
























 Медицина
МедицинаПохожие презентации:










Мышечная дистрофия Дюшенна
1.
НАО «Западно-КазахстанскийМедицинский Университет
имени Марата Оспанова»
Тема: Мышечная дистрофия Дюшенна
Научный руководитель: PhD, Аяганов Д.Н.
Құлжанова А.А., студентка 5 курса, ф-т «Общая
медицина»
2.
Нервно-мышечные заболеванияНервно-мышечные заболевания – условно
выделяемая группа заболеваний, которые
характеризуются нарушением функции мышц, прежде
всего их слабостью.
3.
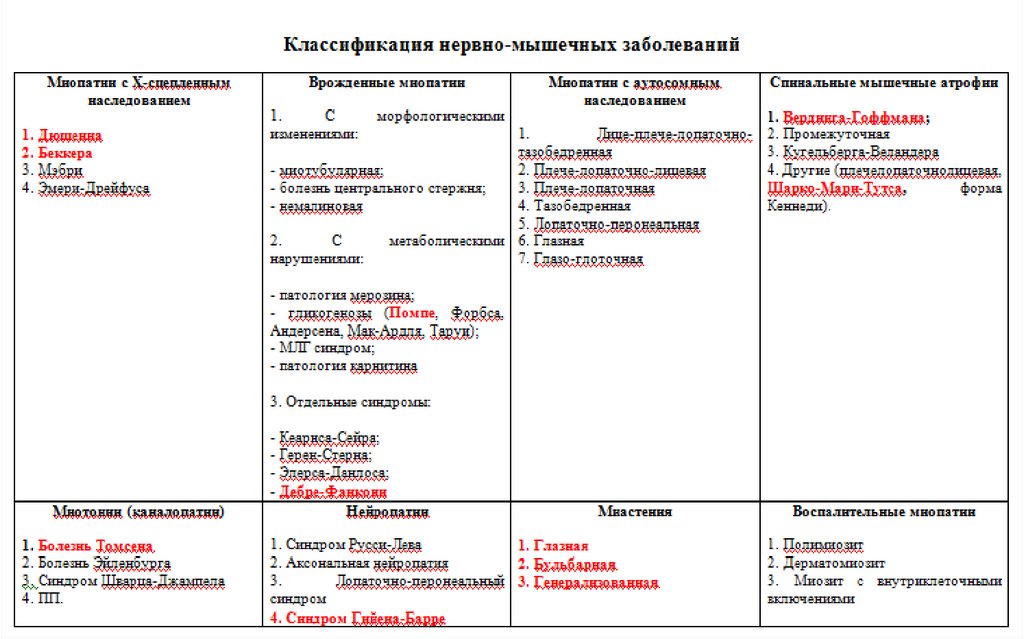
Нервно-мышечные заболевания делятся:1. Первичные миопатии с рецессивным Х-сцепленным
наследованием;
2. Первичные миопатии с аутосомным наследованием;
3. Врожденные миопатии;
4. Спинальные мышечные атрофии;
5. Миотонические синдромы;
6. Нейропатии;
7. Миастения;
8. Воспалительные миопатии.
4.
5.
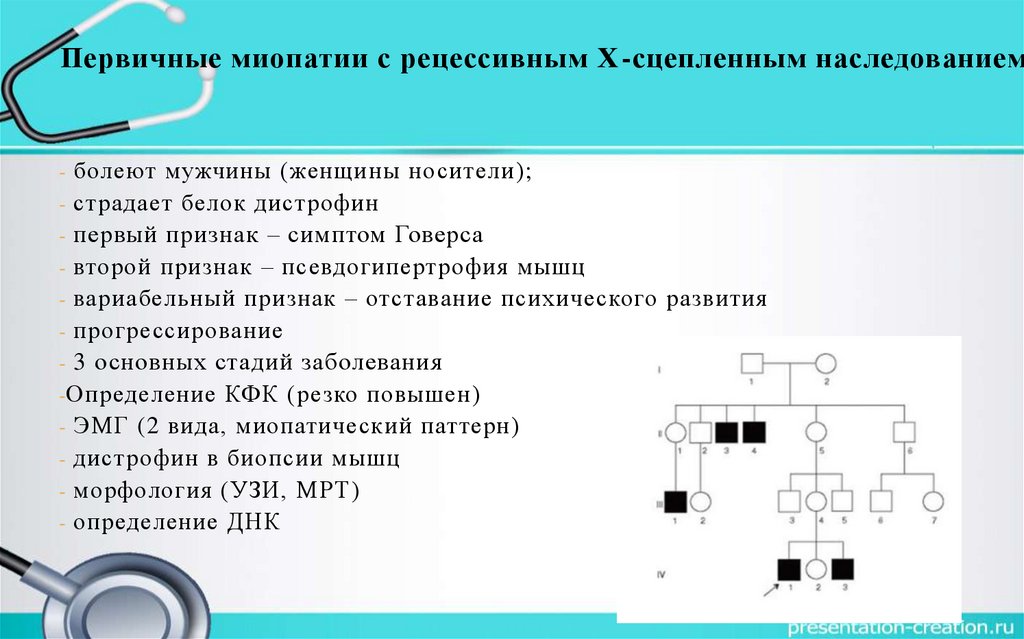
Первичные миопатии с рецессивным Х-сцепленным наследованием- болеют мужчины (женщины носители);
- страдает белок дистрофин
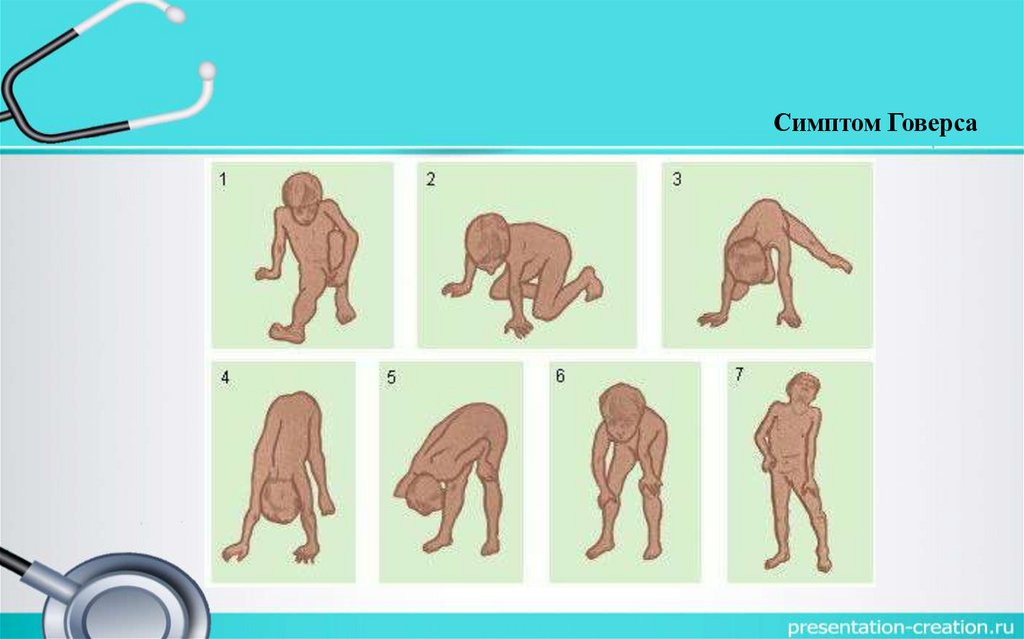
- первый признак – симптом Говерса
- второй признак – псевдогипертрофия мышц
- вариабельный признак – отставание психического развития
- прогрессирование
- 3 основных стадий заболевания
-Определение КФК (резко повышен)
- ЭМГ (2 вида, миопатический паттерн)
- дистрофин в биопсии мышц
- морфология (УЗИ, МРТ)
- определение ДНК
6.
Формы заболеваний1.
2.
3.
4.
Форма
Форма
Форма
Форма
Дюшенна;
Беккера;
Мэбри;
Эмери-Дрейфуса
7.
• Мышечная дистрофия Дюшенна- наследственное, рецессивное,сцепленное с Х-хромосомой, нервно-мышечное заболевание,
вызванное мутациями в гене DMD, приводящими к отсутствию или
недостаточной функции дистрофина, цитоскелетного белка, который
обеспечивает прочность, стабильность и функциональность
миофибрилл. Заболевание описано Дюшенном в 1853 году.
• Распространенность мышечной дистрофии Дюшенна варьирует от 9,7
до 32,6 случаев на 100 000 живорожденных мальчиков.
• Широкие эпидемиологические исследования в Казахстане по
распространенности нервно-мышечных заболеваний, в том числе по
мышечной дистрофии Дюшенна, не найдены в доступных нам
литературах.
8.
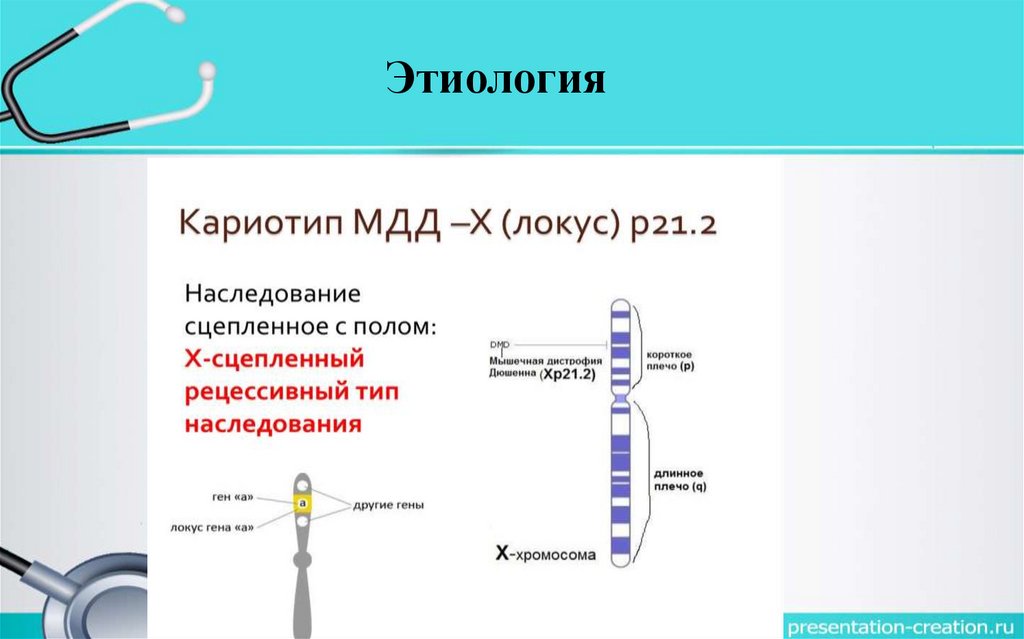
Этиология• Наследуется по рецессивному, сцепленному с Х-хромосомой типу.
• Развитие мышечной дистрофии обусловлено различными генами. То
есть при этом девочки являются носителями патологии, а развивается
заболевание только у лиц мужского пола (так как рецессивный аллель
у них не подавляется доминантной парной генетически нормальной
Х-хромосомой).
9.
Этиология10.
ПатогенезДистрофин - составная часть большого дистрофин-гликопротеидного
комплекса, состоящего из многих белков и связывающего внутриклеточный
актин с ламинином внеклеточного матрикса
Основная функция дистрофина- обеспечение устойчивости и эластичности
мышечного волокна при последующих мышечных сокращениях,
При отсутствии дистрофина вследствие мутации мембрана разрушается, в ней
появляются участки некроза, что приводит к вымыванию содержимого
саркоплазмы в кровяное русло. Происходит постепенная гибель мышечных
волокон и замещение их соединительнотканными структурами.
11.
Классификация• Клиническая: формированием вялых парезов,
параличей и контрактур мышц, ранней
обездвиженности.
• Молекулярно-генетическая: Классификация
нервно-мышечных заболеваний, основанная на
первичном молекулярном дефекте.
12.
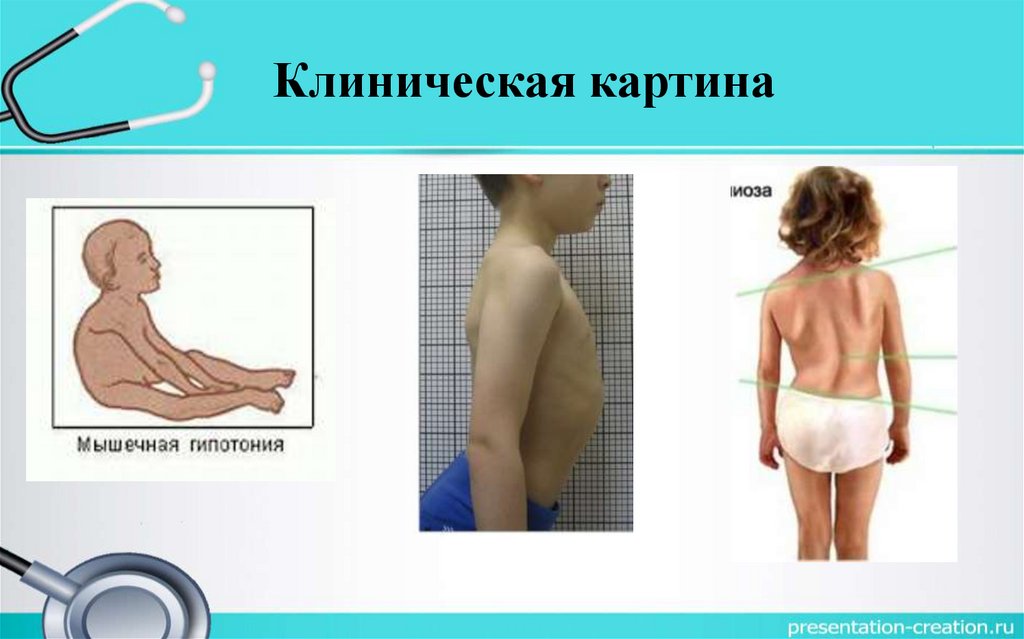
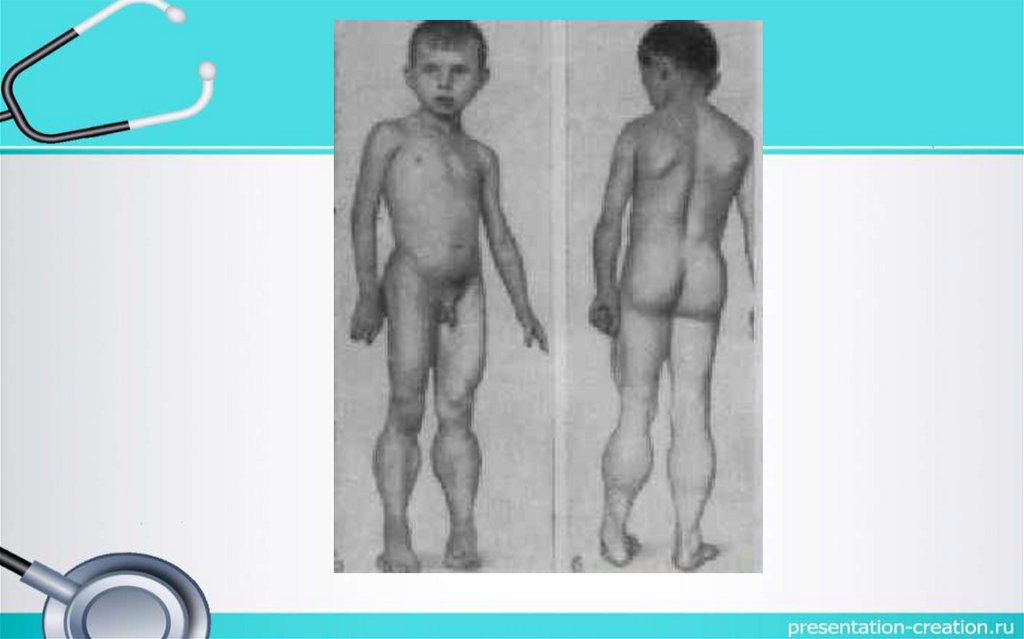
Клиническая картина• Начало заболевания в раннем возрасте – 3 – 4 года, потеря ходьбы в 612 лет, смерть до 30 лет.
• Преимущественно поражаются мышцы тазового пояса.
• Походка «утиная», (переваливающейся).
• По мере прогрессирования увеличивается слабость в конечностях.
• Отмечаются контрактуры.
• Сухожильные рефлексы вначале снижаются, затем исчезают.
• Отмечаются атрофия мышц языка, мягкого нёба, гортани и
жевательных мышц.
• При наличии этих симптомов говорят о бульбарно-паралитической
форме.
13.
Клиническая картина14.
15.
Симптом Говерса16.
ДиагностикаДиагноз ставится на основании:
• данных генеалогического анализа
• клинических особенностей болезни
• данных биохимических исследований (увеличение активности КФК в
30-50 раз, увеличение уровня печеночных трансаминаз)
• данных электромиографии (снижение амплитуды потенциала
действия; снижение длительности потенциала действия; полифазные
(более чем из четырех фаз) потенциалы действия)
17.
Диагностические критерииЖалобы моторные, связанные с движениями:
повышенная утомляемость (снижение выносливости);
частые падения или неуклюжесть;
патологическое изменение походки;
затруднения при подъеме по лестнице, приседании, беге или преодолении препятствия;
использование вспомогательных приемов при подъеме с пола;
неспособность прыгать;
ходьба на носочках;
снижение мышечного тонуса;
боли в мышцах и спине;
уплощение стопы;
уплотнение и/или увеличение мышц голени;
утрата ранее приобретенных двигательных навыков;
задержка развития крупной моторики.
18.
Диагностические критерииЖалобы не моторные, не связанные с движениями:
поведенческие нарушения;
задержка речевого развития или нарушения артикуляции;
трудности с обучением и вниманием;
задержка физического развития и/или недостаточный набор
веса;
• учащенное сердцебиение, одышка, чувство нехватки воздуха;
• боли в области сердца;
• общая слабость, потливость;
19.
ДиагностикаНа сегодняшний день в Казахстане доступен метод мультиплекснолигазнозависимая амплификация (MLPA) для обнаружения крупных
мутаций в гене, такие как делеции и дупликации, но, к сожалению,
данная методика не позволяет выявлять точечные мутации в этом гене.
Поэтому для обнаружения микроделеций и микродупликаций, как
точковые мутации, требуется проведение секвенирования (New
Generation Sequencing) этого гена, где можно оценить всю нуклеотидную
последовательность.
20.
ЦельПредставить
клинические
случаи
характеристики мышечной дистрофии
данным в г. Актобе
генетической
Дюшенна по
21.
Описание клинических случаев. Случай 1Приводим клиническое наблюдение ребенка Р., мальчика, 4 лет,.с жалобами на
слабость в ногах, нарушение ходьбы, быструю утомляемость. Из анамнеза жизни и
заболевания известно, что ребенок от 2-й беременности, протекавшей на фоне
угрозы прерывания, 2-х срочных родов. Семейный анамнез был отягощен по
материнской линии: у двоюродного брата в 3 года диагностирована мышечная
дистрофия Дюшенна. При поступлении в стационар было отмечено, что ребенок
низкого физического развития, интеллект соответствует возрасту. Самостоятельно
не смог подняться с пола, по лестнице. У мальчика наблюдалась «утиная походка».
Приемы Говерса были положительными (применение вспомогательных маневров
при вставании с поверхности). Псевдогипертрофия икроножных мышц. При
электронейромиографии (ЭНМГ) нижних и верхних конечностей признаков
мотосенсорной невропатии не выявлено. Креатинфосфокиназа 1223,8 (норма до
450). Ребенку и матери проведено молекулярно-генетическое исследование
мультиплексно- лигазнозависимая амплификация (MLPA набор MRC-Holland Salsa
MLPA kitP034, P035), у сына и у мамы выявлена гомозиготная делеция 50 экзона в
гене DMD
22.
Описание клинических случаев. Случай 2На частный прием к детскому неврологу обратился ребенок С.,7 лет в сопровождении мамы, с
жалобами на слабость и быструю утомляемость при физических нагрузках. Из анамнеза: мать с
рождения заметила, что ноги в тазобедренных суставах туго двигаются. Голову начал держать в 3
месяца, переворачиваться с 4 месяца, сидеть начал к 9 месяца, ходить самостоятельно начал к 1 году
1 мес. Часто спотыкался. В возрасте 1,5 года мать заметила трудности поднимания по лестнице,
трудности вставания с пола без поддержки заметила в 2 года. Гипертрофия икроножных мышц
отмечена в 4 года. В годовалом возрасте ребенок лепетал, но дальше разговаривать не стал. Родители
водили ребенка на реабилитацию речи с госпитализацией в стационары, проводимые мероприятия к
ожидаемому результату не привели. С нашей стороны было подозрение на МДД.
Походка утиная. Симптом Говерса положителен. Опирается на проксимальную часть бедра при
вставании с пола. Контрактура стоп с обеих сторон. Псевдогипертрофия икроножных мышц. В этой
связи сдали MLPA анализ гена DMD, где результат этого анализа был отрицателен, то есть не
найдены мутации в гене. Уровень КФК в крови 15724 ед/л. Результаты биопсии мышц: участки
некробиотических изменений с участками воспаления, с разрастанием соединительной ткани. МРТ
мышц бедер и голеней: картина, соответствующая дистрофии мышц. После чего был клинически
выставлен диагноз «Миодистрофии Дюшенна». В рамках углубленного изучения сдали ДНК анализ
на секвенирование (New Generation Sequencing) гена, где была мутация по типу нонсенс в одном
экзоне, которая привела к прекращению трансляции белка
23.
Обсуждение1 случай. В связи с отягощенным семейным анамнезом по материнской
линии (болен двоюродный брат) необходимо было проведение молекулярногенетического исследования у данного ребенка, до появления клинической
картины болезни. В целях предупреждения следующих беременностей
целесообразно проведение медико-генетической консультации для выбора
дальнейшей тактики планирования семьи.
2 случай. Демонстрирует необходимость проведения NGS при
отрицательном результате MLPA с характерным клиническим течением
миодистрофии Дюшенна. Для предупреждения рождений детей с МДД при
последующих беременностях необходимо уточнить статус носителя у мамы.
Для этого рекомендуется матери также сдать анализ путем секвенирования
24.
ЛечениеНемедикаментозное лечение:
диета, обогащенная белками, витаминами, микроэлементами, содержащая
микроэлементы (кальций), витамины (группы В, Д, С); иммунизация обязательна по
прививочному календарю до начала гормонотерапии, иммунизация
инактивированными вакцинами допускается на фоне приема гормональной терапии; на
фоне приема гормональной терапии следует избегать иммунизации «живыми»
вакцинами. адекватная состоянию и стадии заболевания физическая активность под
контролем родителей/опекуна, реабилитолога, инструктора ЛФК, медицинского
персонала медицинских и образовательных учреждений. физической активности
пациента и улучшение качества его жизни (лечебная гимнастика, массаж)
Использование протезов (позволяет больным двигаться и замедляет формирование
сколиоза)
Генная терапия (гены дистрофина и утрофина)
Лекарственная терапия для увеличения мышечной массы, замедляя прогрессирование
болезни – преднизолон по 0,75 мг/кг/сут
Симптоматическое лечение
25.
ЛечениеМедикаментозное лечение. Глюкокортикостероидная терапия:
препараты - преднизолон, дефлазакорт. Это метод симптоматического
лечения, позволяющий замедлить утрату мышечной силы и функций,
уменьшить риск развития ортопедических осложнений, стабилизирующий
функциональное состояние легких и сердца;
начало гормональной терапии зависит от фазы развития двигательной
функции ребенка, первоначального физического статуса, наличия ранних
осложнений, формы и течения заболевания. Рекомендуемый возраст начала –
4-5 лет. эффективность гормональной терапии зависит от физиологических
особенностей детского организма, индивидуальной переносимости,
адекватного подбора суточной дозы и режима, мониторинга побочных
эффектов, взаимоотношения врача и семьи пациента.
26.
ЛечениеСпецифическое лечение назначается консилиумом:
препарат аталурен одобрен для лечения амбулаторных пациентов с
миодистрофией Дюшенна в возрасте 2-х лет и старше с подтвержденной
нонсенс-мутацией в гене DMD. Точечная нонсенс-мутация встречается
примерно у 11% больных мальчиков, вызывает образование стоп-кодона и
преждевременную терминацию синтеза белка дистрофина. Мутация
диагностируется методом секвенирования.
препарат этеплирсен одобрен для лечения пациентов с миодистрофией
Дюшенна с подтвержденной мутацией в виде делеции в гене DMD,
корригируемой методом пропуска 51 экзона. Мутация встречается у 13%
пациентов и диагностируется методом MLPA.
препарат голодирсен одобрен для лечения пациентов с миодистрофией
Дюшенна с подтвержденной мутацией в виде делеции в гене DMD,
корригируемой методом пропуска 53 экзона. Мутация встречается у 8%
пациентов и диагностируется методом MLPA.
27.
ЛечениеМетаболическая терапия:
• препараты холекальциферол, препараты кальция
• терапия, направленная на улучшение обменных
процессов в скелетных мышцах,костной ткани, печени,
миокарде;
• нормализация белкового и жирового обмена, угнетение
образование кетокислот,
• снижение лактат-ацидоза;
• для профилактики и устранения побочных эффектов
гормональной терапии.
28.
ЗаключениеПри MLPA можно оценить делеции, дупликации, тогда как для обнаружения
микроделеций и микродупликаций, как точковые мутации, требуется
проведение секвенирования этого гена, где можно оценить всю нуклеотидную
последовательность. Очень важным моментом является то, что определение
статуса носительства матери должно быть в составе комплексного подхода
диагностики этого заболевания, так как вторичная профилактика зависит от
этого показателя.
29.
Литература1.
2.
3.
4.
5.
6.
7.
8.
Gene // GenetTestMolBiomarkers, 2014. Vol.18. P.93‐97.
LeeT. etal. Differencesincarrierfrequencybetweenmothers of Duchenne and Becker muscular dystrophy patients //
J Hum Genet, 2014. Vol.59. P.46‐50.
Sakthivel Murugan S.M., Arthi C., Thilothammal N., Lakshmi B.R. Carrier detection in Duchenne muscular
dystrophy using molecular methods // Indian JMed Res, 2013. Vol.137. P.1102‐1110.
Рекомендации по ведению пациентов с миодистрофией Дюшенна. 2-е и здание. М.:фонд « Мой-Мио».
2018; 63 с. [Rekomendatsii po vedeniyu patsientovs miodistrofiey Dyushenna. (Recommendations for the
management of patients with Duchenne myodystrophy.)2nd ed. Moscow: fond “MoyMio”. 2018; 63 p. (In Russ.)]
Ахмедова П.Г., Угаров И.В., Умаханова З.Р. и др.Распространённость прогрессирующих мышечных
дистрофий Дюшенна/Беккера в Республике Дагестан (по данным Регистра нервно-мышечных
заболеваний). Мед.генетика. 2015;14(1):20–24. [Achmedova P.G., UgarovI.V., UmachanovaZ.R. etal.
Prevalence of progressive Duchenne/Becker muscular dystrophy in Republic of Dagestan (according to the
Register of neuromuscular disease.Meditsinskaya genetika. 2015; 14 (1): 20–24. (In Russ.)]
Влодавец Д.В. Новая таргетная терапия при прогрессирующей мышечной дистрофии Дюшенна.Рос.
вестн. перинатол. и педиатрии. 2015; (4): 220–220. [Vlodavets D.V. New target therapy for progressive
Duchenne muscular dystrophy. Russian bulletin of perinatology and pediatrics. 2015; (4): 220–220. (In Russ.)]
Trabelsi M., Beugnet C., Deburgrave N. et al. Whenamid-intronic variation of DMD gene creates an ESE
site.Neuromusc. Dis. 2014; 24 (12): 1111–1117. DOI: 10.1016/ j.nmd.2014.07.003.
Bladen C.L., Salgado D., Monges S., Foncuberta M.E.The TREAT-NMD DMD Global Database: Analysis of
Morethan 7,000 Duchenne Muscular Dystrophy Mutations. Hum.Mutat. 2015; 36 (4): 395–402. DOI:
10.1002/humu.22758.
30.
Литература9.
10.
11.
12.
13.
14.
15.
16.
D`Amico A., Catteruccia M., Baranello G. et al.Diagnosis of Duchenne muscular dystrophy in Italy in the last decade:
critical issues and areas for improvements.Neuromusc. Dis. 2017; 27: 447–451. DOI: 10.1016/ j.nmd.2017.06.555.
Повереннова И.Е., Захаров А.В., Черникова В.В.Анализ клинических и инструментальных параметров,
характеризующих кардиомиопатии при различных формах прогрессирующих мышечных дистрофий. Саратовский
науч.-мед. ж. 2017; (1): 160–164. [Poverennova I.E., Zakharov A.V., Chernikova V.V. The analysis of the clinical and tool
parameters characterizing a cardiomyopathy at various forms of the progressing muscular dystrophies. Saratovskiy nauchnomeditsinskiy zhurnal. 2017; (1): 160–164. (In Russ.)]
Ke Q., Zhao Z.-Y., Mendell J.R. et al. Progress in treatment and newborn screening for Duchenne muscular dystrophy and
spinal muscular atrophy. World J. Pediatrics. 2019; 15: 219–225. DOI: 10.1007/s12519-019-00242-6.
Guiraud, S. et al. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp Physiol, 2015. 100(12):
p.1458-67.
Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet.
2016 Mar; 53 (3): 145–51. doi: 10.1136/jmedgenet-2015-103387.Epub 2016 Jan 11.
Ingrid E.C Verhaart, Agata Robertson, Jan J. Wilson, Annemueke Aartsma-Rus. Prevalence, incidence and carrierfrequency
of 5q linked spinal muscular atrophy a literature review. Orphanet Journal of Rare Disease. 2017;12:124-126
Finkel R.S., Mercuri E., Meyer O.H., Simonds A.K., Schroth M. K. , Graham R.J., Kirschner J., Iannaccone S.T., Crawford
T.O., Woods S., Muntoni F., Wirth B., Montes J, Main M, Mazzone E. S., Vitale M., Snyder B., Quijano-Roy S., Bertini E.,
Davis H. R., Qian Y., Sejersen T. Diagnosis and management of spinal muscular atrophy: Pulmonary and acute
care;medications, supplements and immunizations; other organ systems; and ethics Neuromuscular Disorders 2018;28
(2):197–207.
Takeshita E, Minami N, Minami K et al. Duchenne muscular dystrophy in a female with compound heterozygous contiguous
exon deletions. Neuromuscul Disord. 2017 Jun; 27(6):569-573.