




































")



")



















")

















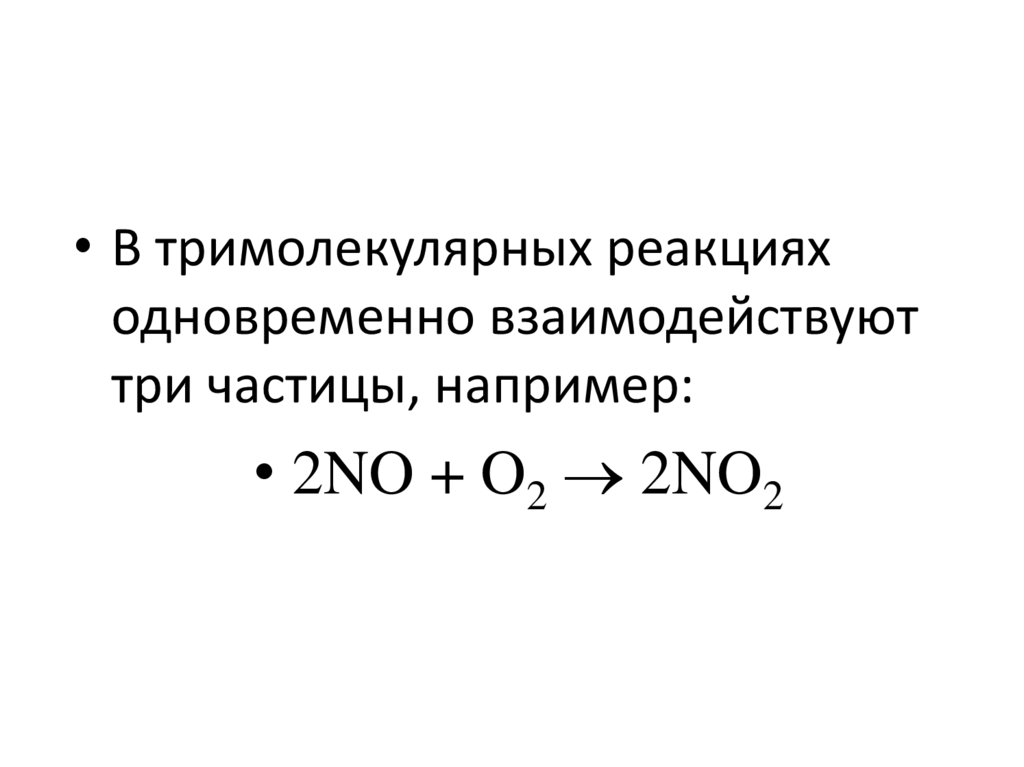










")








")



 кинетики реакций первого порядка")




 во времени в реакциях 1-го порядка")



")


























")








")




















 Химия
ХимияПохожие презентации:






")



Физическая химия
1.
профессорКонюхов Валерий Юрьевич
2. Литература
3.
• Физическая химия//Под ред. К.С.Краснова. М.: Высшая школа, 2001
• Вишняков А.В., Кизим Н.Ф.
Физическая химия. М.: Химия, 2012.
• Эмануэль Н.М., Кнорре Д.Г. Курс
химической кинетики. М.: Высшая
школа.
• Еремин Е.Н. Основы химической
кинетики. М.: Высшая школа
4. Два «кита» – два подхода к исследованию процессов, совершающихся в системах:
5. Термодинамический подход
6. Определяет интенсивность процесса,
• его потенциальные возможности,т.е. насколько далеко от начала
находится состояние равновесия:
• G0 = RT lnKP
7. Кинетический подход
8. С какой скоростью и через какие стадии происходит процесс?
9.
• Термодинамика можетпредсказать принципиальную
возможность осуществления
процесса ( G < 0).
• Но может ли на самом деле этот
процесс происходить (с
конечной скоростью) определяет
кинетика.
10.
• Оба подхода различны посути, но дополняют друг
друга при исследовании
процессов.
11. Глава 7. Химическая кинетика и катализ
12.
• Скорости реакций очень сильноразличаются.
• Почему? И что следует сделать, чтобы
ускорить или замедлить реакцию?
• Имеются две причины для изучения
скоростей реакций.
13.
• Во-первых, практически очень важнознать, как быстро реакционная смесь
достигнет равновесного состояния:
скорость может зависеть от ряда
контролируемых факторов, таких, как
температура, давление, присутствие
катализатора, и в зависимости от наших
целей мы можем проводить реакцию с
оптимальной скоростью.
14.
• Во-вторых, изучение скоростейреакции помогает
исследованию механизмов
реакций.
15. Два подхода к понятию «механизм»
• С одной стороны, «механизм» — этосовокупность элементарных стадий,
составляющих химическую реакцию.
• Например, реакция взаимодействия
водорода и брома протекает через ряд
стадий, предполагающих разрыв Вr2 на
атомы брома, взаимодействие одного из
этих атомов с Н2 и т. д.
16.
• Другое значение термина механизмсвязано с самими элементарными
стадиями и касается их детальной
природы.
• В этом смысле «механизм» объясняет, что
происходит, когда атом брома
приближается и атакует вращающуюся,
колеблющуюся молекулу водорода.
17.
• Первый тип механизмов изучаетклассическая химическая кинетика
– раздел физической химии.
• Второй тип механизмов изучает
химическая динамика – раздел
химической физики.
18.
• Химическая кинетика – этонаука, изучающая
закономерности протекания
химических реакций во
времени, а также - их
механизмы
19. 7.1. Основные понятия и определения химической кинетики
20. Вещества – участники реакции
• Вещества, содержащиеся в реакционнойсистеме, в зависимости от их роли в акте
химического превращения делят на:
• Исходные (реагенты);
• Конечные (продукты);
• Промежуточные;
• Инертные;
• Катализаторы.
21. Реакции: элементарные, простые, сложные
22.
Элементарная реакция элементарный акт химическоговзаимодействия- - единичный акт
превращения частиц (молекул,
атомов, радикалов, ионов)
исходных веществ в частицы
продуктов реакции.
23.
• Если в системе одновременнопротекает несколько
элементарных реакций , то
реакцию называют сложной.
• Т.е. сложная реакция может
быть представлена как
совокупность ряда стадий.
24.
• Стадия может быть либонеобратимой (односторонней),
либо обратимой (двухсторонней).
• В первом случае стадия состоит из
одной элементарной реакции, во
втором – из двух (прямой и
обратной).
• Одностадийные реакции называют
простыми.
25. 1. Скорость химической реакции
26. Скорость реакции – это мера интенсивности её протекания
27. С точки зрения физики:
• Изменение количества ni i-говещества в объеме реакционного
пространства Ф за время t :
t
ni dt ri ( xi , yi ...t )dФ
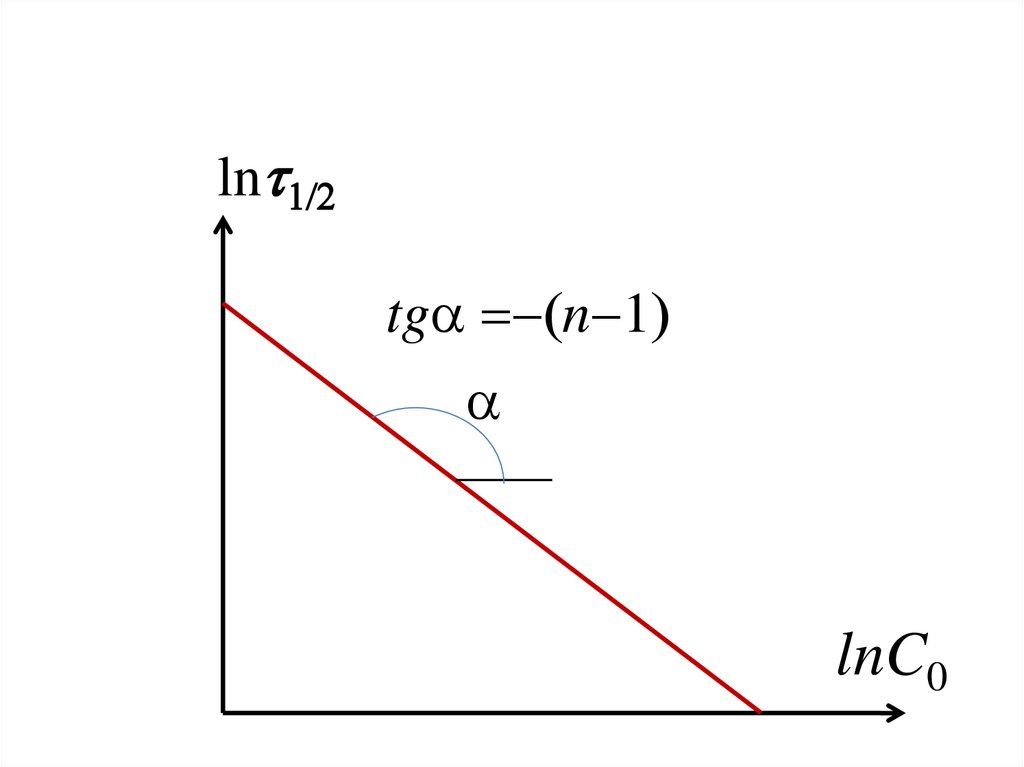
0
Ф
ri – плотность источника (стока) i-го
вещества.
(1)
28.
• Если роль источника (стока)выполняет химическая реакция, то ri
– скорость образования
(расходования) i-го вещества за счет
химической реакции.
• Из (1) следует:
ni
ri
t Ф
2
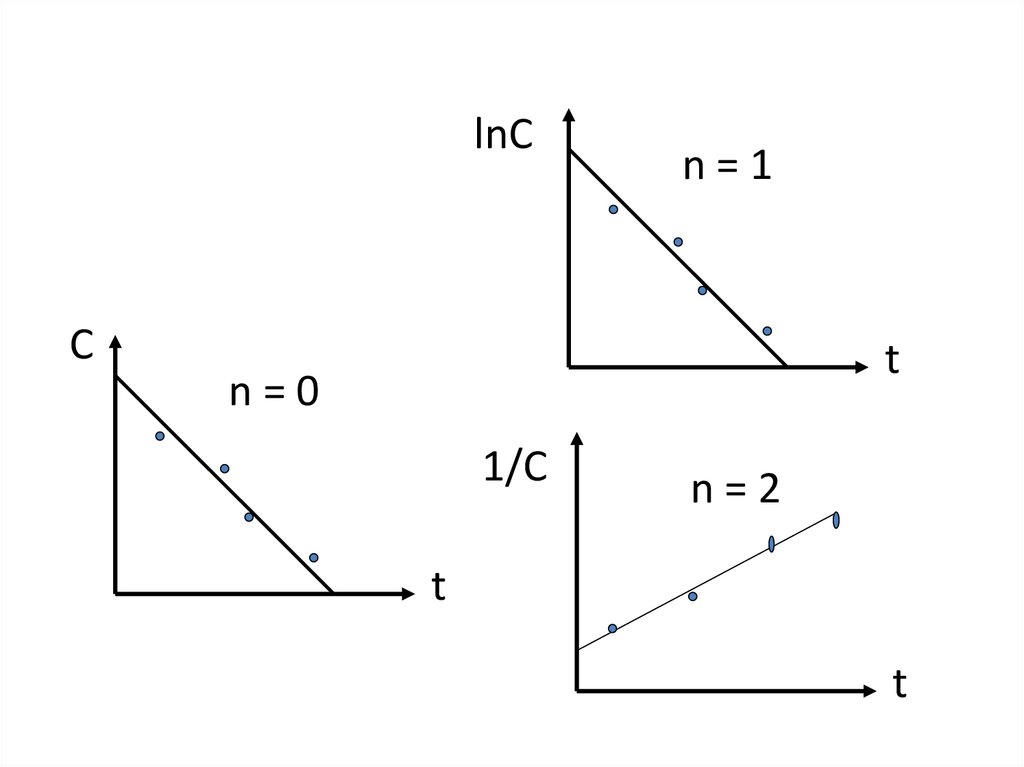
(2)
29.
• В гомогенных реакцияхреакционное пространство
трехмерное, так что Ф = V.
• Гетерогенные процессы
протекают на границе раздела
фаз с поверхностью S.
• В этом случае Ф = S.
30. Разные режимы протекания реакций
31. Стационарный режим реакции
32.
• Если реакция проводится встационарных условиях, то ri не
зависит от времени и из (1) следует:
ni t ri ( x, y,..)dФ
(3)
Ф
и скорость реакции:
1 dni
ri
t dФ
(4)
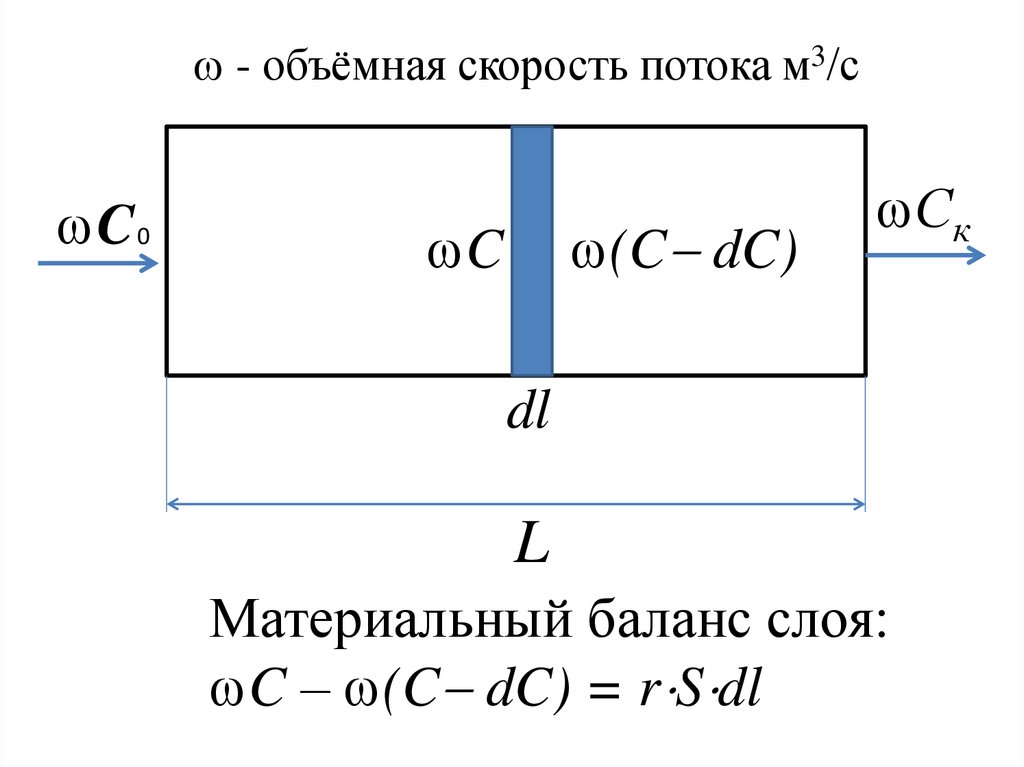
33. Реактор идеального вытеснения
34.
- объёмная скорость потока м3/сC 0
(C dC)
C
Ск
dl
L
Материальный баланс слоя:
C – (C dC) = r S dl
35. Материальный баланс слоя
dC r S dldC r S
dC r
dC
r
d
dl
dV
36. С другой стороны можно записать и так:
dCS
r
dl
dC
1 S
k C
dl
dC
S
k dl
C
37.
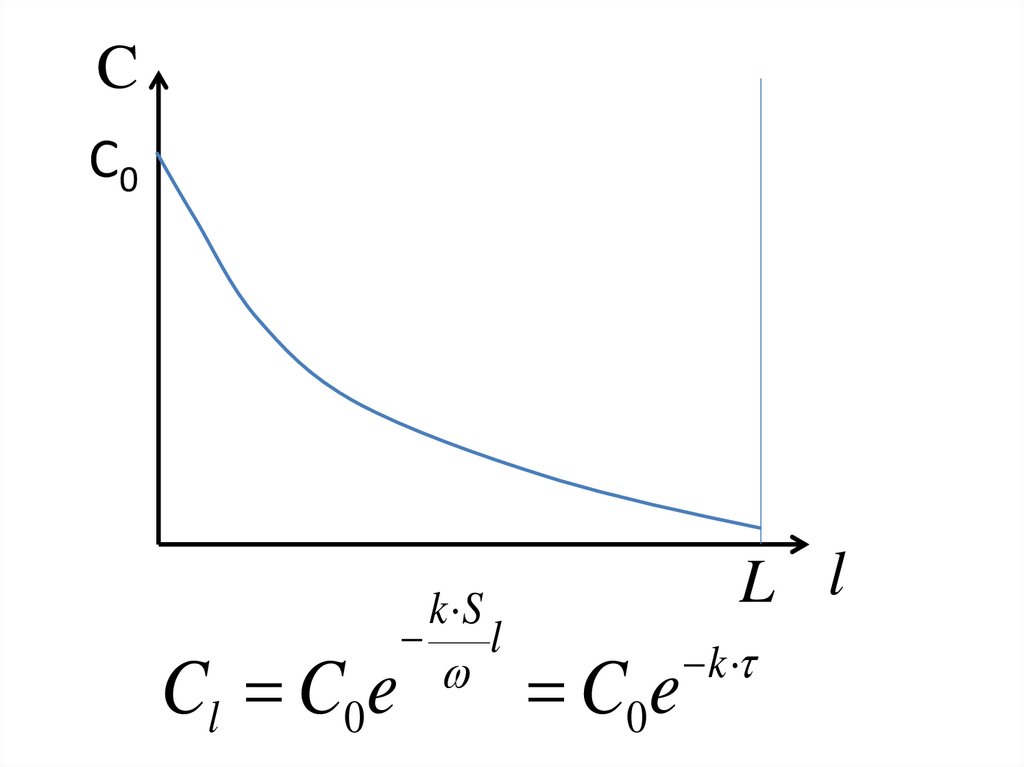
CС0
Cl C0e
k S
l
L l
C0e
k
38.
• Если скорость реакции не зависит отпространственных координат, но зависит
от времени реакции (статические
условия осуществления реакции), то
изменение количества вещества
пропорционально Ф:
ni Ф ri (t )dt
Ф
скорость реакции:
(5)
1 dni
ri
Ф dt
(6)
39.
• Скорость реакции не зависит отвремени и пространственных
координат, то:
ni
ri
t Ф
(7)
В случае гомогенных реакций такой режим
реализуется в РИС,
В случае гетерогенных реакций - в проточноциркуляционной установке – безградиентном
реакторе.
40. Реактор идеального смешения (РИС)
C0A продукты
V
C
41.
В стационарном режиме dCA/dt = 0Материальный баланс реактора:
C A0 C A
rA (C A 0 C A )
V
где = V/ – время пребывания
вещества А в реакторе
42.
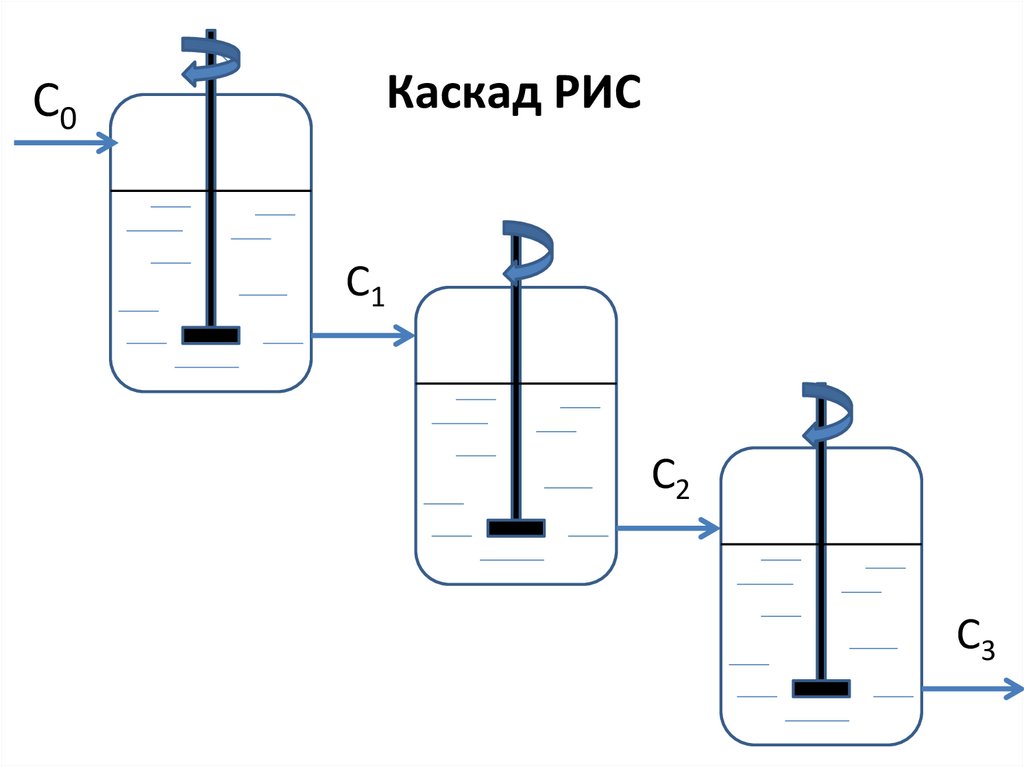
Каскад РИСC0
C1
C2
C3
43.
БЕЗГРАДИЕНТНЫЙ РЕАКТОР44. Проточно-циркуляционная каталитическая установка BI-CATr-EXP
45. Скорость реакции в статических условиях (закрытой системе)
46. Закрытая система
A ВСА0
СА = f(t)
47. Изменение концентрации вещества А во времени
Изменение концентрацииС вещества А во времени
СА0
t
48.
• Скорость химической реакции (по компоненту)численно равна количеству компонента,
реагирующего в единицу времени в единице
реакционного пространства.
• Это средняя скорость реакции на заданном
промежутке времени t.
• Предел этой величины при t 0 дает
выражение для мгновенной скорости по
компоненту А:
1 dn A
rА
V dt
1 dn A
rА
S dt
(9)
49.
• Для продукта реакции Ввыражение скорости химической
реакции имеет вид:
1 dnB
1 dnB
rB
rB
V dt
S dt
В общем виде, для любого участника
гомогенной реакции:
1 dni
ri
V dt
(10)
50.
• Когда стехиометрическиекоэффициенты реагентов различны,
как, например, для реакции:
• N2 + 3H2 = 2NH3
• скорости реакции по компонентам
различаются: в единицу времени азота
реагирует в три раза меньше, чем
водорода.
51.
• Для учёта этого факта в выражения дляскорости реакции по компоненту:
• a1A1 + a2A2 +….aiAi продукты
• следует ввести стехиометрические
коэффициенты ai:
1 1 dn Ai
r
ai V dt
Здесь r - скорость реакции (без
отнесения к конкретному реагенту).
(11)
52.
• Если объём системы в ходе реакциине изменяется (V = const), то
величину V можно внести под знак
дифференциала:
n Аi
d
1 V
r
ai dt
(12)
или
1 dC Аi
r
ai dt
53.
• Если же стехиометрическийкоэффициент аi = 1, то выражение (12)
для скорости упрощается:
r
В общем виде:
dC Аi
dt
r
dC i
dt
(13)
54. Методы экспериментального определения скорости реакции в статической системе
55.
• Для экспериментального определенияскорости гомогенной реакции измеряют
концентрацию хотя бы одного из
реагентов в различные моменты
времени.
• Для этого в ходе реакции отбирают из
реактора пробы (жидкости или газа) и
анализируют их на содержание реагентов
или измеряют величину,
пропорциональную С.
56. Аналитический метод определения скорости реакции
57.
• Полученные экспериментальныеданные по зависимости концентрации
от времени C = f(t) представляют в виде
эмпирического уравнения, например,
полинома:
• С = С0 + а t + b t2 +d t3 +…,
• где C0 - начальная концентрация
вещества (моль/м3); а, b, d –
подбираемые коэффициенты.
58.
• Дифференцированием полученногоуравнения определяют производные
dC/dt и по ним вычисляют скорость
реакции в различные моменты
времени t (т.е. при различных
концентрациях С).
59. Метод графического дифференцирования кинетической кривой
60. Кинетические кривые
61.
• К кинетической кривой в различных еёточках (при различных С) проводят
касательные, тангенсы углов наклона
которых равны скорости реакции:
dC
r
dt
62.
• Для определения скорости реакции необязательно отбирать пробы из
реактора и аналитически измерять в них
концентрации реагирующих веществ.
• Значительно удобнее измерять какоелибо свойство системы, меняющееся в
ходе реакции и пропорциональное С.
63.
• Например, можно использоватьоптическую плотность системы D,
которая согласно закону БугераЛамберта- Беера пропорциональна
концентрации вещества C:
• D = l C.
• Здесь - молярный коэффициент
поглощения (экстинкции); l - длина
поглощающего слоя (кюветы).
64.
• Применяют для определения скоростиреакции и измерение электрической
проводимости, показателя
преломления, а для газовых реакций –
давления во времени.
• Для непрерывного анализа веществ
непосредственно в реакционном
объеме используют и другие методы
физико-химического анализа:
потенциометрию, полярографию и т.п.
65. 2. Основной постулат химической кинетики (закон действующих масс)
66.
• В 1867 году в своей монографии«Исследования сил химического
сродства» Гульдберга и Вааге показали,
что химические реакции протекают как в
прямом, так и в обратном направлении.
67. Согласно закону действующих масс:
• скорость химической реакции rпропорциональна концентрациям
реагентов Сi (их произведению) в
некоторых степенях:
r k C
ni
i
(14)
68.
• Здесь k – константа скорости реакции,она равна r при Сi = 1 моль/л, поэтому её
иногда ещё называют удельной
скоростью.
• Размерность константы скорости зависит
от показателя степени n, она равна:
[объёмная концентрация]1 n
[k ]
время
69.
• Степень ni при концентрации Сiназывают порядком реакции по
компоненту.
• Их сумма ni = n общий порядок
реакции (или просто порядок
реакции).
70.
• Порядок реакции показывает, как сильно (вкакой степени) скорость реакции зависит от
концентрации реагентов.
• Так, например, если n = 1, то скорость прямо
пропорциональна концентрации.
• Если n = 2, то скорость растёт в большей
степени, чем концентрация: если
концентрация увеличивается в 2 раза, то
скорость – в 4 раза и т.д.,
• При n = 0 скорость вообще не зависит от
концентрации.
71.
• Большинство химических реакцийявляются сложными.
• Например, стехиометрическая запись
реакции:
• Н2 + Cl2 2HCl
• отражает лишь материальный баланс
реакции, показывающий что 1 моль
водорода нацело реагирует с 1 моль
хлора с образованием 2 моль
хлористого водорода.
72.
• На самом деле реакция идёт через рядстадий (элементарных реакций).
Начинается она с диссоциации молекулы
Cl2 на два радикала Cl
1. Cl2 2Cl
2. Cl H 2 HCl H
3. H Cl HCl Cl
2
4.....и т.д...........
(I)
Выражение в скобках – схема механизма
реакции.
73.
• Для отдельной элементарной реакциипорядок реакции по веществу
совпадает с его стехиометрическим
коэффициентом и закон действующих
масс для стадий записывается так:
r1 k1 CCl 2
r2 k 2C
r3 k3C
1
Cl
C
1
H
C
1
H2
1
Cl 2
74. Как быть со сложными реакциями? Можно ли написать аналогичное уравнение и для брутто реакции?
75.
• Можно, но порядок реакции повеществу в этом случае не равен
его стехиометрическому
коэффициенту:
r kC
n1
H2
C
n2
Cl 2
(15)
76.
• Здесь n1 и n2 - эмпирические величины,определяемые из кинетических данных
реакции.
• В общем случае они представляют собой
сложную комбинацию из
стехиометрических коэффициентов
веществ различных стадий, и поэтому их
значения могут быть любыми: целыми и
дробными, равными нулю и даже
отрицательными.
77. 3. Молекулярность реакции
78.
• Молекулярность реакцииопределяется числом частиц
(молекул, атомов, радикалов или
ионов), участвующих в
элементарном акте химического
взаимодействия (в элементарной
реакции).
79. Элементарные реакции бывают только трех типов:
1. Мономолекулярные реакции элементарные реакции распада иизомеризации, в которых участвует
только одна частица:
80. Разрыв связей в исходной молекуле происходит под действием света или при нагревании, например: CH3Br CH3 + Br
Разрыв связей в исходноймолекуле происходит под
действием света или при
нагревании, например:
CH3Br CH3 + Br
81.
• В бимолекулярной реакции происходитвзаимодействие (столкновение) двух
частиц
82.
• При этом одни связи разрываются, адругие образуются, например:
• Н + Сl2 НСl + Сl
• Бимолекулярные реакции - самый
распространенный тип элементарных
реакций.
83.
• В тримолекулярных реакцияходновременно взаимодействуют
три частицы, например:
• 2NO + O2 2NO2
84.
• Тримолекулярные реакции встречаютсяв природе крайне редко,
четырёхмолекулярные реакции вообще
не известны – слишком мала
вероятность столкновения четырёх
нужных для реакции молекул,
обладающих необходимым запасом
энергии.
85.
• Вант-Гофф писал в своей монографии«Очерки по химической кинетике»: «Мне
было бы приятно получить на опыте
различные случаи, отвечающие тому
уравнению, если положить в нем n = 3,4
и т.д., т.е. осуществить трех-,
четырехмолекулярные и т.д.
превращения. Я неоднократно пытался
ставить такие опыты, но они удались мне
лишь частично».
86.
• Для элементарной реакции значенияпорядка реакции и молекулярности
совпадают.
• Например, порядок реакции стадии (2)
механизма (I) равен двум и в акте
химического взаимодействия
участвуют две частицы: радикал хлора
и молекула водорода - молекулярность
равна 2.
87.
• Время полупревращения – это время,в течение которое исходное
количество (или концентрация)
реагента уменьшается вдвое, т.е.
реагирует половина взятого количества
вещества.
• Обозначают его обычно 1/2.
88. Кинетические кривые и 1/2
Кинетические кривые и 1/289. 7.2. Формальная кинетика
90.
• Порядок реакции показывает нам, каксильно (в какой степени) скорость
реакции зависит от концентрации
реагентов.
• Для различных порядков реакции
закономерности изменения
концентрации реагентов во времени
различны.
91.
• Выяснением этих общихзакономерностей (без отнесения
к конкретным реакциям – отсюда
и термин «формальная»)
занимается раздел химической
кинетики – «формальная
кинетика».
92.
• Основы формальной кинетикизаложил Я. Вант-Гофф, опубликовав в
1884 г. в Амстердаме монографию
«Очерки по химической динамике»
на французском языке.
93. 7.2.1. Закономерности изменения концентрации реагента в реакциях различных порядков
94. 1. Реакция нулевого порядка (n = 0)
95.
• Выясним, по какому закону изменяетсяконцентрация реагента в ходе реакции, если
её скорость не зависит от концентрации
реагента.
• Закон действующих масс (кинетическое
уравнение в дифференциальной форме) в
этом случае записывается следующим
образом:
dC
0
r
kC k
dt
96.
• Разделяя переменные и интегрируя впределах: от t = 0 до t и от С0 до С (С0
– начальная концентрация реагента
при времени t = 0, С и t – текущие
параметры) получаем:
С
t
С0
0
dC
k
dt
C 0 C kt
(16)
97. С = С0 – kt
CС = С0 – k t
С0
tga k
a
t
98.
• Т.е. концентрация реагента в случаереакции нулевого порядка меняется
во времени линейно (с постоянной
скоростью).
• Уравнение (13) называют
кинетическим уравнением реакции
нулевого порядка в интегральной
форме.
99.
• Справедливо и обратноеутверждение: если опытные точки по
зависимости концентрации реагента
от времени в координатах С от t
группируются вдоль прямой, то
изучаемая реакция – нулевого
порядка.
• На этом утверждении основан т.н.
графический метод определения
порядка реакции.
100.
• Из графика С = f(t) можно вычислитьконстанту скорости реакции k: она
равна тангенсу угла наклона прямой
(со знаком ).
• Её размерность:
моль
моль
моль
,
,
3
л с
л мин м с
101.
• Получим выражение для времениполупревращения реакции нулевого
порядка:
С0
С0
k 1 / 2
2
или
C0
1/ 2
2 k
Т.о. при нулевом порядке реакции 1/2
пропорционально начальной концентрации
реагента: чем больше С0, тем больше
требуется времени на превращение его на
половину.
(17)
102.
• Большинство известных реакцийнулевого порядка представляют
собой гетерогенные процессы,
например разложение на
платиновой проволоке оксида
азота (I):
• 2N2O 2N2 +O2
• или аммиака:
• 2NH3 N2 + 3H2
103. 2. Реакции первого порядка (n = 1)
104.
• В этом случае скорость реакциипропорциональна концентрации
• кинетическое уравнение в
дифференциальной форме имеет
вид:
dC
1
r
kC
dt
105.
• Разделяя переменные и интегрируя впределах: [С0 ; С], [0 ; t], получаем:
С
C0
ln
k t
C
t
dC
k
dt
С C 0
0
или
ln C ln C 0 k t
С С0 e
k t
(18)
106. Таким образом, в случае реакции первого порядка концентрация реагента изменяется во времени по экспоненте:
CC0
t
107. Линейные координаты (анаморфозы) кинетики реакций первого порядка
lnClnC0
tga k
a
Размерность k
[с-1, мин-1 и т.д.]
t
108.
• Т.е. линейными координатами вслучае реакции первого порядка
являются lnС от t.
• Таким образом, если опытные точки
по зависимости С от t в координатах
lnС от t группируются вдоль прямой,
следовательно кинетический
порядок реакции первый.
109. Выражение для времени полупревращения 1/2
Выражение для времениполупревращения 1/2
C0
ln
k 1 / 2
C0
2
1/ 2
ln 2 0,693
k
k
(19)
110.
• Только для реакций первогопорядка время 1/2 не зависит от
начальной концентрации реагента
С0: чем больше его взято, тем
быстрее идёт реакция.
• В итоге, время, необходимое для
полупревращения исходного
количества реагента, всегда одно и
тоже.
111.
• Если записать дифференциальноекинетическое уравнение в форме:
d (C A0 x)
dt
k (C A0 x)
dx
k (C A0 x) решение дает :
dt
k t
x C A0 1 e
112. Изменение концентрации С исходного вещества и продукта (х) во времени в реакциях 1-го порядка
113.
• Известно довольно много реакцийпервого порядка:
• 1. Разложение оксида азота (V) в
газовой фазе
• N2O5 2NO2 +1/2O2
• 2. Гидрирование этилена на
никелевом катализаторе
• C2H4 + H2 C2H6
• 3. Радиоактивный распад.
114.
• Процесс размножения бактерий вограниченной по объёму питательной
среде подчиняется кинетике реакций
первого порядка.
• Рост числа бактерий идёт сначала медленно
(скорость процесса прямо
пропорциональна числу бактерий), но со
временем всё сильнее ускоряется и
наконец приобретает характер «взрыва».
• Этот «демографический взрыв»
описывается экспоненциальной кривой.
115.
• Такая схема развития присуща любойцивилизации, паразитирующей на
окружающей среде.
• При этом по «взрывному закону»
изменяются все главные
характеристики паразитирующей
цивилизации: народонаселение,
потребление пищи, загрязнение.
116. 3. Реакции второго порядка (n = 2)
117.
• Возможны варианты:• 1. А + А D + ….
• Кинетическое уравнение в этом
случае имеет вид:
dC А
2
rА
kCА
dt
или
1 dC А
2
r
kCА
2 dt
118.
• 2. В реакциях типа А + В D +…..• скорость прямо пропорциональна
произведению концентраций:
dC А
dC B
r
kCA C B
dt
dt
119.
• Для простоты рассмотрим случай,когда начальные концентрации
обоих реагентов одинаковы:
• СА0 = СВ0
Кинетическое уравнение:
dC
1 1
2
r
kC A C B kC
dt
(20)
120.
• Интегрирование уравнения (20) впределах [С0 ; С], [0 ; t] даёт:
1 1
k t
С С0
С0
С
1 k С0 t
(21)
121. Линейный график кинетики реакций второго порядка
Линейный график кинетики1/C реакций второго порядка
a
1/C0
tga k
t
122.
• Т.е. линейными координатами вслучае реакции второго порядка
являются 1/С от t.
• Таким образом, если опытные
точки по зависимости С от t в
координатах 1/С от t
группируются вдоль прямой,
следовательно кинетический
порядок реакции второй
123.
• Угловой коэффициент прямой равенконстанте скорости реакции k.
• Единицами измерения k в случае n = 2
служат:
л
л
м
;
;
моль с моль мин моль с
3
124.
• Получим уравнение для времениполупревращения реакции второго
порядка:
1
1
k 1 / 2
С0 С0
2
1
1/ 2
k C0
(22)
125.
• Время, необходимое напревращение половины исходного
количества реагента, уменьшается
обратно пропорционально росту
этого количества.
126.
• Скорость реакции при этомописывается кинетическим
уравнением реакции первого
порядка:
dС A
r
k ' С A k ' (С A0 x)
dt
где k’ = k CB
Такие реакции называют реакциями
псевдопервого порядка.
127.
• К реакциям второго порядкаотносят:
• газофазное разложение HI:
• 2HI H2 + I2
• разложение оксида азота:
• 2NO2 2NO + O2
• димеризация циклопентадиена и
т.д.
128. 4. Реакции n - го порядка
129.
• Дифференциальное уравнениекинетики реакции в этом случае
имеет вид:
dC
n
r
k C
dt
130.
• Разделяя переменные и интегрируяв пределах от 0 до t и от С0 до С,
получаем интегральное
кинетическое уравнение:
1 1
1
n 1 n 1 k t
n 1 C
C 0
(23)
131.
• Выражение для времениполупревращения в этом случае
имеет вид:
1
1
1
n 1 n 1 n 1 k 1 / 2
n 1 C0 / 2
C0
(24)
132.
n 12 1
k
1/ 2
n 1
n 1 C0
1/ 2
n 1
2 1
n 1
n 1 k C0
(25)
Логарифмирование уравнения (25)
даёт:
ln 1/ 2
n 1
2
ln
n 1 ln C0 (26)
n 1 k
133.
• Из уравнения (26) вытекает методэкспериментального определения порядка
реакции.
• С этой целью определяют время
полупревращения реакции при нескольких
начальных концентрациях реагента.
• Далее полученные опытные значения 1/2 и
С0 откладывают на графике ln 1/2 против С0,
из углового коэффициента определяют
порядок реакции tga = -(n 1).
134. 7.2.2. Методы определения порядка реакции
135.
• Порядок реакции являетсяэмпирической величиной и не может
быть установлен теоретически, если
неизвестен механизм реакции.
• Существует несколько методов
экспериментального определения
порядков реакции по компонентам и
общего порядка реакции.
136. Определение общего порядка реакции
137.
• Для определения частных порядковреакции по компонентам (а затем и
общего, как их суммы) применяют
метод избытка (изоляции).
• Для этого все вещества реакции (1),
кроме одного, например A1, берут в
концентрациях настолько больших, что
изменением их со временем можно
пренебречь, и их можно ввести в
константу скорости.
138.
• Этот прием позволяет понизитьпорядок реакции и тем самым
упростить вид кинетических
уравнений. Так, уравнение
r kС С С ...
n1
A1
n2
A2
n3
A3
упрощается до
r k'С
n1
A1
где n1 – порядок по веществу A1
139.
k ' k C C ...n2
A2
n3
A3
• Таким же способом определяют другие
частные порядки по каждому
компоненту.
• Общий порядок равен сумме частных
порядков.
140.
• Все методы определения частныхи общего порядков реакции
можно разделить на
дифференциальные и
интегральные
141. Интегральные методы
142. I. Метод подбора кинетического уравнения
143. Аналитический вариант метода (метод подстановки)
1. Аналитический вариантметода
(метод подстановки)
144.
• Порядок реакции можно установить,проверив какое из кинетических
уравнений лучше описывает опытную
зависимость С = f(t).
• Для этого измеряют концентрации
реагента С в различные моменты
времени t, подставляют их в
кинетические уравнения нулевого
первого, второго и т.д. порядков.
145. Константы скорости реакции различных порядков:
1n=0
k (C 0 C )
t
C
1
0
n=1
k ln
t
C
1 1
1
n=2
k (
)
t C C0
146.
• Для каждой пары С и t вычисляютконстанту скорости.
• Реакция принимается того порядка,
константа скорости которого
практически постоянна при всех С и t
(колеблется около какой-то средней
величины и отклонения от среднего
лежат в пределах погрешности её
определения).
147. 2. Графический вариант метода
148.
• Полученные опытные данные зависимостиконцентрации реагента С от времени
реакции t представляют в линейных
координатах реакций нулевого (С от t),
первого (lnC от t), второго (1/С от t) и т.д.
• Порядок реакции тот, в координатах
которого точки группируются вдоль прямой.
• По тангенсу угла наклона прямой
определяют константу скорости реакции.
149.
lnCn=1
С
t
n=0
1/С
n=2
t
t
150. II. Метод Оствальда-Нойеса
151. Определение порядка реакции из зависимости времени превращения вещества на 1/p его часть от исходной концентрации реагента С0
152.
• Например, время полупревращения(период полураспада) для реакций
различного порядка по-разному зависит от
начальной концентрации реагента С0.
• Так, для реакции первого порядка оно не
зависит от С0 (19), для реакции второго
порядка обратно пропорционально С0 (22),
а для нулевого порядка прямо
пропорционально С0 (17).
153. 1. Аналитический вариант метода (n 1 )
1. Аналитический вариантметода
(n 1 )
154.
• Время превращения на 1/р часть ( 1/p)однозначно связано с начальной
концентрацией исходного вещества
(C0) и порядком реакции (n):
n
ln( '1/ p / 1/ p )
ln( С0 / С '0 )
1
155.
• Например, отношение двух времён1/2 , соответствующих двум
начальным концентрациям C0
равно:
1
С
0
2
1 С 0
2
n 1
156. Для двух точек:
1Для двух
точек:
ln
n 1
2
1
2
C 0
ln
C 0
Таким образом, порядок реакции можно
установить, проведя всего 2 опыта
Понятно, что точность определения n в этом
случае низка.
157. 2. Графический вариант метода
158.
• Для определения порядка реакцииполучают, например, зависимость 1/2
от С0, опытные данные представляют в
координатах ln 1/2 против lnC0 (26).
• Через отложенные точки проводят
прямую, тангенс угла её наклона равен (n 1).
159.
ln 1/2tga n 1
a
lnC0
160. Дифференциальные методы
161. Метод графического дифференцирования кинетической кривой
162.
• Суть метода, предложенного ВантГоффом, заключается в следующем.• Логарифмирование кинетического
уравнения n-го порядка
• r = k Cn
• даёт:
• lnr = lnk + n lnC
163.
• Таким образом, для вычисления nследует определить скорость
реакции r при различных
концентрациях реагента С.
• Скорость же можно определить,
дифференцируя кинетические
кривые (r = дС/дt).
164. Два варианта метода
165. 1. По зависимости начальной скорости r0 от исходной концентрации реагента С0
ln r0 ln k n ln С0166. Определение концентрационного порядка реакции
Определениеконцентрационного
порядка
С
реакции
C03
C02
C01
a
t
167. Определение концентрационного порядка реакции
lnr0tga n
a
lnk
lnC0
168. 2. Дифференцированием одной кинетической кривой при различных t
ln rt ln k n ln Сt169. Определение временного порядка реакции
Определение временногоС
порядка реакции
C0 a
С1
С2
t
170. Определение временного порядка реакции
lnrОпределение временного
порядка реакции
tga n
a
lnk
lnC
171. Концентрационный и временной порядки реакции могут не совпадать
172.
• Если образующиеся вещества тормозятреакцию, то временной порядок выше
концентрационного: скорость по ходу
реакции снижается в большей степени
(степень и есть порядок реакции), чем если
бы она уменьшалась только за счёт снижения
концентрации реагента.
• И, наоборот, если образующиеся вещества
ускоряют реакцию (проявление
автокатализа), то временной порядок
меньше концентрационного
173. Аналитический вариант метода
174. Проводят 2 опыта при двух С0. Для определения n решают систему уравнений:
ln r10 ln k n ln С10ln r20 ln k n ln С20
Тогда:
n
ln( r10 / r20 )
ln( С10 / С20 )