























")






































")




 Медицина
Медицина Биология
БиологияПохожие презентации:










Амилоидная агрегация. Прионы
1. Амилоидная агрегация. Прионы.
Стройлова Юлия Юрьевнас.н.с. лаборатории молекулярной и клеточной
биологии Института молекулярной медицины
2. Сворачивание белка
3. Biology/Chemistry of Protein Structure
PrimaryAssembly
Secondary
Folding
Tertiary
Packing
Quaternary
Interaction
PROCESS
STRUCTURE
Biology/Chemistry of Protein Structure
4. X-Ray Crystallography
• crystallize andimmobilize single,
perfect protein
• bombard with X-rays,
record scattering
diffraction patterns
• determine electron
density map from
scattering and phase
via Fourier transform:
• use electron density
and biochemical
knowledge of the
protein to refine and
determine a model
5. Нормальная упаковка белка
• Происходит в «толпе»(crowded environment, 350
мг/мл)
• Участие шаперонов и фолдаз
• Система контроля качества
• (убиквитинилирование и
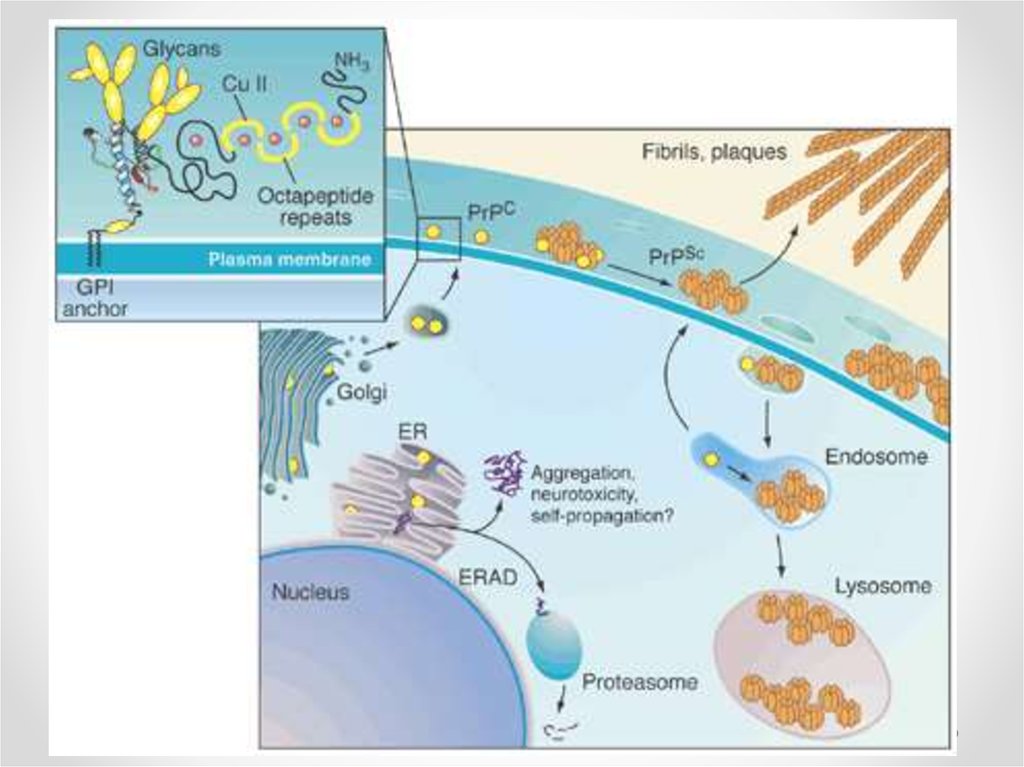
деградация в протеасомах)
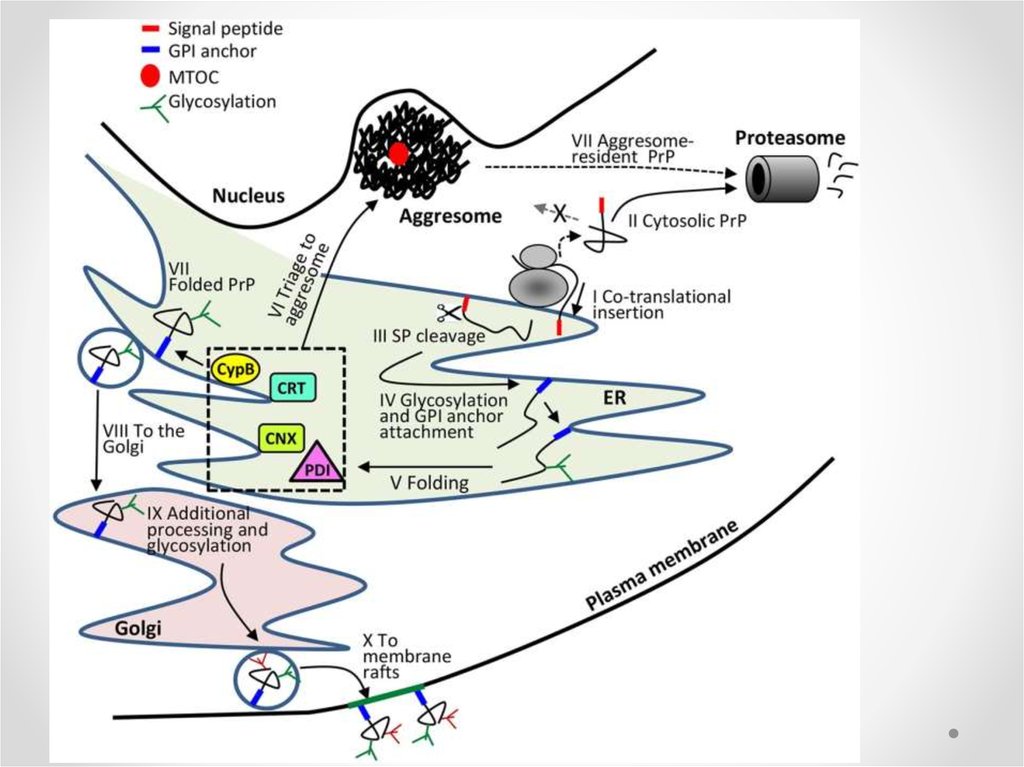
6. Неправильное сворачивание белка - misfolding
• Мисфолдинг – образование структурыбелка со значительным числом ненативных
связей между аминокислотными
остатками
• Ведет к потере функции белка
• Вызывает накопление токсичных агрегатов
в клетках
• Может приводить к гибели клеток
7. Конформационные болезни
• Болезни, которые вызваны наличием белков снеправильной структурой
• Например, в случае мукосвисцидоза – неправильное
сворачивание белка - муковисцидозного
трансмембранного регулятора проводимости (МВТП)
приводит к нарушению его функции
• Амилоидные болезни – когда происходит накопление
патологических агрегатов неправильно свернутого
белка в тканях и органах (мозг, печень, селезенка)
8. Конформационные болезни
9. Амилоидные болезни
• Особенный случай агрегации неправильно свернутыхбелков - амилоидная
• Происходит образование амилоидных фибрилл и
бляшек
• Состояние белка в амилоидной фибрилле
стабильно и конкурирует с нативным
Зачем был нужен такой путь сворачивания белка
эволюционно?
Некоторые амилоиды животных имеют полезные и
важные функции. У дрозофилы переход в амилоидное
состояние белка Orb2 является ключевым событием в
механизме долговременной памяти.
10. Функциональные амилоиды у бактерий
11. Механизм агрегации белка
12. Свойства амилоидных агрегатов
• Образование поперечных β-структур• Формирование длинных
неразветвленных фибрилл диаметром
около 10 нм
• Происходит образование амилоидных
фибрилл и бляшек
13.
• С помощью алгоритмаTANGO предсказали
способность к амилоидной
трансформации
• В образовании амилоидного
«ядра» (core) могут быть
вовлечены всего несколько а.о.
• Изменения (мутации) по
одной или нескольким а.о.
могут резко увеличивать
вероятность амилоидной
агрегации
14. Прионы
Структура, свойства и роль в развитииприонных болезней
15.
Структура, свойства и роль в развитииприонных болезней
16. История открытия
• Prions - от англ. protenacious infection• Прионные болезни были известны с начала
XVIII века
• «Странные» свойства возбудителей: не
размножаются на искусственных
питательных средах, устойчивы к высокой
температуре, формальдегиду, различным
видам излучений, действию нуклеаз.
• В конце 80-х годов было установлено, что
инфекционный агент – белок
• В 1997 г – Нобелевскую премию получил S.
Prusiner
17.
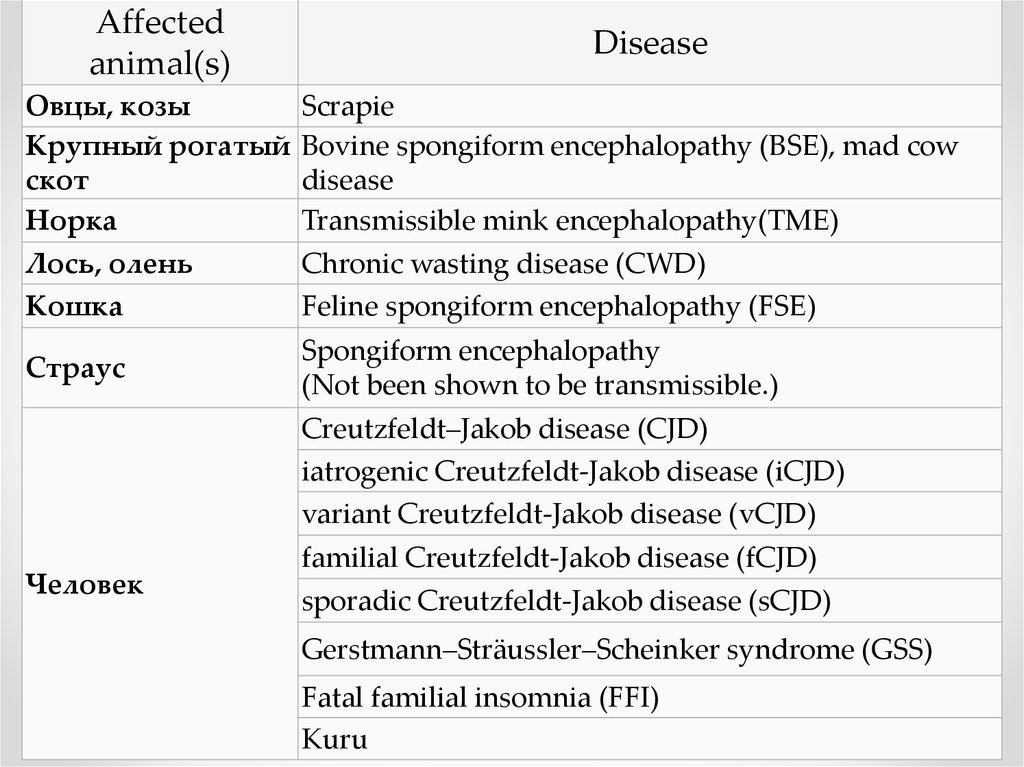
Affectedanimal(s)
Disease
Овцы, козы
Крупный рогатый
скот
Норка
Лось, олень
Кошка
Scrapie
Bovine spongiform encephalopathy (BSE), mad cow
disease
Transmissible mink encephalopathy(TME)
Chronic wasting disease (CWD)
Feline spongiform encephalopathy (FSE)
Страус
Spongiform encephalopathy
(Not been shown to be transmissible.)
Creutzfeldt–Jakob disease (CJD)
iatrogenic Creutzfeldt-Jakob disease (iCJD)
variant Creutzfeldt-Jakob disease (vCJD)
Человек
familial Creutzfeldt-Jakob disease (fCJD)
sporadic Creutzfeldt-Jakob disease (sCJD)
Gerstmann–Sträussler–Scheinker syndrome (GSS)
Fatal familial insomnia (FFI)
Kuru
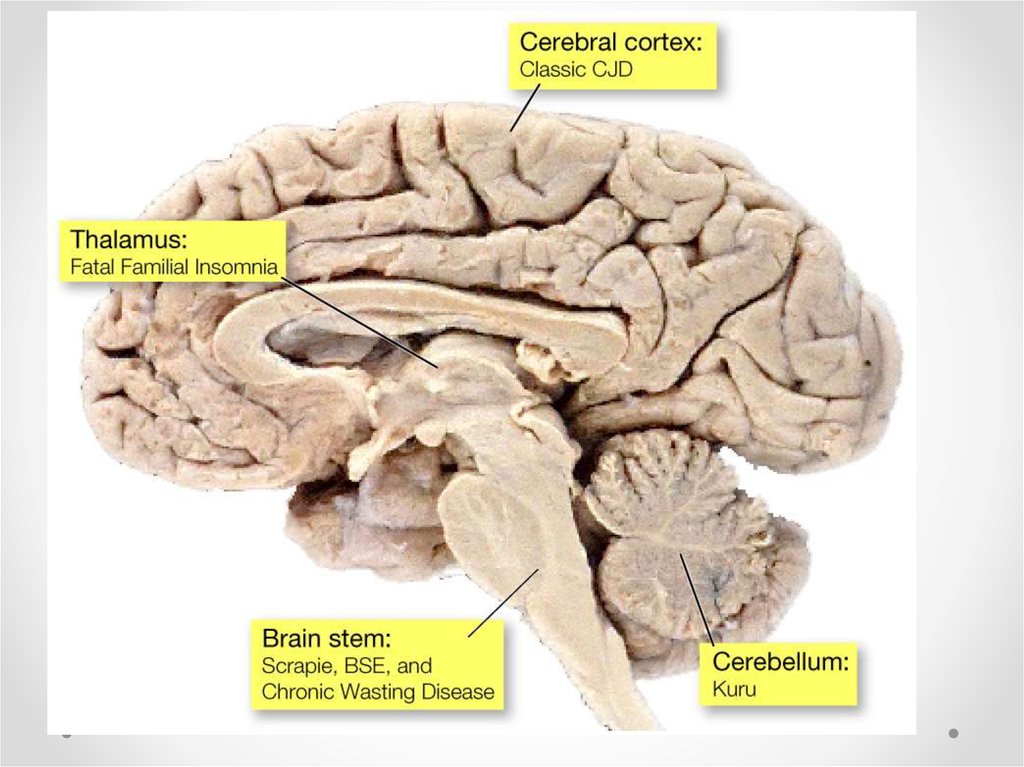
18. Прионные болезни
Главные характеристики прионныхзаболеваний:
• Длительный инкубационный период (нужен
для репликации и накопления прионов)
• Накопление прионов происходит в
лимфоретикулярной системе (лимфоузлы,
пейеровы
бляшки тонкого кишечника)
• Репликация в ЦНС, что приводит к гибели
нейронов, активации астроцитов
• Ткань мозга приобретает губкообразную
морфологию
19.
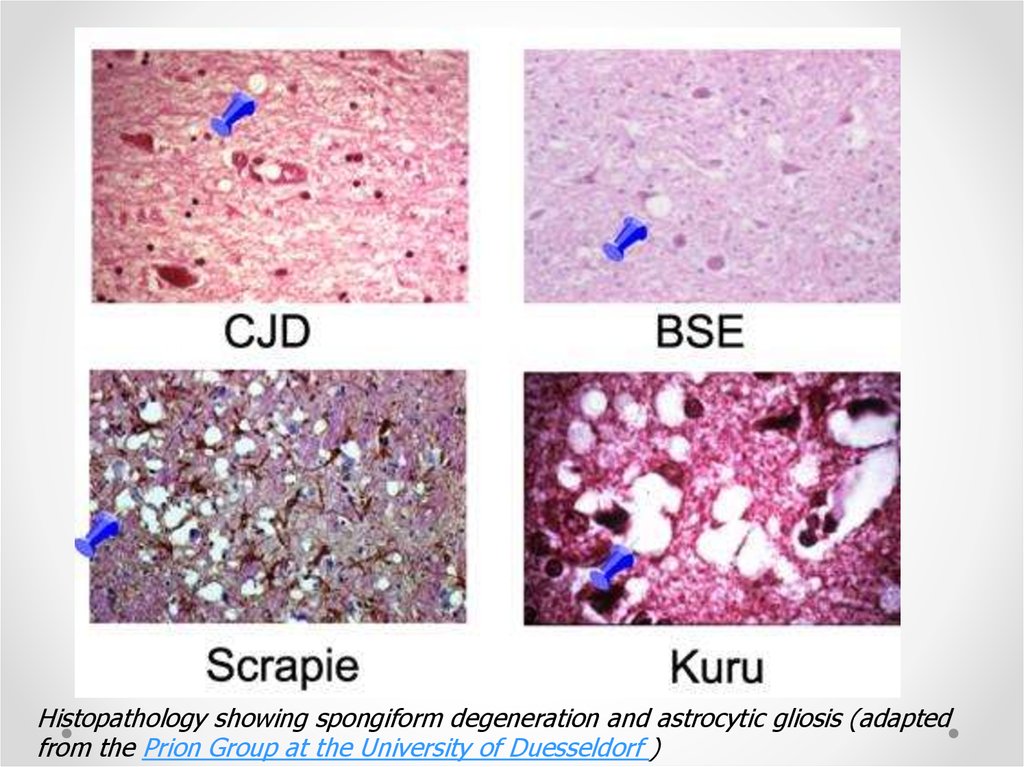
Histopathology showing spongiform degeneration and astrocytic gliosis (adaptedfrom the Prion Group at the University of Duesseldorf )
20.
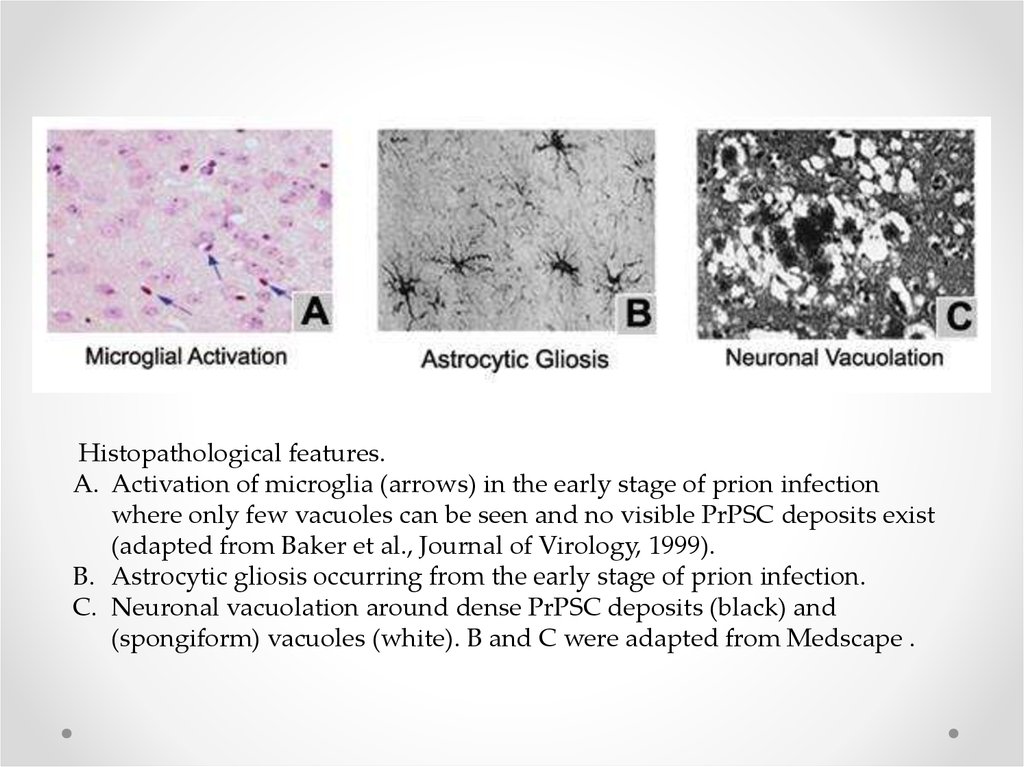
Histopathological features.A. Activation of microglia (arrows) in the early stage of prion infection
where only few vacuoles can be seen and no visible PrPSC deposits exist
(adapted from Baker et al., Journal of Virology, 1999).
B. Astrocytic gliosis occurring from the early stage of prion infection.
C. Neuronal vacuolation around dense PrPSC deposits (black) and
(spongiform) vacuoles (white). B and C were adapted from Medscape .
21. Межвидовой барьер
22. Прионные болезни
Пути возникновения:1) Спорадическая. Спонтанная трансформация здорового белка
головного мозга в инфекционный Болезнь может развиваться в
мозге десятилетиями.
2) Ятрогенная. Прион заносится непреднамеренно во время
медицинских вмешательств — через кровь, ткани глаза,
нейрохирургические, патологоанатомические инструменты.
3) Наследственная
4) нвБКЯ («новый вариант»), когда прион заносится в организм
от мяса животных, зараженных «коровьим бешенством».
5) каннибализм
Инфекционный агент - белок
23.
24. Болезнь Крейтцфельдта-Якоба
Симптоматика болезни КрейтцфельдтаЯкоба характеризуется постепеннойтрансформацией ткани головного мозга
в губчатую массу. Протекает этот
процесс стадийно, по трем стадиям.
• Первая стадия – продромальный
период (больной мучается
бессоницей, у него снижается
аппетит, ухудшается внимание,
память, он теряет в весе, идет процесс
изменения личности).
• Вторая стадия – инициальный период
(больного мучают головные боли,
нарушается зрение, развивается
парестезия).
• И третья стадия – развернутые период
заболевания (этот период проявляется
развитием спастического паралича).
25. Болезнь Крейтцфельда-Якоба
Эта разновидность прионной инфекции довольномногообразна, потому что в настоящее время
принято выделять несколько ее форм:
• спорадическую (классическую);
• семейную (наследственную);
• ятрогенную;
• новую атипичную форму, возникающую в
результате инфицирования мясом крупного
рогатого скота, пораженного «бешенством
коров», то есть эта форма представляет собой
аналог «коровьего бешенства» у людей.
26. Болезнь Крейтцфельда-Якоба
• Семейная форма связанас мутациями в области 20й хромосомы.
• Составляет около 5-6% всех
случаев болезни
Крейтцфельда-Якоба.
• Заболевание носит
аутосомно-доминантный
характер
• Первые признаки
заболевания возникают
приблизительно на 5-10 лет
раньше, чем при
спорадической форме
27.
• Ятрогенная форма возникает в результате заражениячеловека во время медицинских вмешательств.
• Статистика этой формы заболевания отсутствует,
поскольку довольно сложно проследить и доказать
именно такой путь заражения.
• Существуют подтвержденные документально случаи
заражения при использовании твердой мозговой
оболочки, роговицы, стереотаксических операциях
на головном мозге (в основном, эти случаи относятся
к Франции и Австралии).
• Инкубационный период (время от заражения
прионами до появления первых признаков
заболевания) колеблется от 7 месяцев до 12 лет.
28.
• В открытых источниках не описывался ни одинслучай БКЯ в России. В 2011 году был выпущен
ПРИКАЗ от 11 января 2011 г. N 1
Роспотребнадзора, в котором указывался
перечень болезней, о которых больницы должны
сообщать в Роспотребнадзор в течение двух
часов — после «установления факта
чрезвычайной ситуации». Среди них, чума,
холера, оспа, Эбола, крымская
геморрагическая лихорадка, бешенство и БКЯ.
29. Хроническая прогрессирующая спонгиозная энцефалопатия детского возраста (болезнь Альпера)
• Эта разновидность прионных заболеванийразвивается у детей (до 18 лет).
• Наследуется по аутосомно-рецессивному типу
• Поражение нервной системы заключается в
нарушении зрения, развитии эпилептических
припадков. Возможны периодические острые
состояния, протекающие по типу инсультов. Кроме
поражения нервной системы, это заболевание
сопровождается и поражением печени. Довольно
быстро развивается хронический гепатит, который
переходит в цирроз. Больные погибают от
интоксикации из-за выраженной печеночной
недостаточности в течение 12 месяцев от начала
заболевания.
30. Семейная фатальная инсомния
Известно науке с 1986 года. Очень редко встречается.Относится к наследственным заболеваниям с аутосомнодоминантным типом передачи. К 2003 году было описано 26
семей с семейной фатальной инсомнией.
• Имеет довольно вариабельный возраст начала заболевания: от
25 лет до 71 года. Продолжительность жизни с момента появления
первых признаков составляет от 6 до 48 месяцев.
• В 178 кодоне гена PRNP, находящегося в 20-й хромосоме,
происходит замена аспарагина на аспарагиновую кислоту
Симптомы:
• бессонница, нарушение циклов сон-бодрствование
двигательные нарушения в виде миоклоний, дрожания,
мышечной слабости
• вегетативные расстройства, проявляющиеся повышением
артериального давления, повышением температуры тела,
потливостью, учащенным сердцебиением
• психические нарушения, возможны зрительные галлюцинации,
панические страхи, эпизоды спутанности сознания.
31. Куру
Куру была подробно описана в 1957 г.австралийским врачом Зигасом и американцем
словако-венгерского происхождения Карлтоном
Гайдушеком
• За открытие инфекционного характера
болезни куру Карлтон Гайдушек был удостоен в
1976 г. Нобелевской премии по физиологии и
медицине. Деньги премии он пожертвовал
племени форе
32. Куру
Kuru is a rare, incurable neurodegenerative disorder which wasprevalent among the Fore people of Papua New Guinea in the
1950s and 60s
The term kuru derives from the Fore word kuria or guria ("to
shake"), due to the body tremors that are a classic symptom of the
disease. It is now widely accepted that kuru was transmitted among
members of the Fore tribe of Papua New Guinea via funerary
cannibalism. Traditionally, deceased family members were cooked
and eaten, which was thought to help free the spirit of the
dead. Females and children usually consumed the brain, the organ
in which infectious prions were most concentrated, thus allowing for
transmission of kuru. Thus, the disease was more prevalent among
women and children.
33.
34.
Клиническая картина болезни Куру складывается изследующих симптомов:
• Затруднения акта ходьбы в первой стадии заболевания.
• Грубая атаксия, миоклонии, хорея (насильственные
движения) и снижение мышечного тонуса в поздних
стадиях.
• Деменция, сходящееся косоглазие и дизартрия
присоединяются в конце течения заболевания.
• Нарастание симптомов происходит на протяжении от 4
месяцев до 2-3 лет. В 100 % случаев наступает
смертельный исход, в основном от осложнений:
пневмония, пролежни, сепсис и т.д.
Патоморфологически болезнь Куру проявляется в
демиелинизации, гибели нейронов, скоплении лейкоцитов и
мононуклеарных клеток в головном мозге.
Запрет эндоканнибализма в 1950 году привел к снижению
эпидемии. Однако, эта болезнь сохраняется и в нынешнем
веке из-за инкубационного периода, который может
превышать 50 лет. В последнее время (с 2003 по 2008) было
зарегистрировано только два смертельных случая.
35.
• В 2009 году американские учёные сделалинеожиданное открытие: некоторые члены
племени форе, благодаря появившемуся у них в
сравнительно недавнем времени новому
полиморфизму гена PRNP, имеют врождённый
иммунитет к куру. (A novel protective prion protein
variant that colocalizes with kuru exposure. (2009)
The New England Journal of Medicine)
36. Диагностика
Лабораторная диагностика БКЯ• ЭЭГ (двух- и трехфазные острые
волны)
• КТ, МРТ
• Биопсия головного мозга
• Исследование ликвора
• Иммунный блот
• Молекулярно-генетические исследования
• Биологические методы
37.
LABORATORY DIAGNOSIS OF CJD• MRI: Diffusion Weighted Imaging shows high intensity signal in the
striatum and linear lesions. The sensitivity of these findings is
similar to that of elevated CSF 14-3-3. Patients with vCJD have high
intensity signal in the posterior thalamus (pulvinar).
• Cerebrospinal fluide: Protein and glucose concentration is normal
and there is no pleocytosis. The most useful CSF marker of CJD is
elevation of protein 14-3-3. This 30kD protein plays a role in cell
proliferation, differentiation and signal transduction and regulates
neurotransmitter synthesis. Protein 14-3-3 is also elevated in
patients with encephalitis, cerebral infarction, and other conditions.
• Biochemistry: Western blotting of PrPSc following proteinase
digestion reveals electrophoretic patterns that identify different prion
strains.
• DNA sequencing: Mutations of the PRNP gene that cause fCJD and
the codon 129 polymorphism that influences other forms of PrPScs
can be detected by sequencing of DNA extracted from blood, brain,
and other tissues.
• Brain biopsy: A definitive diagnosis can be made by microscopic
examination of brain tissue showing the characteristic spongy
change. PrPTSE can be detected in brain tissue extracts by ELISA
and in tissue sections by immunohistochemistry.
38. Характеристика устойчивости прионов к различным воздействиям
39. Прион
40. PRNP ген
41. Структура приона
42. Взаимодействие приона с мембраной
• Располагается на lipid rafts (холестерин исфингомиелин), но также есть свободная форма
• Для конверсии нужна фосфолипаза С
• Обе формы имеют сродство к отрицательно
заряженным мембранам
43.
44.
45. Механизм нейродегенерации
Aguzzi, 201346.
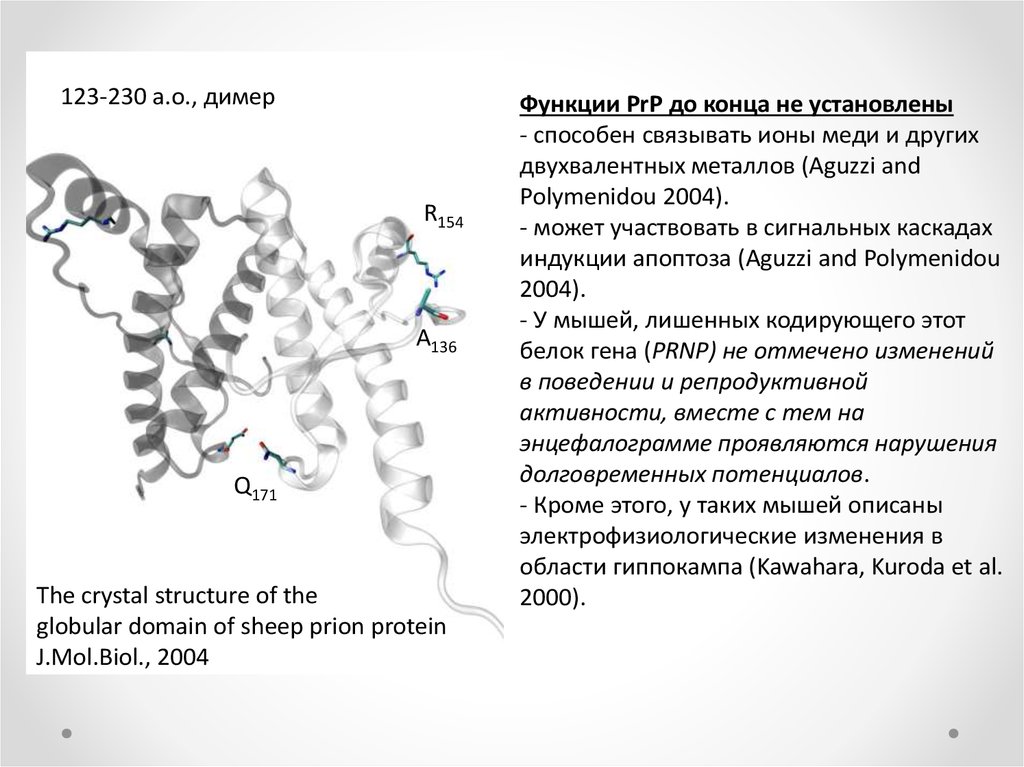
123-230 a.о., димерR154
A136
Q171
The crystal structure of the
globular domain of sheep prion protein
J.Mol.Biol., 2004
Функции PrP до конца не установлены
- способен связывать ионы меди и других
двухвалентных металлов (Aguzzi and
Polymenidou 2004).
- может участвовать в сигнальных каскадах
индукции апоптоза (Aguzzi and Polymenidou
2004).
- У мышей, лишенных кодирующего этот
белок гена (PRNP) не отмечено изменений
в поведении и репродуктивной
активности, вместе с тем на
энцефалограмме проявляются нарушения
долговременных потенциалов.
- Кроме этого, у таких мышей описаны
электрофизиологические изменения в
области гиппокампа (Kawahara, Kuroda et al.
2000).
47. Что происходит с белком при болезни?
Структурный переход PrPc – PrPSc
Вызывает превращение нормального белка
α-спирали переходят в β – слои
Образует нерастворимые агрегаты
(фибриллы)
Появляется устойчивость к протеиназе К
Связывают красители Конго Красный и
тиофлавин Т
48.
49. «Репликация»
50.
Мономер => олигомеры => агрегаты (фибриллы)
51. Как in vitro получить разные формы PrP?
2 M мочевина или гуанидингидрохлорид
Температура
Давление
52. Детекция амилоидных структур
• Электронная микроскопия (длинные инеразветвленные фибриллы)
• ИК-спектроскопия и КД (выявление
поперечно ориентированных β-слоев)
• Окрашивание Конго красным
• Флуоресценция тиофлавина Т
• Устойчивость к протеиназе К
53. Краситель Конго-Красный
54. Флуоресценция тиофлавина Т
55. Подходы к разработке лечения
А – стабилизациянативной структуры
B – ингибирование
протеаз, генерирующих
амилоидные пептиды
С – влияние на синтез
белка
D – активация
механизмов «отсева» и
деградации
E – ингибирование
образования фибрилл
F – предотвращение
накопления
промежуточных
агрегатов
56.
57.
58. Болезнь Альцгеймера
• Открыл Алоис Альцгеймер в 1907 году• Болеет 10% людей в возрасте старше 65 лет и
практически половина тех, кому больше 85 лет
• Клинические проявления –
амилоидные бляшки и
клубки => смерть нейронов
Самая распространенная
деменция
59. Эпидемиология
1. Генетический компонент• Наследственные формы – около 10% (аутосомнодоминантный тип)
• У людей с синдромом Дауна (трисомия по 21 хром.)
болезнь Альцгеймера развивается примерно к 40
годам
2. Коррелирующие факторы окружающей среды:
- Слабое развитие ребенка в раннем детстве
- Тяжелые травмы головы
- Алюминий в питьевой воде
Клинические, нейропатологические и биохимические
проявления идентичны во всех случаях, независимо от
причины возникновения
60. Болезнь Альцгеймера
Основные участники:- белок-предшественник β-амилоида (β-APP)
- Tau белок
- Белки, участвующие в процессинге β-APP (α,
β, γ – secretase, presenilins)
- Apolipoprotein E (APOE)
61. Tau белок
1. Функции:- стабилизирует микротрубочки в нейронах
- Необходим для аксонального транспорта
везикул
- Необходим для формирования аксонов и
дендритов при формировании и хранении
воспоминаний
- Регулируется
фосфорилированием
62. Повреждения нейронов
• В нейронах накапливаетсябольшое количество «клубков»,
они заполняют цитоплазму,
- Ядро смещается
Такие нейроны могут жить
годами, но не функционировать…
63. Функции βAPP
Белок-предшественник пептида бета-амилоида• В норме участвует в синаптическом транспорте
• Нейропротектор
В мутантном АРР происходит
отщепление 42 a.к. фрагмента βамилоида, формирующего бляшки
Мутации в гене АРР располагаются вблизи
сайтов расщепления α, β, γ – секретаз
64. Расщепление АРР в норме и патологии
• В норме происходит расщепление -секретазой=> -APP
• При Альцгеймере происходит расщепление - и
β-секретазами
=> 40 а.к. пептид - безвредный
=> 42 а.к. – пептид β-амилоид – 10% от
расщеплений
Почему такой вариант плох?
• 42 а.к. – пептид β-амилоид экскретируется во
внеклеточное пространство, формирует фибриллы и
бляшки
65. β-APP
66. Агрегаты при болезни Альцгеймера
67. Нейротоксичность бета-амилоида
• Агрегация пептида бета-амилоида приводит к:- Нарушениям кальциевого гомеостаза
- Повреждение митохондрий – окислительный
стресс
- Воспалительный ответ
- Нарушение энергетического обмена
- Потеря функций нейронов
68. Что происходит в мозге?
• Нейродегенерацияначинается примерно лет
за 20-30 до появления
симптомов
• Сначала появляются
клубки
• Патология развивается в
энторинальной коре, затем
распространяется в зоны
гиппокампа, амигдалы и
неокортекса
• Бета-амилоид сначала
появляется в неокортексе
Отложения тау-белка и бетаамилоида развиваются
независимо
69. Как выглядит пораженный мозг?
70. Этапы развития болезни
1. Первые признаки незначительны:- Легкая потеря кратковременной памяти
- Небольшие изменения в личности (обычно
принимаются за нормальное старение)
2. Прогресс ухудшения памяти:
- Невозможность поддержать беседу
- Невозможность решать задачи на несколько
этапов
3. Значительные повреждения памяти:
- Потеря долговременной памяти
- Невозможность совершать простые действия
(прием пищи, одевание и т.д.)
4. Смерть
71. Позитронная эмиссионная томография (PET)
• Позволяет визуализировать активностьмозга в момент совершения им
когнитивных функций – запоминаниявспоминания, речи, чтения и т.д.
• Позволяет выявить повышенный риск до
появления симптомов
• Проводится введение малых
безопасных доз радиоактивной
формы глюкозы
• Можно наблюдать, как мозг использует
глюкозу (красный > желтый > синий)
72. Лечение амилоидных болезней
• В настоящий момент неизлечимы• Для некоторых амилоидозов применяют
трансплантацию органов (печень)
• Иммунологический подход (вакцина АТ к
β-амилоиду), но не все так просто
• Необходимо искать способы ранней
детекции ДО развития клинических
симптомов
73. Подходы к разработке лечения
А – стабилизациянативной структуры
B – ингибирование
протеаз, генерирующих
амилоидные пептиды
С – влияние на синтез
белка
D – активация
механизмов «отсева» и
деградации
E – ингибирование
образования фибрилл
F – предотвращение
накопления
промежуточных
агрегатов
74. Подходы к лечению
1. Анти-ацетилхолинэстеразы замедляют развитие2. Хелаторы железа и меди – уменьшают
окислительный стресс
3. Ингибиторы β- и -секретаз = меньше бетаамилоида
4. Вакцины для развития иммунного ответа на бетаамилоид
75. Болезнь Паркинсона
• Вторая пораспространенности
среди
нейродегенеративных
болезней
• α-синуклеин формирует
амилоидные фибриллы
• Накапливается в тельцах
Lewy в нейронах мозга –
отличительная черта