












































 Химия
ХимияПохожие презентации:
")






")


КАТАЛИЗ Металлокомплексный 2
1. КАТАЛИЗ
1.4 Металлокомплексный иферментативный катализ
В последнее десятилетие все более широкое значение
получает гомогенный металлокомплексный катализ.
Комплексообразователем является атом или ион
переходного металла. Химические соединения, входящие
в координационную сферу комплекса переходного
металла, называются лигандами. Число лигандов,
входящих в координационную сферу комплекса, коордиационным числом.
По характеру химической связи металла с
лигандами
в
комплексах
переходных
металлов, имеющих значение для катализа,
лиганды можно подразделить на четыре
группы.
2. 1. Гомогенный катализ 1.1 Основные понятия. Причины каталитического действия.
Лиганды, имеющие только одну молекулярнуюорбиталь (МО) с неподеленной парой электронов, способную
взаимодействовать с атомом или ионом переходного металла
(например, NH3, Н2О). Они образуют связь посредством
комбинации с вакантными d-, s- или р-орбиталями металла. При
этом образуется донорная σ-связь.
1-я
группа.
3.
2-я группа. Лиганды, имеющие одну орбиталь содним электроном на ней (H, CH3, С2Н5 и другие
алкилы),
способную
взаимодействовать
с
переходным металлом. При этом также образуется σсвязь, но для образования двух элекронной связи
нужна орбиталь металла с одним электроном на ней.
Для образования такой связи требуется перенос
электрона
металла
с
несвязывающей
на
связывающую орбиталь, т. е. окисление металла.
4.
3-я группа. Лиганды с двумя заполненнымиорбиталями (CI-, Br-,I- ОН-), которые могут
взаимодействовать с двумя вакантными валентными
орбиталями металла. Образуются две связи: σ-связь
между –орбиталью металла и рx-орбиталью лиганда;
π-связь между dxy -орбиталью металла и рyорбиталью лиганда. При этом обе связи являются
донорными, т. е. обе пары электронов принадлежат
лиганду и при образовании ковалентной связи с
металлом плотность электронов на металле
увеличивается. Поэтому эти лиганды очень прочно
удерживаются в координационной сфере металла и
редко принимают непосредственное участие в
металлокомплексном катализе.
5.
4-я группа. Лиганды, имеющие наряду с заполненными такжевакантные орбитали, которые могут взаимодействовать с металлом
(монооксид углерода, олефины, фосфины и др.). Эта группа лигандов
имеет наиболее важное значение для реакций гомогенного
металлокомплексного катализа, используемых на практике.
6.
Наиболее важными элементарными процессами,которые протекают при осуществлении процессов
промышленного металлокомплексного катализа,
являются
обмен
лигандов,
окислительное
присоединение и реакция внедрения.
7.
Важнейшей элементарной реакцией, протекающей в координационнойсфере при гомогенном металлокомплексном катализе, является
реакция внедрения. Это реакция, происходящая в координационной
сфере переходного атома или иона, в результате которой один лиганд
внедряется между металлом и другим лигандом. Внедрение СО можно
описать с помощью элементарной стадии с трехцентровым переходным
состоянием:
где □ - вакантное место в координационной сфере. Другие лиганды,
кроме CR и СО, для простоты не указываются.
Внедрение С2Н4 описывается с помощью четырехцентрового
переходного состояния
Эти элементарные стадии называются реакциями цис-внедрения.
8.
Гомогенный металлокомплексный катализ имеет рядпреимуществ перед обычным гетерогенным катализом хорошую селективность, высокую общую активность. Однако
применение гомогенного катализа требует введения в
технологический процесс стадии разделения продуктов и
катализатора, что увеличивает трудоемкость, металлоемкость и
не позволяет проводить процесс непрерывно. Низка
стабильность гомогенных металлокомплексных катализаторов
и продолжительность их работы. Это связано с их большой
чувствительностью к кислороду и влаги, с их склонностью к
агломерации, что приводит к их постепенной дезактивации.
9.
Большие сложности вызывает регенерация гомогенныхметаллокомплексных
катализаторов.
Она
требует
включения в технологический процесс специальных
дополнительных операций и аппаратов. Для изготовления
гомогенных металлокомплексных катализаторов, как
правило, требуются благородные металлы, что повышает
их
стоимость
по
сравнению
с
гетерогенными
катализаторами.
10.
Очень высокой специфичностью (селективностью) обладают природныегомогенные катализаторы - ферменты. Это специальные белковые
молекулы, в которых имеются полости и несколько активных центров,
благодаря чему взаимодействующие молекулы строго ориентируются в
пространстве относительно друг друга и активных центров фермента. В
состав большинства ферментов входят металлы переменной валентности.
Поэтому ферменты можно формально относить к гомогенным
металлокомплексным катализаторам.Все процессы жизнедеятельности в
живой природе регулируются специальными ферментами.
11. 1.2 Каталитическая активность и селективность
1.5 Кинетика металлокомплексногои ферментативного катализа
Гомогенно-каталитические реакции являются, как правило, сложными,
многостадийными. При этом лимитирующими могут быть разные стадии. В
связи с этим математические модели также могут быть разными. Рассмотрим
несколько типичных случаев для закрытых систем в квазистационарном
приближении.
12.
Первый случай. Гомогенно-каталитическойреакции состоит из двух элементарных
односторонних реакций, причем продукт Е
взаимодействия катализатора К с первым
реагентом А во второй стадии реагирует со
вторым реагентом В с образованием второго
продукта Р, а катализатор К регенируется:
(26)
(27)
где D - первый продукт, который получается в
первой
стадии.
При
квазистационарном
протекании реакции скорость получения
промежуточного вещества Е равна нулю:
(28)
Общая концентрация
катализатора в системе
остается
постоянной
и
слагается
из
концентрации cE катализатора, связанного в
промежуточное соединение (или перешедшего в
промежуточную форму), и из концентрации cK
свободного катализатора в исходной форме:
= cE + cK или cK = - cE .
Из уравнения (28) получаем
(29)
(30)
Скорость реакции равна скорости образования
продукта Р:
С учетом выражения (30) находим отсюда
искомое кинетическое уравнение
(31)
Это выражение упрощается, если одним из
слагаемых в знаменателе можно пренебречь,
например, если k2cB » k1cA (или k2 » k1), т. е. когда
первая стадия является лимитирующей. При
этом
и уравнение (31)
преобразуется к виду
(32)
Эта
модель
кинетического
уравнения
встречается в окислительно-восстановительных
реакциях между ионами.
13.
Второй случай. Гомогенно-каталитическая реакция состоит из трех элементарныхреакций. В первых двух взаимно противоположных элементарных реакциях
(двусторонняя стадия) получается промежуточный продукт присоединения исходного
вещества А к катализатору К (промежуточный комплекс АК), который далее
распадается на продукты Р, R, … , а катализатор К регенерируется:
(33)
(34)
Подобная кинетическая модель часто встречается в металлокомплексном и
ферментативном катализе. В квазистационарном приближении концентрация
промежуточного комплекса определяется из соотношения
(35)
Учитывая, что общая концентрация катализатора постоянна и равна
получаем из (35)
откуда
(36)
= cK + cAK ,
14.
При этом считаем, что концентрация исходного вещества значительно большеконцентрации катализатора и поэтому можно пренебречь тем количеством вещества А,
которое вошло в состав комплекса.
Скорость образования целевого продукта Р равна
Разделив числитель и знаменатель на k1 , получим искомое уравнение
(37)
где KM = (k-1+k2)/k1 – константа Михаэлиса.
При достаточно большой концентрации исходного вещества cA»KM и KM + cA ≈ cA . При
этом
(38)
т.е. скорость реакции при увеличении концентрации исходного вещества стремится к
некоторому предельному значению wmax.
Подставляя значение wmax из уравнения (39) в (38), получим так называемое уравнение
Михаэлиса, которое первоначально использовалось для описания только
ферментативных реакций:
(39)
15.
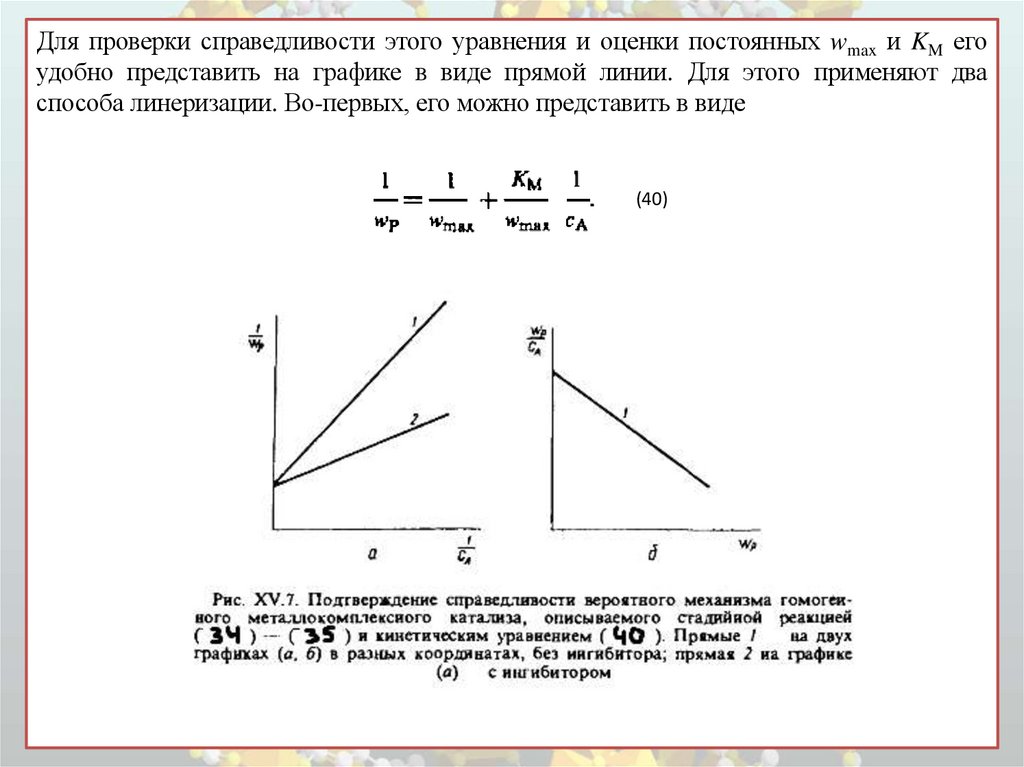
Для проверки справедливости этого уравнения и оценки постоянных wmax и KM егоудобно представить на графике в виде прямой линии. Для этого применяют два
способа линеризации. Во-первых, его можно представить в виде
(40)
16. 1.3 Соотношения Бренстеда-Поляни
Если опытные точки отложить на графике в координатах 1/wР -1/сА , то в случаесправедливости рассматриваемого механизма и его математической модели,
описываемой уравнением (40), должна получиться прямая линия с угловым
коэффициентом р=tg Θ = KM/wmax и отрезком на оси ординат q= 1/ wmax (рис. VX.7,
а). Зная из графика, построенного по опытным данным, wР при разных начальных
сA, а также q и р, вычисляем коэффициенты КM и wmax и далее k2 по уравнению
(38), если известно
Во-вторых, уравнение (39) можно переписать в виде
и помножить обе его части на 1/ сА КM :
(41)
При этом в случае адекватности данной математической модели реальному механизму
процесса опытные точки должны расположиться на прямой линий на графике в координатах
wР /сА - wР (рис. XV.7, б). Угловой коэффициент этой прямой равен р´=tg Θ´ = 1/KM и отрезок
на оси ординат q'= wmax/ KM. Найдя из графика р´ и q´, можно вычислить параметры wmax и
КM. Второй график считается более надежным для проверки механизма и оценки
кинетических параметров, так как опытные точки располагаются на нем более равномерно.
17.
Перспективным направлением в развитии катализа является гетерогенизированныйметаллокомплексный катализ. Гетерогенизация металлокомплексных катализаторов
заключается в том, что они закрепляются на твердом носителе (матрице). Это
позволяет устранить один из основных недостатков гомогенного металлокомплексного
катализа - необходимость отделения катализатора от реагентов после проведения
процесса. Таким образом, гетерогенизированный металлокомплексный катализ
занимает промежуточное положение между гомогенным и традиционным
гетерогенным катализом. Гетерогенизированные металлокомплексные катализаторы
называют также закрепленными, нанесенными, иммобилизованными, привитыми,
гибридными.
Влияние носителя на каталитическую активность и селективность комплексов нельзя
предсказать заранее. Носитель увеличивает стерические затруднения для
координируемых молекул реактантов. Результатом закрепления катализатора на
носителе может быть как повышение специфичности, так и понижение общей
активности. Носитель можно рассматривать как макролиганд. При этом для каждого
конкретного случая необходимо учитывать его электронное влияние на свойства
гетерогенизируемого комплекса. Носитель должен удовлетворять ряду требований.
Связь комплекса с носителем должна быть максимально прочной, чтобы
предотвращать его переход в реакционную среду при проведении каталитического
процесса. Он должен быть химически и термически стабильным; иметь достаточно
однородную поверхность; удельная поверхность носителя должна быть достаточно
велика. К числу таких носителей относятся различные кремнеземы и полистирол или
его сополимеры.
18.
Основным недостатком гетерогенизированных (закрепленных) металлокомплексных катализаторовявляется их сравнительная (с традиционными гетерогенными катализаторами) нестабильность.
Причиной нестабильности может быть постепенное смывание в раствор, которое связано, например, с
установлением различных равновесий в растворе с участием комплекса переходного металла или
протекание химической реакции между компонентами реакционной смеси и фиксирующим комплекс
лигандом. Гетерогенизированные металлокомплексные катализаторы более чувствительны (по
сравнению с традиционными гетерогенными катализаторами) к отравлению молекулами продуктов и
примесей.
Еще одно специфическое ограничение применимости этих катализаторов обусловлено недостаточным
отводом теплоты от поверхности катализатора при проведении экзотермических реакций, так как
носители, как правило, являются теплоизоляторами, а скорости реакций велики; может произойти
перегрев катализатора и его деструкция.
Недостатком является также довольно сложная их регенерация, которая в каждом конкретном случае
должна решаться особым способом.
Несмотря на некоторые недостатки, этот вид катализа в последние годы успешно развивается.
Ферменты также могут быть гетерогенизировани, т. е. химически связаны с полимерным носителем.
Такие катализаторы называют иммобилизованными ферментами. Изучение иммобилизации
ферментов стимулируется их большой практической важностью. Широко используются такие методы
иммобилизации, как образование химической ковалентной связи, ионное взаимодействие,
солеобразование, внедрение в полимер. Можно ожидать дальнейшего усовершенствования этих
методов с целью создания новых ценных катализаторов.
19.
1. 6. Кислотно-основной катализ. Другиевиды гомогенного катализа.
Различают общий и специфический кислотно-основной катализ. Согласно общей
(протонной) теории кислот и оснований кислотой является вещество, способное
отщеплять протон, а основанием - вещество, способное присоединять протон
(Бренстед). В дальнейшем для конкретности будем рассматривать водные
растворы.
Общий кислотно-основной катализ. Диссоциацию кислоты НА и основания В
можно представить в виде следующих протолитических реакций с участием
растворителя (H2O):
В общем кислотном катализе каталитическим действием кроме иона гидроксения
(протона) обладают также недиссоциированная молекула кислоты (НА) и
молекула растворителя (Н2О). Поэтому скорость лимитирующей стадии при
общем кислотном гомогенном катализе состоит из трех слагаемых
20. 1.4 Металлокомплексный и ферментативный катализ
Аналогично, в общем основном катализе получаемгде cS - исходное вещество. Общий кислотно-основной катализ имеет место в том
случае, когда переход протона является замедленной стадией и сам катализатор
(кислота или основание) участвует в лимитирующей стадии.
Рассмотрим двустадийную реакцию
Если 2-я стадия реакции (45) является лимитирующей, то
При этом первая стадия практически равновесна:
21.
С учетом (47) получим из (46)(48)
где kэфф = kHAcHA ; kHA= k2K. В данном случае в выражение для общего кислотного
катализа входит только одно слагаемое из трех, входящих в общее выражение (42).
Из уравнения (48) следует, что в общем кислотном катализе имеет значение не
только рН раствора, но и природа кислоты-катализатора.
Рассмотрим способ определения kHA в уравнении (42), которое перепишем в таком
виде:
(49)
Если провести серию опытов при разной концентрации кислоты - катализатора НА и
при постоянном рН раствора (буферный раствор), то величина L в уравнении (49)
будет постоянной и на графике в координатах kэфф - cHA согласно (49) получится
прямая линия с угловым коэффициентом, равным kHA. (Раствор предполагается
разбавленным, поэтому Сн2о - практически постоянная величина. ) Для разных
кислот - катализаторов угловой коэффициент и, следовательно, величина будут
иметь разное значение. Имеются способы определения также
, входящих
в уравнение (42). В частном случае выражения для скорости реакции (48) величина L
из графика kэфф - cHA будет равна нулю, так как kэфф = kHAcHA и слагаемые
и
равны нулю.
22.
Для общего кислотно-основного катализа между константой скорости kкаталитической реакции и константой ионизации Кa, кислоты - катализатора (в
случае общего кислотного катализа) или константой основности Кв, основания катализатора (в случае общего основного катализа) выполняется соотношение
линейности, так называемое корреляционное соотношение Бренстеда:
(50)
или в логарифмической форме
(51)
Эти соотношения хорошо подтверждаются опытом. Например, если для данной
гомогенно-каталитической реакции использовать разные однотипные кислоты катализаторы, то на графике в координатах Ink- In Кa опытные точки располагаются
на прямой линии в соответствии с уравнением (51). Аналогичное соотношение
наблюдается для оснований - катализаторов на графике Ink- InKв .
23.
Специфический кислотно-основной катализ. Если катализатором-кислотойявляется только ион гидроксония Н3О+, а катализатором-основанием - только ион
гидроксила ОН-, катализ называют специфическим кислотно-основным. Он имеет
место, когда в лимитирующей стадии реакции не принимают участие другие формы
катализатора, кроме Н3О+ и ОН- Например, специфический кислотный катализ (S субстрат)
(52)
(53)
и специфический основной катализ
(54)
(55)
Иногда механизм кислотно-основного катализа включает несколько стадий,
например, содержит стадию дегидратации
(56)
(57)
(58)
24. 1.5 Кинетика металлокомплексного и ферментативного катализа
Рассмотрим сначала специфический кислотный катализ по схеме (52) и (53) вразбавленном водном растворе. Если вторая стадия (53) лимитирующая, то
(59)
Активная форма SH составляет некоторую долю χ =
от общей концентрации
исходного вещества (субстрата) в растворе. Если равновесие в первой стадии (52)
устанавливается быстро, то соотношение активностей практически не отличается от
равновесного и определяется константой основности
(60)
Если раствор разбавленный, то активности
можно заменить
концентрациями, а активность воды принять равной единице. Кроме того, если
концентрация исходного вещества (S) значительно больше концентрации
катализатора (Н3О+), то равновесную концентрацию исходного вещества можно
считать практически равной его общей концентрации:
С учетом этих упрощений выражение (60) примет вид
(61)
25.
Подставляя уравнение (61) в (59), получаем для кинетического уравненияспецифического кислотного катализа выражение
(62)
(63)
где kэфф – эффективная константа специфического кислотного катализа.
Аналогичные математические преобразования для специфического основного
катализа (в разбавленном растворе) приведут к кинетическому уравнению (62) :
(64)
где Kи =
– константа ионизации в первой равновесной стадии.
26.
1. 7. Гомогенный катализ в газовой фазеГомогенно-каталитические реакции в газовой фазе протекают обычно по механизму
цепных реакций. Примером может служить гомогенный катализ цепной реакции
окисления монооксида углерода в диоксид углерода в присутствии следов водяного пара.
Реакция начинается с образования атома кислорода (активного центра):
При отсутствии паров воды цепная реакция не развивается, так как активный центр Ȯ не
регенерируется:
где М - инертная частица (например, стенка сосуда или примесь в газе), которой передается
избыточная энергия. Могут быть и другие реакции с участием активного центра Ȯ,
приводящие к обрыву цепи.
27.
В присутствии следов водяного пара в газовой смеси активный центр Ȯвступает в реакцию с молекулой Н2О, в связи с чем протекает
разветвленная цепная реакция с регенерацией активных центров ȮН, Ḣ и
Ȯ:
В стадиях (а) и (б) возникают частицы
Ӧ и ȮН, которые участвуют в
разветвленной
цепной
реакции
[стадии (в), (г), (д), (в)…]. В стадиях
(e), (ж), (з), (и) происходит обрыв
цепи. В стадии (и) регенерируются
молекулы катализатора Н2О, который,
таким образом, к концу реакции
остается без изменения. Механизм
гомогенного
катализа
рассматриваемой ценной реакции
можно считать в основных чертах
доказанным, так как промежуточные
активные частицы ȮН, Ḣ, Ȯ
обнаружены при помощи новых
физических методов (ЭПР, ИКспектроскопия).
28.
2. Гетерогенный катализ• При гетерогенном катализе катализатор представляет собой
твердое тело, а реагирующие вещества могут находиться в
газовой фазе или в растворе.
• Механизм гетерогенного катализа в принципе не отличается от
гомогенного. Атомы или группы атомов на поверхности
твердого катализатора образуют с реагирующими веществами
активированные комплексы или неустойчивые промежуточные
соединения. Вследствие этого снижается энергия активации и
реакция ускоряется в том или в другом термодинамически
возможном направлении. Нужно учитывать также изменение
энтропии активации активированного комплекса с участием
катализатора. Однако механизм процесса в гетерогенном
катализе сложнее, чем в гомогенном. Это объясняется, в
частности, адсорбцией реагирующих веществ на поверхности
твердого тела и десорбцией продуктов с его поверхности при
гетерогенно-каталитическом процессе.
29.
2.1 Гетерогенные катализаторыПо способу осуществления различают гетерогенно-каталитические процессы с
неподвижным катализатором, когда его используют в виде достаточно крупных гранул
(0,3 - 1,0 см), и с подвижным (плавающим, диспергированным или
псевдоожиженным), когда его применяют в измельченном виде, причем катализатор
способен перемещаться под влиянием потока реагентов.
Рассмотрим некоторые особенности гетерогенных катализаторов.
1) Прибавление к катализатору вещества, которое само по себе является каталитически
недеятельным, может иногда сильно повысить активность катализатора. Такие
вещества называют промоторами или модификаторами. Как правило, эффективность
действия промотора зависит от его количества: максимальная активность катализатора
наблюдается при определенном составе промотированных катализаторов. Промоторы
влияют на состояние поверхности катализатора - они способствуют сохранению
активных мест на поверхности и изменяют их распределение по энергиям адсорбции.
Промоторы добавляют к катализатору на соответствующей стадии его приготовления.
30.
2) Различают структурообразующие и модифицирующие промоторы.Структурообразующие промоторы стабилизируют активную фазу катализатора по
отношению к нагреванию или другим воздействиям; они препятствуют
термической рекристаллизации (укрупнению кристаллов) катализатора.
Модифицирующие промоторы (модификаторы) изменяют строение и химический
состав активной фазы. В зависимости от концентрации одни и те же добавки могут
оказывать как промотирующее, так и отравляющее действие.
31.
• От промотирования (илимодифицирования) следует отличать
смешанные катализаторы. Смешанные
катализаторы состоят из компонентов,
каждый из которых обладает
каталитической активностью к данной
реакции. Они могут существенно
отличаться по каталитической активности
от исходных компонентов.
32. 1. 6. Кислотно-основной катализ. Другие виды гомогенного катализа.
3) Срок службы катализаторов зависит от многих факторов:главные из них - отравление и старение. Присутствие
реагирующей смеси некоторых веществ, часто в совершенно
ничтожном количестве, способно понижать или полностью
подавлять активность катализатора. Такие вещества называют
каталитическими ядами, а само явление - отравление
катализатора. К типичным каталитическим ядам относят
соединения серы H2S, CS2, тиофен, тиоспирты, синильная
кислота; монооксид углерода, свободные галогены, ртуть и
соли ртути (HgCl2, Hg(CN)2); соединения фосфора, мышьяка,
свинца и др. Действие ядов зависит от природы катализатора.
33.
Снижение активности катализатора может вызываться нетолько каталитическими ядами, но и старением катализатора,
которое обусловлено рядом причин: образованием из тонко
раздробленной, термодинамически неустойчивой активной
структуры в процессе рекристаллизации стабильной
крупнокристаллической структуры с меньшей поверхностью;
процессами перекристаллизации в поверхностном слое – в
особенности при проведении реакции при высоких
температурах; отложением на поверхности катализаторов
продуктов, полученных при протекании каких-либо побочных
процессов; изменением структуры или химического состава
носителя.
34.
4) При практическом использовании катализаторы часто наносят на поверхностьтвердых тел с развитой поверхностью, так называемых носителей. В качестве
носителей обычно употребляют древесный уголь, кокс, силикагель, алюмогель,
асбест, пемзы, кизельгур, стекло, фарфор, сульфат бария и др. Функции носителя
не ограничиваются только экономией катализатора, что имеет значение для таких
дорогих катализаторов, как платина, палладий, золото, серебро, осмий и иридий.
Носитель способен влиять на активность и избирательность катализатора,
проявляя определенный промотирующий эффект, а также способен резко
повышать устойчивость нанесенных или адсорбционных катализаторов к
спеканию при температурном воздействии и к отравлению ядами по сравнению с
массивными металлическими катализаторами.
35.
Спеканием катализатора называется уменьшение истинной поверхностикатализатора и се удельной активности. Носитель препятствует спеканию,
повышая срок и температурный интервал действия катализатора. Это позволяет
повышать температуру проведения процесса, ускорять реакцию и повышать выход
продукта реакции.
36.
5) По типу реакций, которые на них осуществляются,гетерогенные катализаторы подразделяют на окислительновосстановительные и кислотно-основные. По своей природе
наиболее важные промышленные гетерогенные катализаторы
представляют собой металлы, сплавы металлов, оксиды и
смеси оксидов (шпинели). Используются и другие химические
соединения.
37.
Важным классом оксидных кислотно-основных гетерогенныхкатализаторов являются цеолиты, которые по химическому
составу являются алюмосиликатами. Их особенностью
являются очень большие параметры кристаллической решетки,
благодаря чему молекулы реагирующих веществ могут
диффундировать внутрь кристалла катализатора и
каталитическая реакция осуществляется при участии почти
всех его внутренних атомов.
38.
• Путем специальной обработки катионынатрия, входящие в состав цеолита, могут
быть заменены на ионы водорода, что
придает ему сильно кислотные свойства и
делает активным катализатором кислотноосновного взаимодействия. Это позволяет
создать
на
основе
кристаллических
цеолитов очень активные катализаторы
крекинга углеводородов. В состав цеолитов
легко
вводить
катионы
переходных
39. 1. 7. Гомогенный катализ в газовой фазе
2.8 Теория гетерогенного катализаИзвестно несколько общих теорий гетерогенного катализа. Различие между ними
заключается в основном во взглядах на природу поверхностных соединений и на природу
активных мест поверхности катализатора, участвующих в образовании поверхностных
соединений. Наибольшее распространение в прошлые годы получили три теории:
мультиплетная, активных ансамблей и электронная.
Согласно мультиплетной теории гетерогенного катализа (А. А. Баландин) предполагается,
что в образовании поверхностного соединения (мультиплитного комплекса) участвуют
группы активных атомов поверхности - мультиплеты (дуплеты, триплеты, квадруплеты и т.
п.), обладающие определенными геометрическими и энергетическими свойствами. В
мультиплетной теории рассматриваются принципы геометрического и энергетического
соответствия. Согласно принципу геометрического соответствия твердое тело может быть
гетерогенным катализатором, если расположение активных мест на его поверхности
находится в геометрическом соответствии с расположением атомов в молекулах
реагирующих веществ. Кроме того, расстояние между атомами в мультиплете должно
соответсттовать расстоянию (длине химической связи) между атомами в реагирующих
молекулах, образующих на поверхности катализатора мультиплетный комплекс.
Необходимость соответствия между длиной химической связи в реагирующей молекуле и
расстоянием между атомами в мультиплете можно пояснить на примере реакции
гидрирования этилена на никеле:
40.
При образовании мультиплетного комплекса двойная связь в молекуле этилена переходитв одинарную и свободными валентностями оба атома углерода присоединяются к двум
атомам дуплета на поверхности никеля:
Углы между одинарными (гибридными) связями углерода составляют 120° и длина
одинарной связи С-С равна 0,154 нм. Принцип геометрического соответствия, как видно из
схемы, требует, чтобы расстояние между атомами дуплета было больше расстояния между
атомами С - С и при образовании мультиплетного комплекса не было большого искажения
углов между связями. Поверхность никеля удовлетворяет этому условию.
На основе принципа геометрического соответствия рассматривался, например, вопрос о
подборе металла (катализатора) для реакции дегидрирования циклогексана: С6Н12 →
С6Н6+3Н2. Из принципа геометрического соответствия следует, что мультиплет для катализа
этой реакции должен быть секстетом. Можно предполагать, что катализаторами реакции
дегидрирования циклогексана должны быть металлы с гексагональной или
гранецентрированной кубической решеткой и с расстоянием между атомами секстета
около 0,25 нм. Такими геометрическими свойствами обладают 11 металлов: Ni, Co, Zn, Ru,
Rh, Pd, Pt, Ir, Os, Re, Си, у которых постоянная решетки равна от 0,25 до 0,28 нм. Опыт
показывает, что 10 из этих 11 металлов (кроме меди) действительно могут служить
41. 2. Гетерогенный катализ
Принцип энергетического соответствия утверждает, что кроме геометрическогосоответствия должно быть также определенное соответствие между энергиями связи
атомов в молекулах реагирующих веществ и в мультиплетном комплексе, чтобы данное
твердое тело могло быть катализатором рассматриваемой реакции. При этом энергии
связей реагирующих молекул с активными центрами на поверхности катализатора должны
быть оптимальными и находиться в определенном соответствии с энергиями связей между
атомами в молекулах реагирующих веществ. Энергетический уровень мультиплетного
комплекса должен быть расположен приблизительно посредине между энергетическими
уровнями исходных молекул и продуктов реакции, а энергии активации его образования и
распада должны быть минимальными.
Мультиплетная
теория
неприменима
к
окислительно-восстановительным
(гомолитическим) реакциям с участием простых молекул, так как процессы получения и
отдачи электронов могут происходить в разных пространственно разделенных участках
поверхности; поэтому для гомолитических реакций не требуется такого строгого
геометрического соответствия между строением молекул и расположением атомов
мультиплета на поверхности катализатора. Кроме того, при применении принципа
энергетического соответствия к этому типу реакций нужно учитывать дополнительно еще
ряд других энергетических величин (работу выхода электрона с поверхности катализатора,
сродство к электрону или к атому реагирующих молекул).
Накопление новых опытных данных показало, что в мультиплетной теории содержится
определенное рациональное зерно. Однако значение геометрического принципа, повидимому, преувеличено. Например, процесс гидрирования бензола успешно
осуществляется в гомогенном металлокомплексном катализе, где нет никакого
геометрического соответствия. Принцип энергетического соответствия сохраняет свое
значение и в настоящее время.
42. 2.1 Гетерогенные катализаторы
В теории активных ансамблей гетерогенного катализа (Н. И. Кобозев) предполагается, чтоактивными центрами служат атомы, беспорядочно расположенные на поверхности
кристаллического тела (аморфная, докристаллическая фаза). Теория применима, если на
поверхности носителя находится небольшое число атомов металла, обычно меньше 1%
того количества, которое требуется для заполнения всей поверхности мономолекулярным
слоем этого материала (так называемые адсорбционные катализаторы).
Рассмотрим строение поверхности адсорбционного катализатора, когда на поверхность носителя нанесено
небольшое количество металла (например, платина на силикагеле) (рис. XVI. 4, a) Согласно современным взглядам
твердое кристаллическое тело (носитель) состоит из большого числа микроскопических участков - блоков или
областей миграции, разделенных геометрическими и энергетическими барьерами. При нанесении на носитель
небольшого числа атомов металла на каждую такую область миграции попадает несколько атомов металла. Под
влиянием теплового движения атомы металла могут перемещаться внутри этих областей миграции, но переход из
одной области миграции в другую затруднен наличием между ними геометрических (рис.XVI. 4, б) и энергетических
(рис. XVI. 4, в) барьеров. Несколько атомов металла-катализатора внутри области миграции называют ансамблем. В
разных областях миграции может находиться разное число атомов металла. Но каталитическое действие согласно
этой теории проявляют только ансамбли с определенным числом атомов металла внутри области миграции. Такие
ансамбли получили название активных ансамблей.
43.
Число атомов в активном ансамбле можно определить из зависимостиактивности адсорбционного катализатора от среднего числа атомов
металла в одной области миграции, так как при этом активность должна
иметь максимальное значение. Это объясняется тем, что согласно теории
вероятности при случайном распределении атомов на поверхности
носителя наибольшее число областей миграции будет содержать среднее
число атомов металла.
Часть атомов на поверхности кристаллов находится в аморфном состоянии
(не входит в состав кристаллической решетки). Согласно теории активных
ансамблей именно эти атомы являются каталитически активными. Однако,
как показали расчеты, эта теория неприменима к кристаллическим
катализаторам, так как рассчитанная на ее основе активность катализатора
оказывается во много раз меньше опытного значения. Это указывает на то,
что в случае кристаллических катализаторов активными центрами
являются в основном атомы кристаллической решетки, а не аморфная фаза.
44.
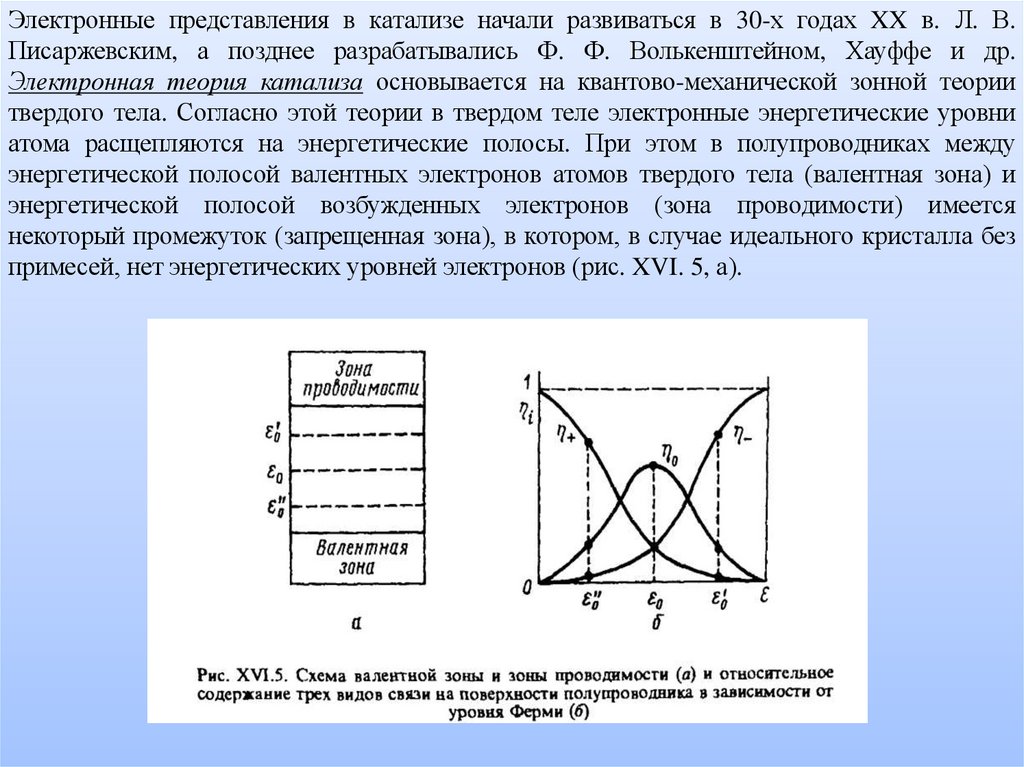
Электронные представления в катализе начали развиваться в 30-х годах XX в. Л. В.Писаржевским, а позднее разрабатывались Ф. Ф. Волькенштейном, Хауффе и др.
Электронная теория катализа основывается на квантово-механической зонной теории
твердого тела. Согласно этой теории в твердом теле электронные энергетические уровни
атома расщепляются на энергетические полосы. При этом в полупроводниках между
энергетической полосой валентных электронов атомов твердого тела (валентная зона) и
энергетической полосой возбужденных электронов (зона проводимости) имеется
некоторый промежуток (запрещенная зона), в котором, в случае идеального кристалла без
примесей, нет энергетических уровней электронов (рис. XVI. 5, а).
45.
Чтобы перейти из валентной зоны в зону проводимости, электрону нужно иметьнекоторую минимальную избыточную энергию активации, равную ширине запрещенной
полосы. Переход электрона из валентной зоны в зону проводимости соответствует
перескоке электрона с аниона на катион и его перемещению по катионной подрешетке
(электронная, или n-проводимость). После перехода электрона в зону проводимости в
валентной зоне остается дырка, которая соответствует аниону, лишенному электрона, и
перемещению этой дырки по анионной подрешетке (дырочная, или р-проводимость). В
идеальном кристалле полупроводника имеется одинаковое число свободных электронов
и дырок (n- и р-носителей тока) и имеет место смешанная n- и р- проводимость.
Если в кристалле имеются донорные или акцепторные примеси (например, изоморфно
замещающие ионы в узлах кристаллической решетки), то в объеме и на поверхности
полупроводника появляются избыточные электроны в зоне проводимости или
избыточные дырки в валентной зоне и соответствующие локальные уровни энергии
внутри запрещенной зоны. В зонной теории относительное количество электронов и
дырок в полупроводнике характеризуется так называемым уровнем энергии Ферми (или
просто уровнем Ферми), который имеет смысл химического потенциала электрона в
полупроводнике.
В идеальном кристалле полупроводника (в отсутствие примесей) уровень Ферми
расположен примерно посредине между зонами валентной и проводимости (уровень ε0
на рис. XVI. 5, а). При наличии донорной примеси (имеющей лишние электроны) уровень
Ферми подымается вверх (уровень ). Это соответствует полупроводнику n-типа
(преимущественная электронная проводимость). При наличии акцепторной примеси
(имеющей недостаток электронов) уровень Ферми опускается вниз (уровень ε"); это
соответствует полупроводнику р-типа (дырочная проводимость).
46. 2.2 Адсорбция на поверхности катализатора
Электронная теория катализа допускает существование разных видов связи хемосорбированныхчастиц из газа на повверхности полупроводника: слабой одноэлектронной связи и двух видов прочной
двухэлектронной связи - акцепторной и донорной, которые в свою очередь могут иметь ковалентный
или ионный характер в зависимости от природы адсорбируемой частицы.
Свободные валентности (электроны и дырки) могут перемещаться (блуждать) по кристаллу. Поэтому
прочная связь может переходить в слабую, а слабая в прочную. При данных условиях (температура,
содержание примесей и т. п.) на поверхности полупроводника имеются определенные равновесные
доли слабой связи η0, прочной акцепторной или n-связи η_ , и прочной донорной, или р-связи η+,
причем η0 + η_ + η+ = 1. Относительное содержание этих трех видов связи на поверхности
полупроводника определяется уровнем Ферми (рис. XVI. 5, б). В идеальном кристалле без примесей
уровень Ферми равен ε0 и доля η0 слабой связи является преобладающей. Если уровень Ферми
смещен вверх в сторону зоны проводимости, то преобладает доля η_ прочной акцепторной связи (nсвязи). Если уровень Ферми смещен вниз в сторону валентной зоны, то преобладает доля прочной
донорной связи (р-связи). Таким образом, в зависимости от характера полупроводника (n- или рполупроводник) на нем будет протекать адсорбция прсимущественно на прочной акцепторной или
прочной донорной связи и cоответственно будет меняться избирательность (селективность)
полупроводникового катализатора.
Электронная теория катализа на полупроводниках позволила в ряде случаев установить корреляцию
между активностью катализатора и электрофизическими свойствами полупроводника и таким
образом объяснить влияние различных факторов на изменение активности катализатора. Однако
электронная теория катализатора имеет и ряд недостатков. Ионы адсорбата и ионы полупроводника
рассматриваются как бесструктурные несжимаемые точечные ионы. Участием конкретных атомных
орбиталей (s-, p-, d- или др.) в образовании связей между катализатором и реагентом пренебрегают.
Основное внимание уделяется лишь коллективным свойствам твердого тела – положению в нем
уровня Ферми и влиянию его изменения при воздействии разными факторами (например, введение
добавок) на концентрацию адсорбированных молекул, находящихся в состоянии прочной или слабой
связи. Ряд экспериментов, проведенных в последнее время, показал, что роль коллективных свойств
системы катализатор - адсорбат преувеличена.
47.
Основной недостаток электронной теории катализа на полупроводниках заключается втом, что свойства поверхности полупроводника сопоставляются с физическими свойствами
твердого тела, хотя между ними имеется только косвенная связь. И те и другие зависят от
химического состава и структуры вещества катализатора, но зависимость эта может быть
разная. Подход с позиции коллективного взаимодействия позволяет оценивать в ряду
катализаторов изменения лишь той части промежуточного взаимодействия, которая
отвечает смещению уровня Ферми. А эти изменения во многих случаях не основные, что
снижает ценность электронной теории для предвидения каталитического действия
полупроводниковых катализаторов.
В последнее время развивается квантово-химическая теория катализа, в которой к
системе адсорбат -- катализатор применяются приближенные квантово-химические
методы расчета. В связи со сложностью проблемы полученные результаты еще очень
невелики, но в принципе это направление перспективно и в будущем оно позволит
предсказывать каталитическую активность. Однако трудность расчета и сложность
системы катализатор - реагенты не позволяют использовать квантово-химическую теорию
для выбора оптимального катализатора.
Предложена цепная теория катализа (Семенов, Воеводский), согласно которой
катализатор, обладая свободными валентностями, может действовать, как свободный
радикал, возбуждая образование цепей и участвуя в их развитии. Цепную теорию катализа
можно рассматривать как распространение электронной теории катализа на
полупроводниках (и металлах) на класс цепных реакций. Цепной механизм встречается
сравнительно редко при гетерогенно-каталитических реакциях, так как образование
радикалов -- эндотермический процесс, требующий большой затраты энергии для
разрыва связей.
Рассмотренные универсальные теории катализа в настоящее время в значительной мере