








 Медицина
МедицинаПохожие презентации:










Лизосомные патологии. Болезни Гоше, Помпе и другие
1.
Лизосомные патологии.Болезни Гоше, Помпе и
другие
2.
Лизосо́мные боле́зни — общее название группы весьма редкихнаследственных заболеваний, вызванных нарушением функции
внутриклеточных органелл лизосом.
3.
Лизосомы — это клеточные органеллы,содержащие сильные ферменты —гидролазы,которые расщепляют внутриклеточные субстанции, а также белки,
проникающие в клетку путем эндоцитоза (внутриклеточное пищеварение).
Отсутствие одной из гидролаз приводит к накоплению внутри клеток химических
соединений, обычно крупных размеров, что нарушает все функции клетки и
обусловливает возникновение лизосомного заболевания.
4.
Лизосомные болезни накопления относятся к редким(орфанным) заболеваниям. Частота отдель- ных форм
колеблется от 1:40 000 до 1:1 000 000 и реже; суммарная
частота составляет 1:7000–1:8000 новорожденных. В
некоторых странах пытаются проводить массовый
скрининг на эти заболевания.
В настоящее время выделяют следующие группы лизосомных
болезней накопления: 1) мукополисахаридозы;
2) липидозы (сфинголипидозы — ,болезнь Гоше,
галактосиалидоз, гранулематоз Фарбера, лейкодистрофии,
болезнь Ниманна—Пика и др.);
3) муколипидозы,
4) гликопротеинозы (фукозидоз, сиалидоз, маннозидоз, болезнь
накопления гликогена II типа — болезнь Помпе, и др.);
5) нейрональные цероидные липофусцинозы;
6) другие болезни накопления (болезнь Ниманна—Пика тип С,
болезнь Вольмана, болезнь накопления холестерина, цистиноз,
болезнь Сала, пикнодизостоз и др.).
5.
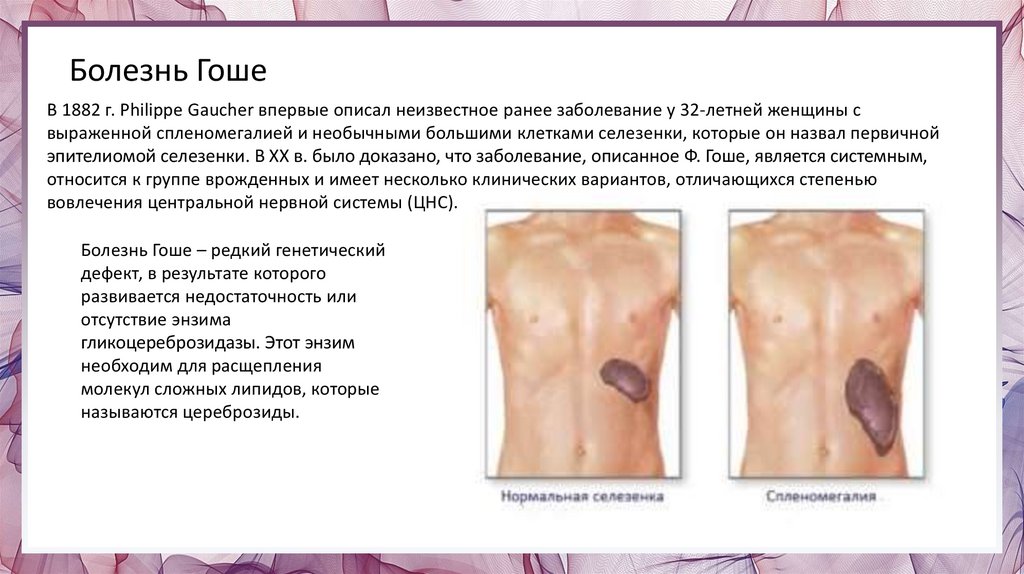
Болезнь ГошеВ 1882 г. Philippe Gaucher впервые описал неизвестное ранее заболевание у 32-летней женщины с
выраженной спленомегалией и необычными большими клетками селезенки, которые он назвал первичной
эпителиомой селезенки. В ХХ в. было доказано, что заболевание, описанное Ф. Гоше, является системным,
относится к группе врожденных и имеет несколько клинических вариантов, отличающихся степенью
вовлечения центральной нервной системы (ЦНС).
Болезнь Гоше – редкий генетический
дефект, в результате которого
развивается недостаточность или
отсутствие энзима
гликоцереброзидазы. Этот энзим
необходим для расщепления
молекул сложных липидов, которые
называются цереброзиды.
6.
В зависимости от клинического течения выделяют 3 типа болезни Гоше:· тип 1 – ненейронопатический (самый частый).
· тип 2 – инфантильный или острый нейронопатический,
· тип 3 – подострый нейронопатический.
Клинические признаки болезни Гоше первого типа,
который является самым распространенным:
Усталость
Боли в костях
Легко образуются синяки
Увеличение печени
Увеличение селезенки
7.
Основные симптомы заболевания при БГ типа 2 возникают в первые 6 мес жизни:• гепатоспленомегалия;
· нарушение глотания, поперхивание, часто осложняющиеся аспирационной пневмонией;
· билатеральное фиксированное косоглазие
прогрессирующая задержка психомоторного развития и потеря ранее приобретенных навыков;
· тонико-клонические и другие типы судорожных приступов
Главной особенностью клинических проявлений БГ типа 3
является то, что наряду с поражением паренхиматозных
органов (гепатоспленомегалия) наблюдаются
неврологические
проявления, возникающие, как правило, в возрасте от 6
до 15 лет и позже:
снижение интеллекта (от незначительных изменений до
тяжелой деменции);
экстрапирамидная ригидность;
мозжечковые нарушения;
расстройства речи, письма;
поведенческие изменения, эпизоды психоза;
8.
Ферментозаместительная терапия – единственный эффективный патогенетический метод лечения болезниГоше, который купирует основные клинические проявления заболевания, улучшает качество жизни больных
и не оказывает выраженных побочных эффектов.
Ферментозаместительная терапия – метод лечения
генетических заболеваний, являющихся результатом
биохимической дисфункции вследствие отсутствия какоголибо фермента. В настоящее время ферменты можно
получать методом рекомбинации ДНК и вводить
внутривенно, что успешно применяется для лечения
болезни Гоше – наследственного глюкоцереброзидоза,
вызванного отсутствием лизосомного фермента
глюкоцереброзидазы.
9.
Болезнь ПомпеЭто заболевание носит имя голландского патолога Иоганна Кассиануса Помпе, который в 1932 году описал
ранее неизвестную патологию у 7-месячной девочки. Оно и сегодня встречается очень нечасто — его
распространенность составляет всего 1 случай 40 тысяч человек1
При болезни Помпе возникает дефицит кислой мальтазы, из-за
чего в клеточных органеллах лизосомах накапливается
нерасщепленный гликоген. Поэтому заболевание относят к
лизосомным болезням накопления. Клинические проявления
заболевания в основном определяются именно накоплением
гликогена.
10.
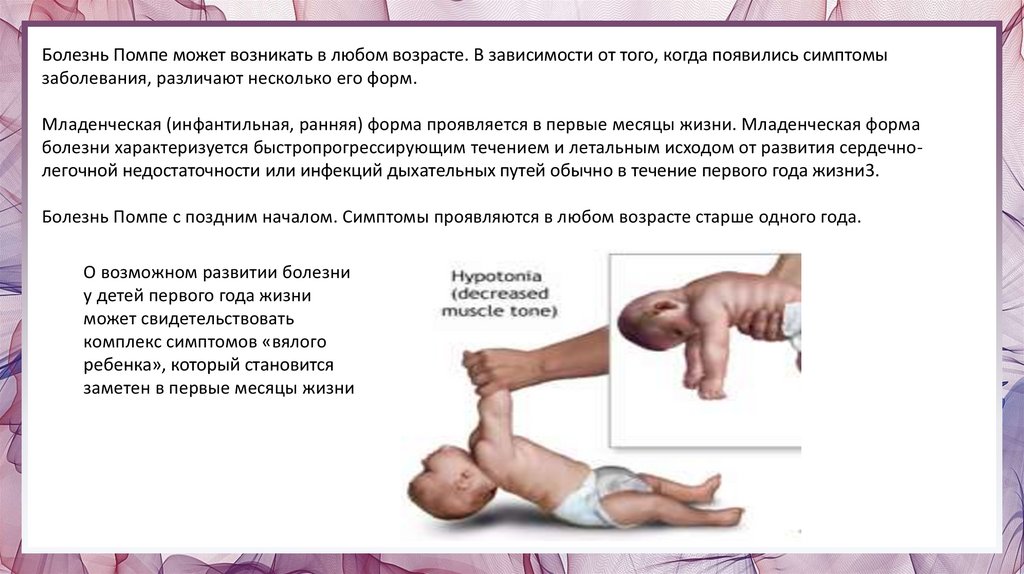
Болезнь Помпе может возникать в любом возрасте. В зависимости от того, когда появились симптомызаболевания, различают несколько его форм.
Младенческая (инфантильная, ранняя) форма проявляется в первые месяцы жизни. Младенческая форма
болезни характеризуется быстропрогрессирующим течением и летальным исходом от развития сердечнолегочной недостаточности или инфекций дыхательных путей обычно в течение первого года жизни3.
Болезнь Помпе с поздним началом. Симптомы проявляются в любом возрасте старше одного года.
О возможном развитии болезни
у детей первого года жизни
может свидетельствовать
комплекс симптомов «вялого
ребенка», который становится
заметен в первые месяцы жизни
11.
Иная клиническая картина возникает при болезни Помпе с поздним началом. Нередко первыми признакамистановятся слабость мышц конечностей и параспинальных мышц. Обычно болезнь прогрессирует медленно,
часто — на протяжении многих лет.
Еще недавно лечение болезни Помпе ограничивалось
назначением симптоматических лекарств и профилактикой
осложнений. Однако сегодня существует терапия, которая
позволяет влиять не на следствия заболевания, а на его
причину, компенсируя наследственный дефицит кислой
мальтазы. Для улучшения прогноза заболевания критически
важен ранний старт ферментозаместительной терапии2.
Своевременное и непрерывное лечение заболевания
позволяет замедлить прогрессирование болезни, улучшить
состояние, качество жизни и прогноз.