




















 Медицина
МедицинаПохожие презентации:




 сипаттамасы, жіктелуі, таралуы")





Дәстүрлі емес тұқым қуалайтын аурулар. Мультифакторлы аурулар
1. Дәріс
Дәстүрлі емес тұқым қуалайтынаурулар.Мультифакторлы
аурулар.
2.
Мультифакторлы аурулар3.
Көп факторлы аурулар тұқым қуалайтын факторлардың(моно- немесе полигенді ) сыртқы орта факторларымен
әрекеттесуінің нәтижесі болып табылады . Бұл аурулардың
жынысы мен жасына байланысты көріністері мен ауырлық
дәрежесінде айырмашылықтар бар.
• Адамның көптеген фенотиптік белгілері гендердің үлкен
санымен бақыланады. Бұл гендердің әрқайсысы басқалардан
тәуелсіз әрекет етеді.
4.
Тұқым қуалайтын бейімділігі бар аурулар – дамуы тұқымқуалайтын және қоршаған орта факторларының өзара
әрекеттесуімен анықталатын аурулар тобы. Мұндай аурулардың
негізі - адам популяциясының ферменттер жүйесіндегі,
ақуыздардағы және антигендердегі генетикалық полиморфизмі.
Тұқым қуалайтын бейімділігі бар аурулар 2-ге бөлінеді:
1. Моногенді – бір геннің патологиялық мутациясымен
байланысты, оның көрінісі сыртқы орта факторының әрекетін
талап етеді. Олар аутосомды рецессивті немесе Х-тәрізді
рецессивті үлгіде тұқым қуалайды.
2. Полигенді аурулар көптеген гендермен анықталады, олар
өздерінің арасындағы және қоршаған орта факторларымен
белгілі бір әрекеттесу арқылы аурудың көрінісіне бейімділік
тудырады. Мұндай аурулар мультифакторлық деп аталады .
Оларға: псориаз, қант диабеті, шизофрения, ойық жара ауруы,
отбасылық көріністері бар гипертония жатады.
5.
Полигенді аурулардыңХалық арасында аурудың жоғары деңгейі. Осылайша,
халықтың шамамен 1% -ы шизофрениямен, 5% -ы қант
диабетімен, 10% -дан астамы аллергиялық аурулармен және
30% -ға жуық гипертониямен ауырады.
Аурулардың
клиникалық
полиморфизмі
жасырын
субклиникалық формалардан айқын көріністерге дейін өзгереді.
Аурулардың тұқым қуалау ерекшеліктері мендельдік
заңдылықтарға сәйкес келмейді .
Аурудың көріну дәрежесі науқастың жынысы мен жасына,
оның эндокриндік жүйесінің қарқындылығына, сыртқы және
ішкі ортаның қолайсыз факторларына, мысалы, нашар
тамақтануға және т.б.
6.
7.
Бронхиалды астма•Локализация – 4, 5, 6, 7, 11, 13, 14 хромосомаларда 30-дан астам гендер қатысады.
• Таралу – дүние жүзінде 0,2-18%.
• Бронх демікпесімен ауыратын балалардың 50-86%-да аллергиялық аурулардың
отбасылық тарихы анықталады. Монозиготикалық егіздер дизиготикалық егіздермен
салыстырғанда астманың даму қаупі жоғары .
• Этиологиясы, патогенезі
а) генетикалық факторлар (бейімді гендер);
б) сыртқы факторлар ( аллергенді , инфекция, темекі шегу, тамақтану және т.б.).
Белгілі болғандай, бронх демікпесі иммундық механизмдерге негізделген. Бронх
демікпесімен ауыратын адамдардың көпшілігі әртүрлі аллергендерге арнайы
антиденелер көрсетеді. Бұл тыныс алу жолдарының аллергиялық қабынуына
негізделген ауру, жедел бронхоспазмның дамуына және шырышты секрецияның
жоғарылауына әкеледі. Бұл өзгерістер бронхиальды обструкцияның бұзылуын
тудырады - бронхиальды обструкция.
• Фенотиптік белгілер – бронх демікпесінің белгілері: жөтел – жиі, ауырсынатын және
тұрақты болуы мүмкін; экспираторлық (негізінен дем шығарудың қиындауы бар) ентігу
– сырылдар;
• тұншығу.
8. Қант диабеті
Қант диабеті – инсулиннің (абсолюттіжәне салыстырмалы) жетіспеушілігінің
салдарынан болатын тұрақты
гипергликемия бұзылыстарымен
сипаттталатын, созылмалы
полиэтиологиялық ауру.
9.
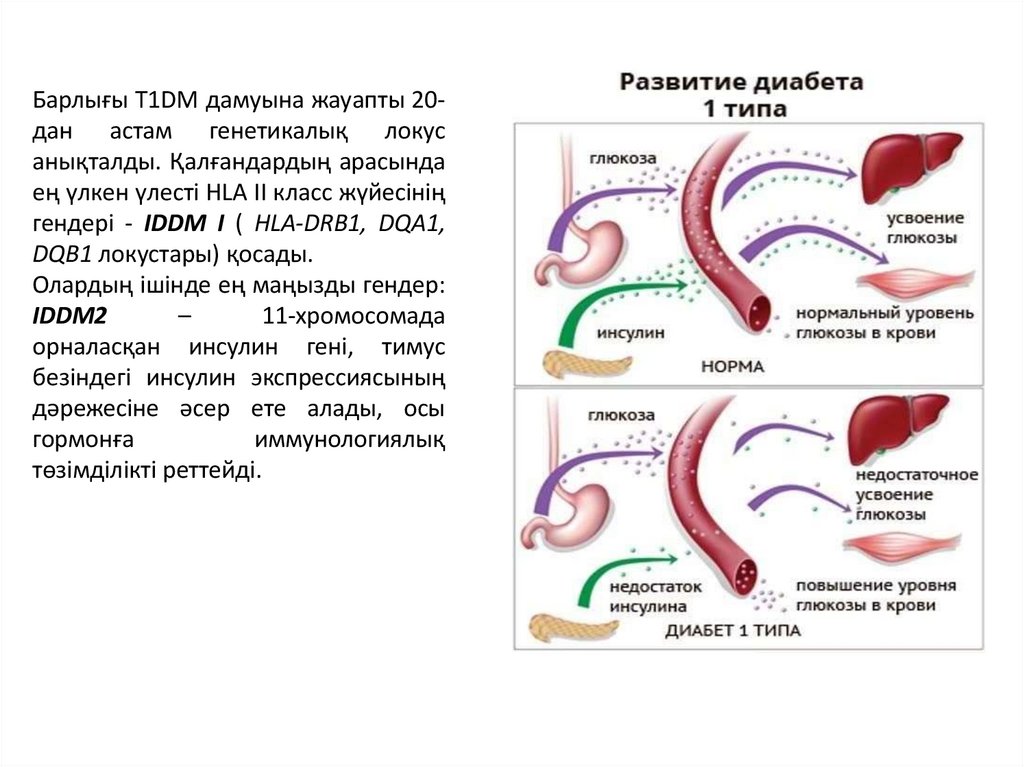
Барлығы T1DM дамуына жауапты 20дан астам генетикалық локусанықталды. Қалғандардың арасында
ең үлкен үлесті HLA II класс жүйесінің
гендері - IDDM I ( HLA-DRB1, DQA1,
DQB1 локустары) қосады.
Олардың ішінде ең маңызды гендер:
IDDM2
–
11-хромосомада
орналасқан инсулин гені, тимус
безіндегі инсулин экспрессиясының
дәрежесіне әсер ете алады, осы
гормонға
иммунологиялық
төзімділікті реттейді.
10. қант диабеті
Локализация – ақаулар гендер 6-шы хромосомада орналасқан
Таралуы – 4%
Этиологиясы, патогенезі – инсулин өндіруге жауапты геннің мутациясы , жаппай өлім эндокриндік ұйқы безінің жасушалары
бездер болуы мүмкін орын егер вирустық инфекциялар, онкологиялық аурулар, панкреатит, улы зақымданулар ұйқы безі бездер,
стресс жағдайлары , иммундық жасушалар болатын әртүрлі аутоиммундық аурулар жүйелер ұйқы безінің β-жасушаларына қарсы
антиденелер жасап , оларды бұзады.
11.
12.
Инсулинге тәуелсіз ІІ түрі - ұлғайған
адамдарда кездесетін және инсулиннің салыстырмалы
жеткіліксіздігінен дамиды (70%).
Қанда инсулиннің мөлшері аздап қана төмендеген
немесе
тіпті
қалыпты
деңгейде
болады.
Сырқаттардың арасында дененің толып кетуі жиі
байқалады және кетоацидоз дамымайды.
12.
• Гипергликемияға байланысты белгілері, қандағы қантмөлшері 10ммоль/л көп болғанда: полиурия, полидепсия, шөлдеу,
аштықты сезіну/қарны ашу/, жүдеу, терісі қышу, бас ауру,есі
тұмандану.
• Иммунитет төмендегендегі белгілер-зәр шығару жолдарының
аурулары, фурункулез.
• Микроангиопатияға байланысты белгілер-аяқ қол ұшында
сезімталдықтың бұзылуы, табанында, тобық буынында ісінеді.
• Диабетті-энцефалопатия –тәртібі бұзылады, бақылау қабілеті
бұзылады.
• Диабетті ретинопатия –сырқаттың көзі көрмей қалады.
• Жүрек қабынады- миокард инфарктісі жиі болады, өлім жиі
кездеседі және де ауыр ағымды әрі түрлі байқалады.
13.
ВИДЕО ОБЯЗАТЕЛЬНО К ПРСОМОТРУ ПРО ДИАБЕТhttps://www.youtube.com/watch?v=QZ0Amp4iLBo
https://www.youtube.com/watch?v=iEVl5UCjPfg
14.
Митохондриялық аурулар15.
16. МЕЛАС синдромдар
17. Геномды импринтинг аурулары
18.
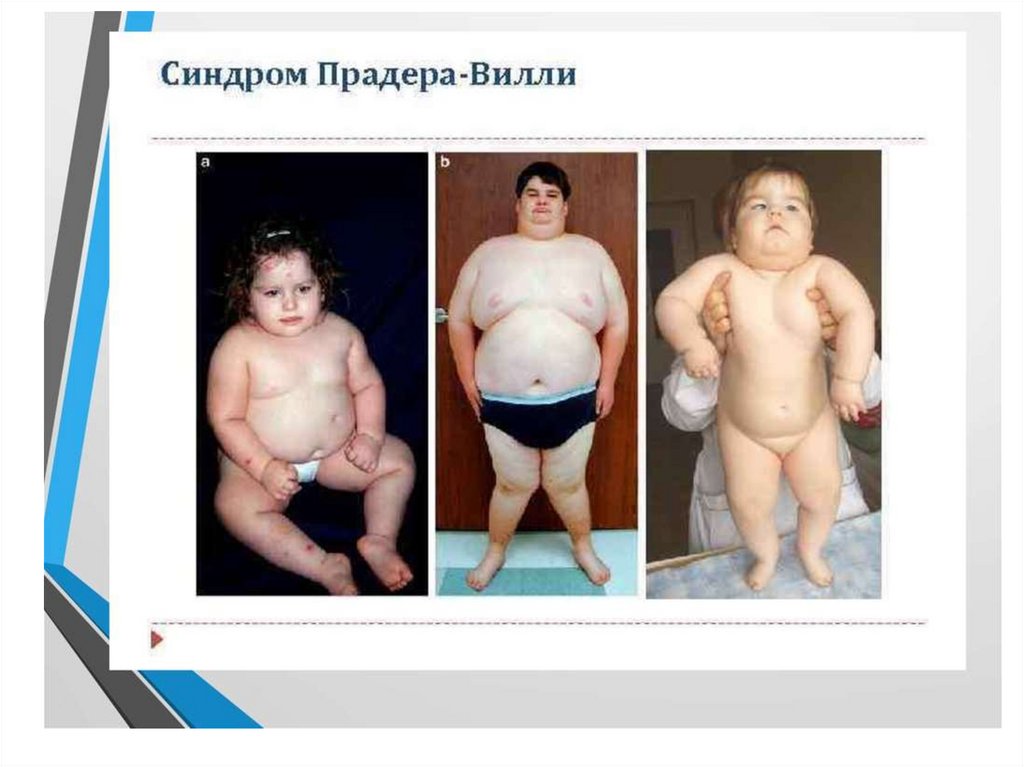
19. Синдром Прадера-Вилли
• Түр мұрагерлік- геномдық басып шығару.• Локализация – 15-ші хромосома.
• Таралуы - пайда болу жиілігі - 1 : 12 000-15 000 тірі туылған нәрестелер. Көпшілік
жағдайлар спорадикалық , үшін сирек сипатталған отбасы жағдайлар тән.
• Этиологиясы, патогенезі – сирек тұқым қуалайтын ауру, оның себебі 15q11-13 хромосома
аймағының аталық көшірмесінің болмауы . 15 -хромосоманың осы аймағында болып
табылады гендер, В реттеу қай қатысты геномдық басып шығару. Қосулы Бір ата-аналық
дисомияның пайда болуына ананың жасы әсер етеді ( аффект тұжырымдама бірге
трисомия немесе моносомия 15 хромосомалар). Мұндай жағдайлар анасы түсіндірді
алшақтық, сағ қай гомологтық хромосомалар жұптар Жоқ бөлініп , жасушаның бір
полюсіне барады . Мұндай гамета ұрықтанғаннан кейін хромосомалардың тақ санынан
тұратын зигота түзіледі . Бастамас бұрын қосымша хромосома дамыту эмбрион жылы көп
жағдайлар ашылған жою.
• Фенотиптік белгілері – Егер әкелік жоқ, белсенді аналық –
синдромы Прадера-Вилли.
• Бүйірінен қысылған маңдай бөлігі , бадам тәрізді көздері, салбыраған бұрыштары бар бас
сүйегі ауыз, кішкентай аяқ Және щеткалар, артта қалу психикалық даму, мінез-құлық
бұзушылықтар, физикалық кешігу дамуы, бойының қысқалығы, гипотензия,
гипогонадизм.
20.
бар аурулар дәстүрлі емес (классикалық емес) түрі мұрагерлік.Аурулар геномдық басып шығару
бір аталық дисомия жоқ әкелік хромосома - белсендіаналық
21.
22. Синдром Ангелман
• Түр мұрагерлік – геномдық басып шығару• Локализация – жою Және инактивация гендер қосулы 15 хромосома. IN аймақ q11—q13
15-шіхромосомалар. Кариотип 46 XX немесе XY, 15q
• Таралуы – 1:10 000
• Этиологиясы, патогенезі – Ангелман синдромы геномның классикалық мысалы болып
табылады басып шығару. Аурудың себептері унипатернальды дисомия болуы мүмкін ,
транслокация немесе мутация бір ген В бұл аймақ.
• Фенотиптік белгілері – В 75 % проблемалар бірге тамақтану, әсіресе бірге нәресте
тамақтандыру, осындай сәбилер Нашар жұмысқа қабылдауда салмақ;
тапшылық назар және гиперактивтілік;
эпилепсия (80 жағдайлардың %-ы), бұзушылықтар ашылады Сондай-ақ
электроэнцефалографиямен ;
жиі күлкі онсыз себебі;
жаяу жүру қосулы қатты аяқ — себебі бұл ерекшеліктері балалар бірге бұл синдромы
Кейде
салыстырылды бірге қуыршақтар;
• Ауыр кешіктіру психикалық Және мотор даму, атаксия, гипотензия, құрысулар
• бақыланбайтын күлкі, шапалақтау В алақан, микробрахицефалия бірге тегістелген бастың
артқы жағы, үлкен түбі жақ, жар ауыз бірге спикерлер тіл, макростомия, сирек өсіп келе
жатыр тістер, гипопигментация