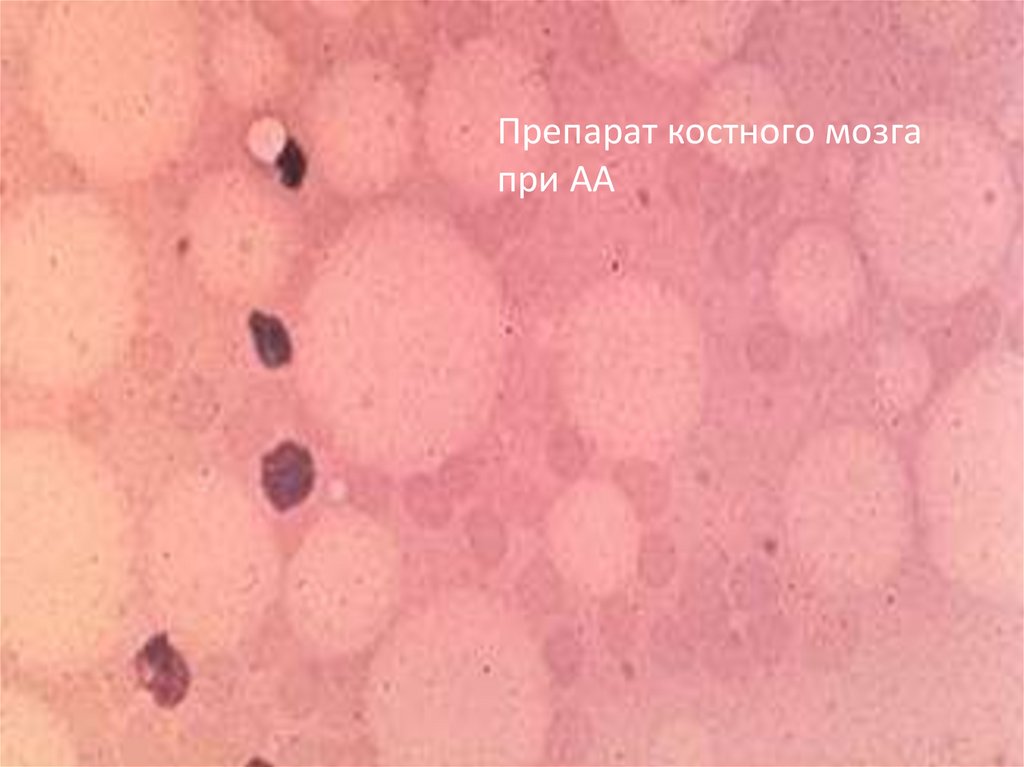
10.
Препарат костного мозгапри АА
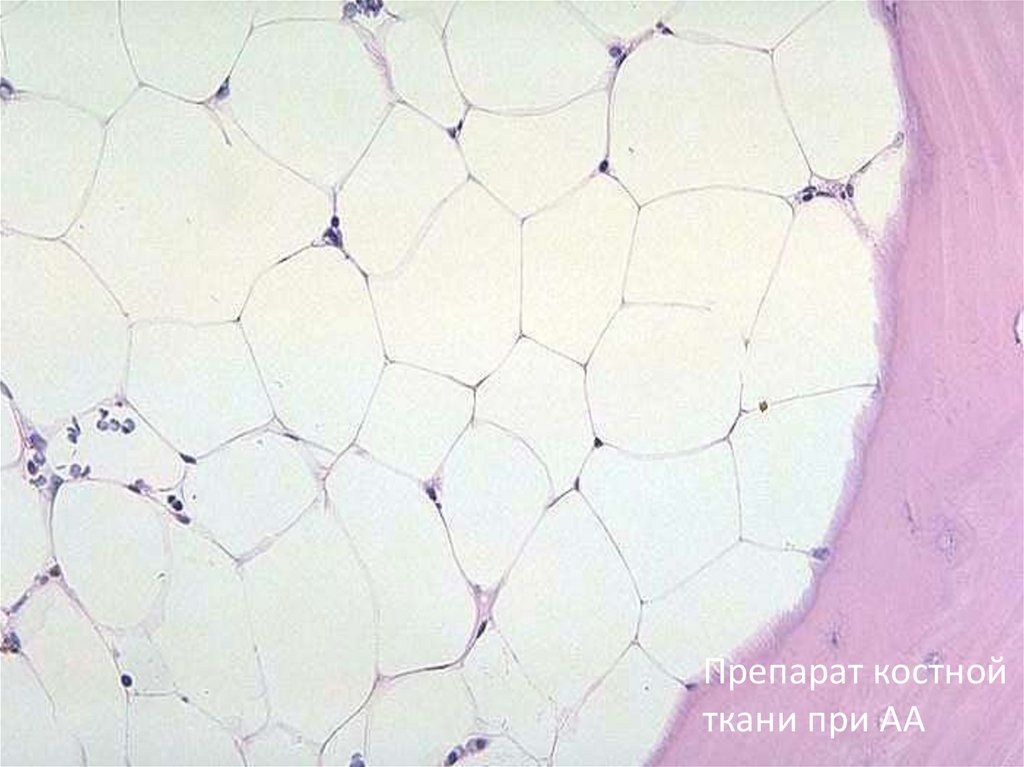
11.
Препарат костнойткани при АА
12.
НАЦИОНАЛЬНОЕ ГЕМАТОЛОГИЧЕСКОЕОБЩЕСТВО
Клинические рекомендации по лечению
апластической анемии
(комбинированная иммуносупрессивная
терапия)
Рекомендации утверждены
на IV Конгрессе гематологов России
(апрель 2018г)
2018
13.
Принципы терапии приобретённойапластической анемии
Диагноз установлен:
«Приобретённая апластическая анемия»
HLA-типирование
Тяжелая АА
Нет донора
5-6 месяцев
2-3 недели
Есть донор
Нетяжелая АА
Наблюдение
Прогрессия
ТКМ
ИСТ
14.
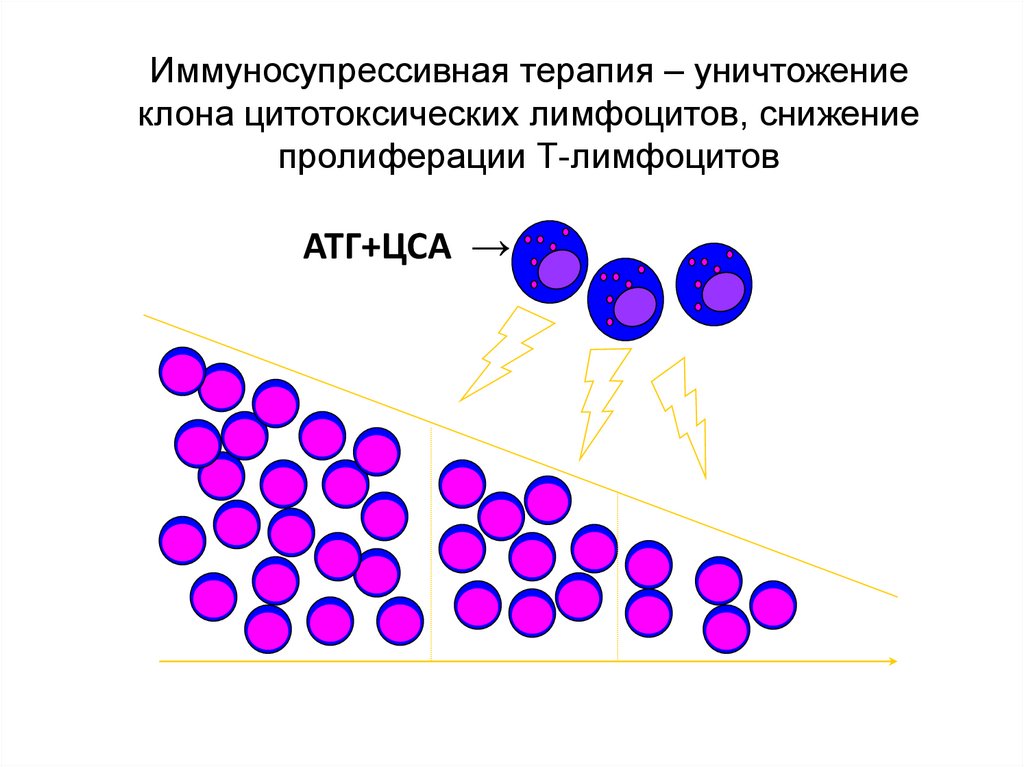
Иммуносупрессивная терапия – уничтожениеклона цитотоксических лимфоцитов, снижение
пролиферации Т-лимфоцитов
АТГ+ЦСА →
15.
Антитимоцитарныйглобулин (АТГ)
Профилактика анафилактического шока - преднизолон
16.
Циклоспорин АКонтроль за лечением – концентрация препарата в
крови
17.
Сопроводительная терапия• Стимуляторы кроветворения:
- гранулоцитопоэза - гранулоцитарный
колониестимулирующий фактор (Лейкостим,
Нейпоген, Граноцит и др.).
- тромбоцитопоэза (Элтромбопаг)
Заместительная терапия: эритроцитная масса,
тромбоконцентраты, СЗП
• Хелаторная терапия (выведение избытка
железа) - десферал
18.
• Элтромбопаг19.
Прогноз, исходы• Алло ТСКК – 85 - 90% длительная
выживаемость
• ИСТ – 85 – 90% 5-летняя выживаемость
• Рецидивы
• Острый лейкоз
• Миелодиспластический синдром – у людей
с мутацией в генах теломеразного
комплекса
20.
Мегалобластные анемии.Определение понятия
Гетерогенная
по
этиологии
группа
заболеваний, общим признаком которых
является наличие в костном мозге
мегалобластов.
Дефицит вит. В12
и/или фолатов
21.
Патогенез• Отсутствие В12 → нарушение синтеза ДНК
• Поражение клеток различных тканей →
полисистемность поражения
• Гемопоэз, пищеварение, нервная система
• Кровь: снижение пролиферации ГК → анемия,
лейкопения, тромбоцитопения, развитие
мегалобластного типа кроветворения,
диспластических измений в клетках крови
• Периферической нервная система: накопление
токсичной для нервных клеток метилмалоновой
кислоты → нарушение миелинизации нервных
волокон
22.
Причины дефицита В12• Экзогенный: вегетарианская диета
• Эндогенный:
• - врожденный дефицит внутреннего фактора болезнь Аддисона-Бирмера, пернициозная
анемия;
• - болезни, резекция кишечника и/или
желудка;
• - инвазия ленточными
глистами
23.
Причины дефицита фолиевойкислоты
• Экзогенные:
• - недоношенность;
• - искусственное вскармливание
козьим молоком без коррекции
фолиевой кислоты;
• - лечение цитостатиками, фенобарбиталом
• Эндогенные:
- синдром мальабсорбции;
- врожденные нарушения метаболизма фолатов.
24.
Клиническиесиндромы
Анемический.
Диспепсический:
хроническая диарея;
“лакированный” язык;
анорексия
25.
Периферическая нейропатия: атаксии,парастезии, гипорефлексия, симптом
Бабинского +
26.
Клинические синдромы• Ранний возраст: задержка роста, БЭН,
снижение резистентности к инфекциям
27.
Лабораторная диагностика• Гемограмма: анемия, ретикулоцитопения
(ниже 0,5%).
• Анемия макроцитарная (MCV ≥ 120 фл),
гиперхромная
• Эритроциты: полихроматофилия,
пойкилоцитоз (макроовалоциты, кольца
Кебота, тельца Жолли)
• Умеренная тромбоцитопения, склонность к
лейкоцитопении, гиперсегментированные
нейтрофилы
28.
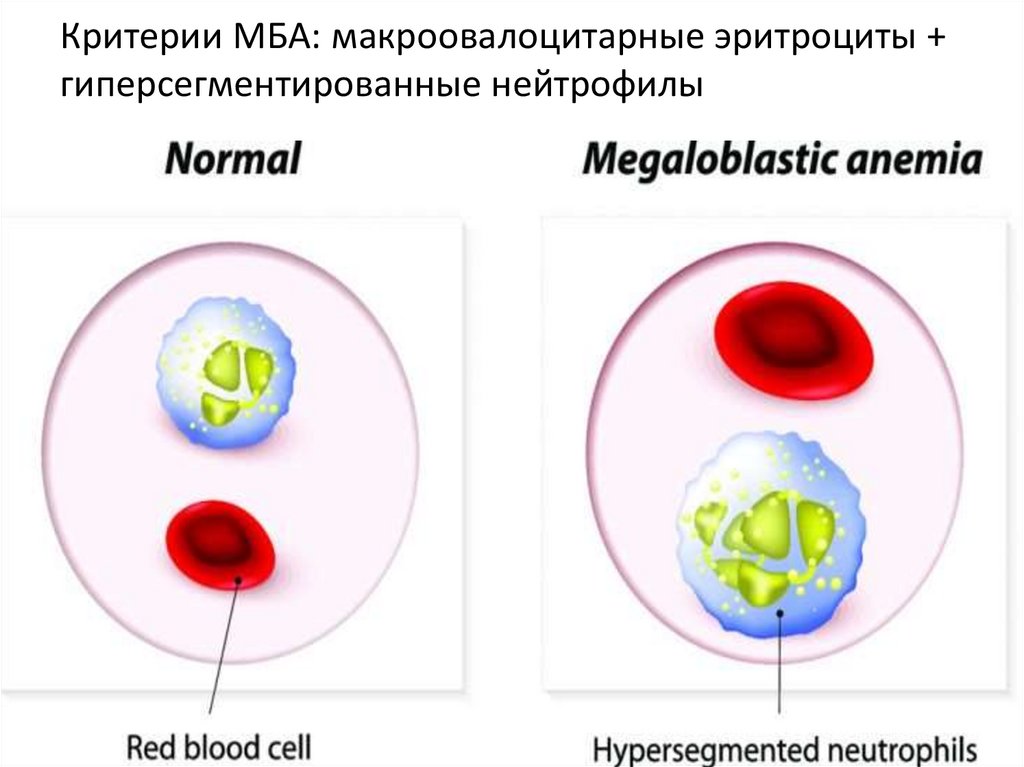
Критерии МБА: макроовалоцитарные эритроциты +гиперсегментированные нейтрофилы
29.
Пойкилоцитоз, тельца ЖоллиНейтрофил
30.
Кольца Кебота31.
Миелограмма: мегалобласты32.
Специфические диагностические тестыСнижение содержания В12
Снижение содержания фолиевой кислоты
Повышение содержания метилмалоновой
кислоты
Повышение содержания гомоцистеина
33.
ЛечениеВыявление причины и ликвидация
Коррекция питания
34.
Лечение дефицита В12100 - 500 мкг в/м на 1 - 2 недели, далее 1
раз в неделю до нормализации
гематокрита, затем 1 раз в месяц (3-6
мес)
35.
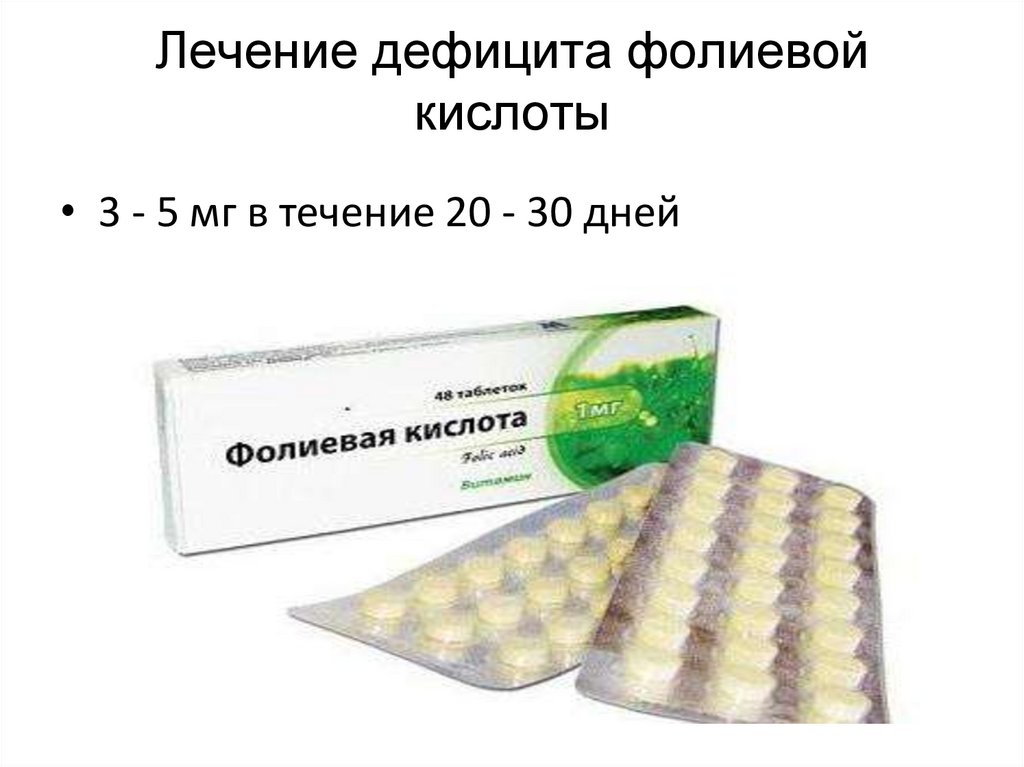
Лечение дефицита фолиевойкислоты
• 3 - 5 мг в течение 20 - 30 дней
36.
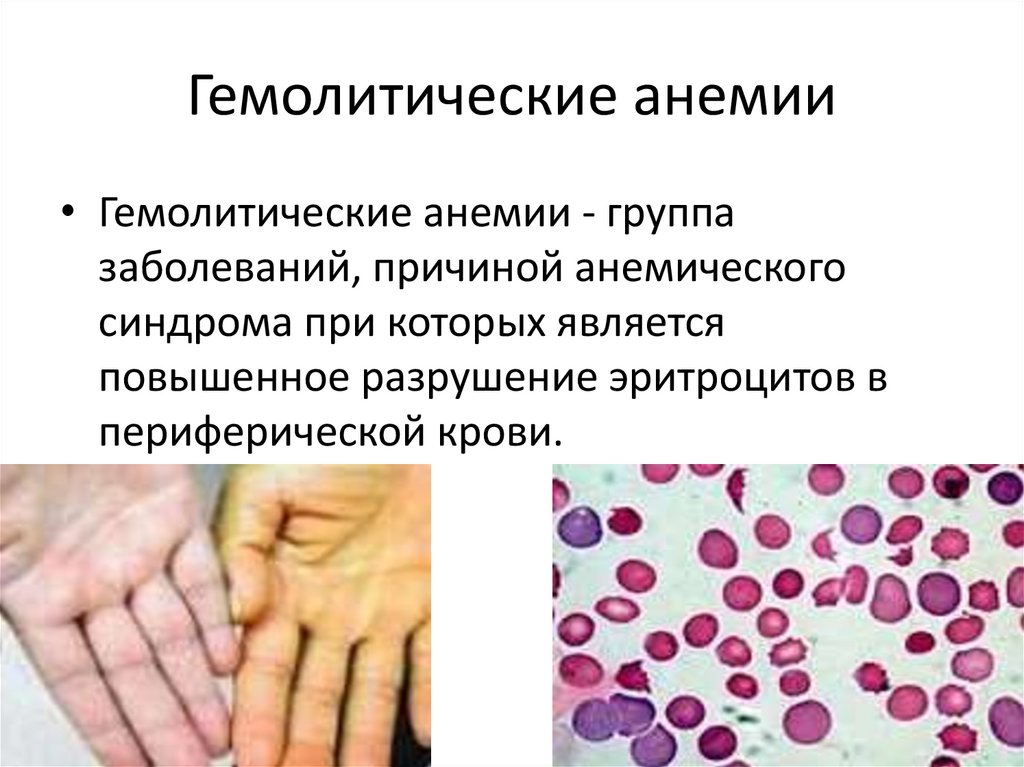
Гемолитические анемии• Гемолитические анемии - группа
заболеваний, причиной анемического
синдрома при которых является
повышенное разрушение эритроцитов в
периферической крови.
37.
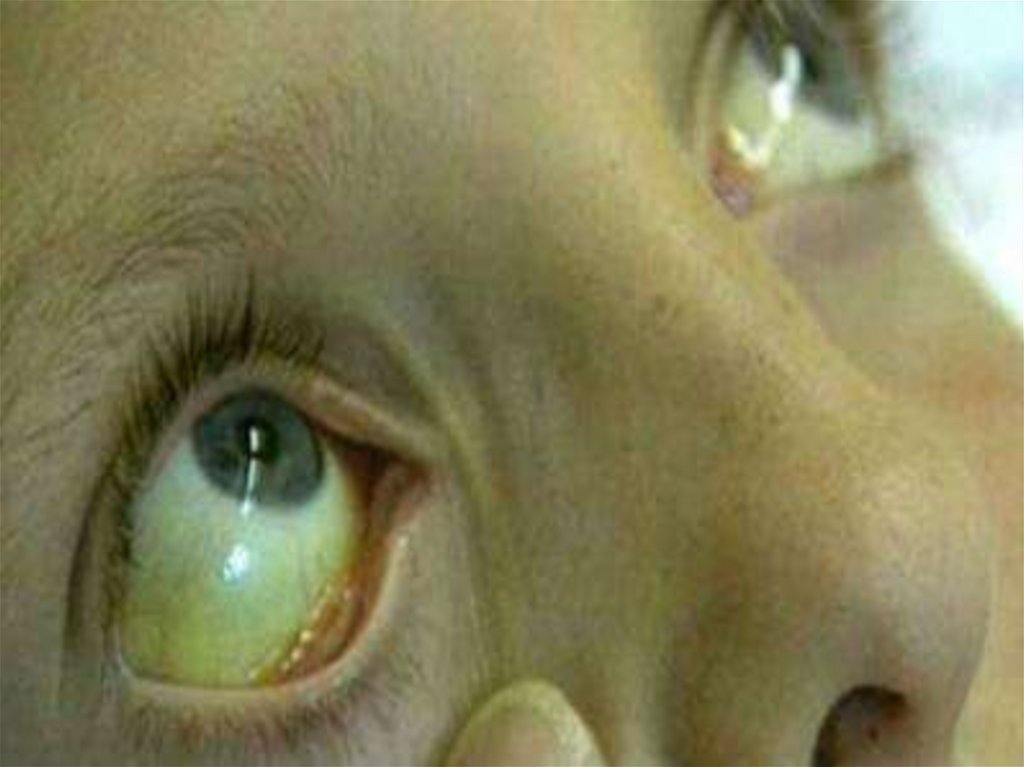
Диагностические критерии• Клинические:
• - желтушность кожи и слизистых
оболочек на бледном фоне;
• - спленомегалия;
• - гепатомегалия реже;
• - тёмный цвет мочи (при
внутрисосудистом гемолизе)
38.
39.
40.
Лабораторные критерииРетикулоцитоз (> 3%)
Гипербилирубинемия
преимущественно за счет непрямого
билирубина
Повышение ЛДГ – признак цитолиза
При внутрисосудистом гемолизе повышение свободного гемоглобина
плазмы и мочи (гемоглобинурия)
41.
Классификация• Врожденные
(наследственные)
• Мембранопатии
• Ферментопатии
• Гемоглобинопатии
• Приобретенные
• Иммунные
• Неиммунные:
• - механические
(ГУС, ТТП)
• - ПНГ
• - при дефиците
витамина Е
42.
Наследственный сфероцитоз• Наследственная сфероцитарная
гемолитическая анемия МинковскогоШоффара - наследственная гемолитическая
анемия, обусловленная молекулярным
дефектом мембраны эритроцитов,
проявляющаяся гемолизом, наличием
микросфероцитов в мазке периферической
крови, уменьшением мин. ОРЭ и
клиническим улучшением после
спленэктомии
43.
Причина• Мутация генов, ответственных за
образование белков мембраны
эритроцитов
• Мутация гена SPTB (lq21), (14q22-q23)
→ дефицит спектрина
• 70% - аутосомно-доминантный тип
• 30% - аутосомно-рецессивный тип
• Спонтанные мутации
44.
Патогенез• Дефект структуры мембраны потеря
подвижности Э, деформабельности
затруднение прохождения Э по узким
сосудам селезёнки травма Э, захват
макрофагами селезенки
• Преждевременное разрушение Э
макрофагами селезенки
укорочение длительности жизни до 10
- 12 дней
45.
Клинические синдромы• Наследственный анамнез
• Анемия + желтуха
• Спленомегалия
• Рецидивирующее течение (криз обострение)
• Задержка развития
• Костные деформации черепа: лобные и
теменные бугры (расширение плацдарма
кроветворения)
• ЖКБ - осложнение
46.
Врожденный дискератоз. Тяжёлыеполиморфные поражения кожи
47.
48.
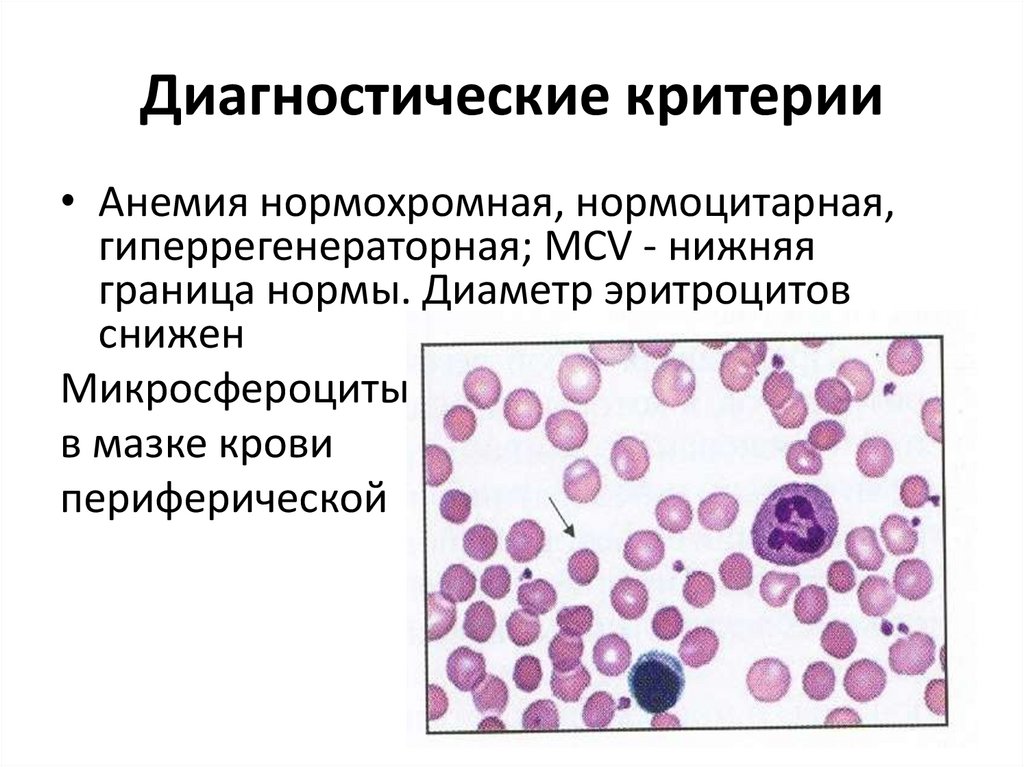
Диагностические критерии• Анемия нормохромная, нормоцитарная,
гиперрегенераторная; MCV - нижняя
граница нормы. Диаметр эритроцитов
снижен
Микросфероциты
в мазке крови
периферической
49.
Диагностические критерии• Уменьшение ОРЭ мин. (% р-ра NaCl, в
котором начинается гемолиз эритроцитов)
50.
Дополнительные диагностическиекритерии
• Плазмаферез белков мембраны
эритроцитов
• Мутация гена SPTB (молекулярногенетический анализ – нет семейного
анамнеза)
51.
Лечение• Легкая формы: наблюдение, УЗИ печени,
ЖВП
• Д№5
• Гемолитический криз: обильное щелочное
питье, в/в 5% глюкоза 10 - 20 мл/кг/с
• Спленэктомия
52.
Спленэктомия• Показания:
• - тяжелая форма;
• - гиперспленизм (тромбоцитопения,
лейкопения);
• - ЖКБ
• - частые кризы: задержка развития,
склонность к инфекционным
заболеваниям, костные деформации;
• - нарушение качества жизни
53.
СпленэктомияВозраст старше 5 лет, при тяжелом
течении – после 3 лет
• Предварительная вакцинация
пневмококковой вакциной
54.
Диспансеризация• В течение всей жизни
• Осмотр 1 раз в 6 мес
• УЗИ
• Д№5
• Прогноз после операции – клиническое
выздоровление
55.
Энзимопатии• Причина: наследственный дефект
ферментной системы анаэробного
гликолиза
• Дефициты: Г-6-фосфатдегидрогеназы
(Г6ФД), пируваткиназы (ПК)
56.
глюкоза↓Г-6-ФД
НАДФ, ГР, ГП АОЗ Г-6-Ф
фосфофруктокиназа
фруктозо-1,6-дифосфат
фосфоэнолпируват
АДФ АТФ
(структура, функция
мембраны - насосы К, Na) ПК
пируват
НАДФ ЛДГ
лактат
57.
Энзимопатии• Мутация генов изменения активности
ферментов эритроцитов недостаток
образования АТФ и глутатиона
нарушение структуры и функции,
преждевременная гибель эритроцитов при
контакте с перекисями
58.
Дефицит Г-6-ФД• Ген Г-6-ФД - Хq28
• Чаще болеют мужчины
• I. Острый гемолитический криз
• II. Хроническая несфероцитарная
гемолитическая анемия
59.
Причины острого гемолитическогокриза
Лекарственные препараты: нитрофураны,
метиленовый синий, сульфаниламиды,
противотуберкулезные средства,
налидиксовая кислота, аспирин,
аскорбиновая кислота, викасол, перекись
водорода и др.
• Острое инфекционное заболевание
60.
Острый гемолитический криз• Желтуха + анемия
• Интоксикация
• Тяжелый криз – ОПН, тёмная моча
(гемоглобинурия)
• Отмена препарата - быстрое купирование
гемолиза
61.
Лабораторная диагностика• Анемия нормоцитарная, нормохромная,
гиперрегенераторная
• Диагностические критерии:
• - тельца Гейнца в эритроцитах;
• - тяжелый криз - свободный гемоглобин в
плазме и моче;
• - активность фермента Г6ФД ;
• - молекулярно-генетический анализ.
62.
Тельца Гейнца:преципитированный,
денатурированный
гемоглобин
Гемоглобинурия
→
63.
Лечение• Вне криза: избегать приема препаратов,
вызывающих гемолиз
• Нетяжелый гемолитический криз:
• - отмена препарата;
• - ксилит, рибофлавин - восстановление
глютатиона в эритроцитах;
• - фенобарбитал билирубинконъюгирующее действие
64.
Лечение• Острый внутрисосудистый гемолиз:
• - госпитализация в ОИТР;
• - профилактика и лечение ОПН;
• - Нв <50 г/л - трансфузия эритроцитов
65.
Иммунные гемолитическиеанемии
Изоиммунные: Ат к Аг другого человека ГБН
Трансиммунная:
трансплацентарная
передача Ат от
матери, страдающей
АИГА
66.
Иммунные гемолитическиеанемии
Аутоиммунная: Ат (IgG или IgM) к
поверхностным Аг собственных
эритроцитов.
67.
Аутоиммунная гемолитическаяанемия. Клиника
Начало
внезапное,
с
острого
гемолитического криза смешанного типа
(внутриклеточный + внутрисосудистый
гемолиз)
Слабость, вялость, тахикардия, одышка,
лихорадка, желтушное окрашивание кожи и
слизистых оболочек, коричневатый или
бурый цвет мочи (гемоглобинурия); может
быть гепатоспленомегалия
68.
Лабораторная диагностикаГемограмма.
Нормо-/гиперхромная,
макроцитарная, гиперрегенераторная
анемия.
Нейтрофильный лейкоцитоз со сдвигом
влево, ускоренная СОЭ.
Количество
изменяется
тромбоцитов
не
АИГА + ИТ – синдром Фишера-Эванса
69.
Лабораторная диагностикаДиагностические критерии
(обнаружение Ат к эритроцитам):
+ прямая реакция Кумбса и/или
гемагглютинационная проба
70.
Лечение гемолитического криза• Преднизолон – 2-3 мг/кг/с
(метилпреднизолон 30 мг/кг/с) с
последующей постепенной отменой
• ВВИГ 0,5 – 1,0 г/кг/с в течение 5 дней
• Тяжёлая анемия + нарушение
гемодинамики – трансфузия подобранных
от индивидуального донора эритроцитов
• Профилактика и лечение ОПН при остром
внутрисосудистом гемолизе
71.
Диспансерное наблюдениеГематолог не менее 5 лет после
перенесенного гемолитического криза
Хроническое течение – постоянно
Наблюдение гематолога 1 раз в 6 мес
Вакцинация противопоказана в течение
двух лет
72.
С наступающим праздником,коллеги!