





















 Маркетинг
Маркетинг Менеджмент
МенеджментПохожие презентации:










Regulatory committee
1.
Association ofInternational
Pharmaceutical
Manufacturers
REGULATORY COMMITTEE
March 13,
2023
For internal use
only Not for
distribution
2.
Association ofInternational
Pharmaceutical
Manufacturers
AGENDA
09:30 – 10:30
1. Administrative issues related to the activities of the Committee in
2023
2. Current legislative initiatives — amendments to 61-FZ
3. Development of the EAEU laws and regulations and law enforcement
4. Issues related to GMP
inspections,
regulatory environment
and law enforcement
10:30 – 12:30
Issues related to
law enforcement in the sphere of
authorizationand
expertise of medicinal products
according to 61-FZ and the EAEU Law
Questions
and
answers
session
with
participation of
the Head of the
Management and Control Division of the FSBI "SCEEMP" — Y.
M. Rychikhina
3.
Association ofInternational
Pharmaceutical
Manufacturers
INTRODUCING
AMENDMENTS
TO 61-FZ
March 13,
2023
For internal use
only Not for
distribution
4.
Association ofInternational
Pharmaceutical
Manufacturers
MAJOR INNOVATIONS (1) — Draft Law of
the Ministry of Health —
current status
The mechanism to define the possibility of considering the medicinal product (MP) as an
orphan one can be realized both in terms of the MP authorized according to the national
procedure or the EAEU Law, and the unauthorized product;
To obtain a permission to conduct a clinical trial, an IMPD should be submitted –
full compliance with the European requirments
Unification of the Advanced Therapy Medicinal Products (ATMP) terminology between the
national legislation and the EAEU
Introduction of a transitional period — a possibility to "complete the circulation" of the
medicinal products and substances before the end of the shelf life after the termination of
the Marketing Authorization (MA)/register entry
Exclusion of a 3-year sunset close
Possibilities of cross-sectoral exchange between the Ministry of Health (MoH) and the
Federal Service for Surveillance in Healthcare (RZN), and the Ministry of Industry and Trade
(MPT) with an access to the registration dossier
Possibility to submit a GMP certificate of the MPT or EAEU in any state, or referral for
inspection by the MPT
Complete transition to the electronic document management according to the registrybased model Unified account as an interaction window between the MoH, FSBI "SCEEMP"
and the applicant Exclusion of paper workflow, any other documents (MA, reports,
instructions, etc.), implementation of a registry-based model (starting from January 1, 2025);
Scientific counselling by the FSBI "SCEEMP"— under further approval
Protection of the data of clinical and pre-clinical studies — under further
5.
Association ofInternational
Pharmaceutical
Manufacturers
Current version of the Law
MAJOR INNOVATIONS (2)
RELEASE INTO CIVIL
CIRCULATION
Revision of the draft Law
Before the MP imported to the Russian Before the MP imported to the Russian
Federation is released into civil Federation is released into civil
circulation, a manufacturer's certificate circulation, a document of the MP
shall be submitted that certifies
manufacturer shall be submitted that
the compliance of the confirms the MP quality
imported
MP
with
the
requirements of the pharmacopoeial
monograph, and in absence thereof,
with the Normative Document (ND)
requirements
First 3 batches being imported
First 3 batches being imported
for the first time
for the first time
a testing protocol shall be submitted with no regard to the dosage strength
that confirms the compliance of the within the framework of one
MP batch or lot with the quality marketing authorization — one dosage
attributes
strength
6.
Association ofInternational
Pharmaceutical
Manufacturers
MAJOR INNOVATIONS (3)
RELEASE INTO CIVIL
CIRCULATION
Current version of the
Revision of the draft Law
Federal Law (FZ)
For the first three batches or lots It is allowed to submit the testing protocol
of
the
MP only for the first MP batch or lot
manufactured for the first time in ,
if
the data on the
the RF, or being imported to the
national or EAEU
RF for the first time,
a testing GMP certificate have been presented,
protocol shall be submitted that provided that for 3 years before the testing
confirms the compliance of the protocol date the manufacturer, in the
MP batch or lot with the quality name of which the expert opinion
attributes stipulated in the ND
(compliance certificate) is issued, had no
cases of non-compliances related to the MP
authorized in the RF with respect to the
manufacturing sites involved in the finished
MP manufacturing and release quality
control.
7.
Association ofInternational
Pharmaceutical
Manufacturers
MAJOR INNOVATIONS (4)
RELEASE INTO CIVIL
CIRCULATION
Current version of the FZ
Revision of the draft Law
There are no individual requirements
for orphan MPs
For orphan MPs, it is allowed to carry out the
testing of 1 batch remotely once per 3 years.
Current version of the FZ
Revision of the draft Law
There are no individual requirements
for ATMP
For ATMP, it is allowed to submit the data on
the
MPT or EAEU
GMP
certificate
(autologous MPs), and other ATMP — 1 batch
being imported for the first time in a remote
mode Additionally, it is allowed to carry out the
testing of 1 batch remotely once per 3 years.
8.
Association ofInternational
Pharmaceutical
Manufacturers
Current version of the FZ
MAJOR INNOVATIONS (5)
RELEASE INTO CIVIL
CIRCULATION
Revision of the draft Law
5. Annually, not later than on 5. Annually, not later than on April 1, the
February 1,
the testing quality testing protocol shall be presented
protocol shall be presented for the for the MP that has been released into
MP
of
the civil circulation during the preceding year
particular manufacturer that has according to the marketing authorization
been released into civil circulation (for one batch of each trade name taking
during the year (for one batch of into account the drug formulation of one
each trade name taking into account dosage)
the drug formulation and dosage)
9.
Association ofInternational
Pharmaceutical
Manufacturers
MAJOR INNOVATIONS (6)
INSPECTION — a draft Law of the Minpromtorg
(similar regulatory provisions being in the draft Law
of the MoH)
Article 16: possibility of submitting a copy of the decision of the
authorized FAEA on conducting a pharmaceutical inspection of the
human medicinal products manufacturing for compliance with the GMP
Rules of the EAEU;
Article 29: possibility of submitting a certificate of compliance of the MP
manufacturer with the GMP Rules of the Union or a copy of the decision
of the Federal Agency of Executive Authority (FAEA) on conducting a
pharmaceutical inspection of the MP manufacturing for compliance with
the GMP requirements of the Union;
Article 30: information about the submission date and registration
number of the decision of the authorized FAEA on conducting a
pharmaceutical inspection of the MP manufacturing for compliance with
the GMP requirements of the Union if the human medicinal product is
manufactured outside the RF, or a copy of the certificate of compliance
of the MP manufacturer with the GMP requirements of the Union.
10.
Association ofInternational
Pharmaceutical
Manufacturers
INSPECTION — draft Law of the
Minpromtorg — current status of discussion
- Unification of inspection standards for all GxP practices — inspection terminology
and requirements
- Cross-sectoral exchange of information about the inspection results between
the FAEAs (certificates)
- Requirements for pharmaceutical inspectors attestation
11.
Association ofInternational
Pharmaceutical
Manufacturers
CONDUCTING GCP INSPECTIONS (1)
Grounds for announcement of a GCP inspection stipulated in
the Decision No. 78:
1. As an alternative to local clinical trials (CT) at the territory of the Union — clause 36—
37 ofWhen
the Decision
No. 78
conducting
the expertise, the Authorized Body takes a decision on the
necessity of an extraordinary inspection, including the bioequivalence studies, for
compliance with the GMP requirements of the Union within the period of the MP
authorization or on the inclusion of the CT inspection into the plan of inspections for the
first 3 years after the MP authorization.
2. Pursuant to clause 27 of the Decision No. 78 — inaccuracy of information within the
dossier.
3. Within the framework of a complex assessment of risk factors according to clauses
38—39 of the Decision No. 78.
At the moment, within the framework of the EAEU, draft Rules for
Conducting Pharmaceutical Inspections for Compliance with the Rules of
Good Clinical Practice of the Eurasian Economic Union are discussed.
12.
Association ofInternational
Pharmaceutical
Manufacturers
CONDUCTING GCP INSPECTIONS
A draft Order of the MoH "On approval of the method to determine the
amount of payment for services related to inspections (examinations) of
clinical trials of the human medicinal products for compliance with the
Rules of Good Clinical Practice of the Eurasian Economic Union"
o Maximum amount of payment for services related to the inspections of CT or
non-CT of the MPs with respect to the applicants engaged in the activities at
the territory of the Russian Federation and for compliance with the Rules of
Good Clinical Practice of the EAEU or the Laboratory Practice of the EAEU —
1,651,140.11 rubles
o Maximum amount of payment with respect to the applicants engaged in the
activities outside the Russian Federation — 2,112,379.31 rubles
Currently, the "regulatory guillotine" Working Group (WG) in cooperation with the
FSBI "SCEEMP" and the MoH is developing draft rules for conducting inspections
for compliance with the GCP to present a revised version to the EEC WG.
Approval of the document — not earlier than at the end of the year
13.
Association ofInternational
Pharmaceutical
Manufacturers
CURRENT LEGISLATIVE INITIATIVES
The draft Order of the MoH "On approval of the amendments to the
classification of variations to the documents of the registration dossier for
the authorized MP that has been approved by the Order of the Ministry of
Health of the Russian Federation No. 959n dated December 13, 2016" will
allow for reducing the number of laboratory expertises assigned while
amending the documents at the national level
Development and updating of General Pharmacopoeial Monographs
(GPM) and Monographs to be included into the State Pharmacopoeia
version XV — any industry-specific proposals (?)
Based on the results of the discussion with the Ministry of Health and the
FSBI "SCEEMP", the possibility to use clause 2 of the Governmental
Decree No. 593 has been confirmed so far as relevant to the application
of the remote methods for national procedures provided there is the
appropriate justification by the applicant and the decision of the expert
institution.
14.
Association ofInternational
Pharmaceutical
Manufacturers
EAEU regulation
15.
DEVELOPMENT OF THE EAEU LAWS ANDREGULATIONS
AND LAW
Proposals of the Association to the regulatory bodies: ENFORCEMENT
Association of
International
Pharmaceutical
Manufacturers
Supplement the Decision of the EEC Council "On temporary measures to
establish the peculiarities of the human medicinal products circulation" No.
96 dated June 10, 2022 with the following clause: "Until December 31,
2024, entitle the applicants to authorize the MP when it is not possible to
provide the data of the CT performed at the territory of the EAEU Member
States, and to provide the data of the international multicenter clinical
trials performed according to the Good Clinical Practice standards as a part
of the registration dossier instead of the specified data";
Prolong the effective period of the Decision of the EEC Board No. 135
dated September 28, 2022 until December 31, 2024 (with respect to
maintaining zero rates of import customs duties related to the MPs, raw
and other materials required for MPs manufacturing, at least according to
the List stipulated in the EEC Decision No. 150 dated September 23, 2022).
16.
Association ofInternational
Pharmaceutical
Manufacturers
DEVELOPMENT OF THE EAEU LAWS AND
REGULATIONS
AND LAW
ENFORCEMENT
Simplification of the standard procedure of bringing
into
compliance Proposal of the MoH of the RF to the
EEC
1. MAs are granted to all MPs in the EAEU*
2. Reduction of the expertise volume*
3. Reduction of the number of documents to be submitted for all
types of procedures
Module 2 is not required; Modules 1 and 3 have been shortened
4. No user testing is required*
5. There is no need for notarial certification of documents
17.
Association ofInternational
Pharmaceutical
Manufacturers
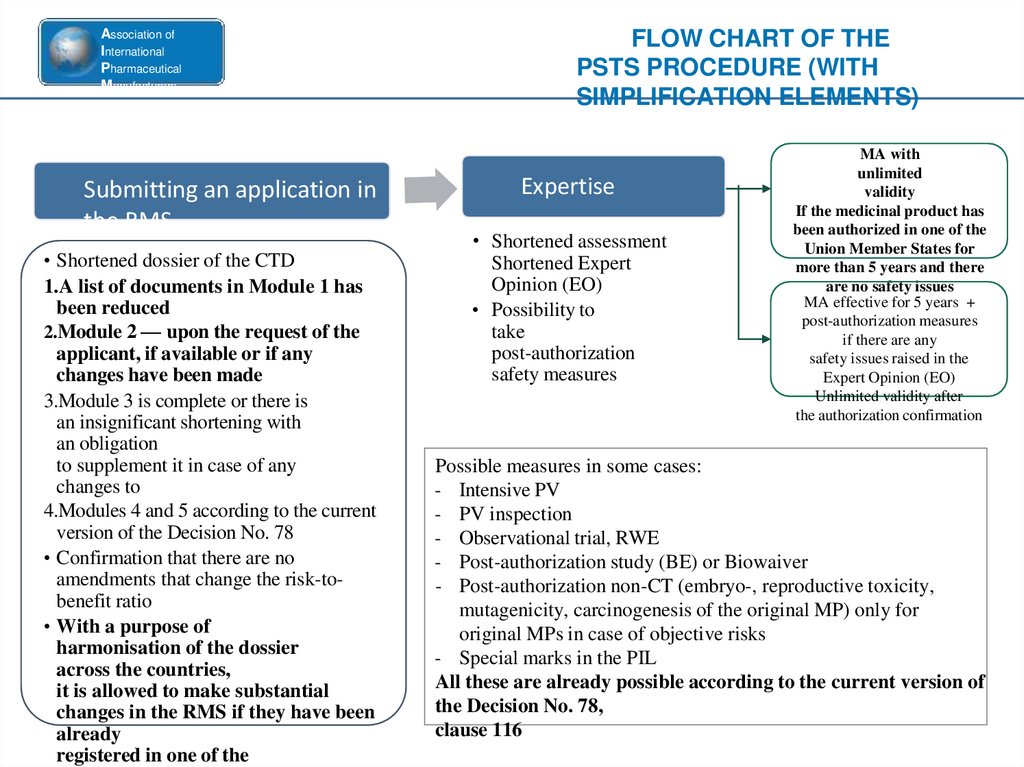
Submitting an application in
the RMS
• Shortened dossier of the CTD
1.A list of documents in Module 1 has
been reduced
2.Module 2 — upon the request of the
applicant, if available or if any
changes have been made
3.Module 3 is complete or there is
an insignificant shortening with
an obligation
to supplement it in case of any
changes to
4.Modules 4 and 5 according to the current
version of the Decision No. 78
• Confirmation that there are no
amendments that change the risk-tobenefit ratio
• With a purpose of
harmonisation of the dossier
across the countries,
it is allowed to make substantial
changes in the RMS if they have been
already
registered in one of the
FLOW CHART OF THE
PSTS PROCEDURE (WITH
SIMPLIFICATION ELEMENTS)
Expertise
• Shortened assessment
Shortened Expert
Opinion (EO)
• Possibility to
take
post-authorization
safety measures
MA with
unlimited
validity
If the medicinal product has
been authorized in one of the
Union Member States for
more than 5 years and there
are no safety issues
MA effective for 5 years +
post-authorization measures
if there are any
safety issues raised in the
Expert Opinion (EO)
Unlimited validity after
the authorization confirmation
Possible measures in some cases:
- Intensive PV
- PV inspection
- Observational trial, RWE
- Post-authorization study (BE) or Biowaiver
- Post-authorization non-CT (embryo-, reproductive toxicity,
mutagenicity, carcinogenesis of the original MP) only for
original MPs in case of objective risks
- Special marks in the PIL
All these are already possible according to the current version of
the Decision No. 78,
clause 116
18.
Association ofInternational
Pharmaceutical
Manufacturers
INTRODUCING AMENDMENTS —
DRAFT DOCUMENT
OF THE MOH IN THE EEC
New terms are introduced (changed):
"changed information about the medicinal product submitted by means of post factum notification" —
a document that does not require a preliminary approval by the authorized body (expert institution) of
the Reference State, which is provided only for changes in the notification-based nature of the
summary of product characteristics, labelling text, or package leaflet, as well as the quality normative
document as a full document in a new version containing an appendix with a table of agreed (approved)
and proposed (new) versions or packaging mock-ups within a new version
. In the upper left footer
of each page of the stated document (changed information about the medicinal product submitted by
means of post factum notification), there should be a dossier version number (sequence), within the
framework of which it is submitted to the authorized body (expert institution).
• Grouping the changes:
The procedure for submitting the same multiple changes to different products of one and the same
holder is stipulated; to describe multiple changes or all type- IA variations, an Addition VII is
implemented.
• Deviations in notifications of type-IA:
In the event of any deviation in the notification of a variation (changes stated in paragraphs 2 and 3 of
sub-clause 1.7.2), implementation of such variation is not interrupted until the end of 365 days from
the start date. The applicant is obliged to submit a new application taking into account the reason for
such deviation before the end of the above period.
19.
Association ofInternational
Pharmaceutical
Manufacturers
INTRODUCING
AMENDMENTS
• Variations made to the draft document (changed):
1. With the change in the product information (clause 1.6), CMS have been added to the type-IA
and -IAin variations
2. Sequential procedures of the dossier submission have been deleted for the Reference Member
State (RMS) and Concerned Member State (CMS) + Expert Committee in case of type II
variation
3. Clarifications have been added throughout the text that are related to the registries
updating
in the Concerned Member State,
submission of the countryspecific documents M1 in the CMS, as well as other inconsistencies
4. The obligation to introduce changes within 90 working days after rejection has been deleted
5. The possibility to implement within the period of up to 365 days even in case of refusals for
type-IA and -IAin
6. Submission of the country-specific documents has been added to the responses to requests
in the CMS for all procedures
7. A letter to the Expert Committee is stipulated in the event there are any divergences of
views between the RMS and the CMS in relation to IB
8. Procedure realization periods have been reduced compared to the previous version
9. The procedure of urgent safety restrictions presentation has been improved — a possibility
to submit the applicant's justification instead of a variation is added
• Further optimization of the procedures:
1. Addition of information about the Expert Committee (EC) for type-II variations
20.
Association ofInternational
Pharmaceutical
Manufacturers
INTRODUCING
AMENDMENTS
1. without a r e v i s i o n of t h e data in clause 1.6 and the data in the registry —
to be submitted and published only in the RMS
SCHEME: 20 working days
Notifications of type-IA
and -IAin
2. with a revision of the data i n c l a u s e 1 . 6 and the d a t a in the registry —
t o be submitted and published i n t h e R M S a n d i n t h e C M S Realization
is possible only after the information is introduced into the registry.
An access to the dossier for the CMS is granted within 5 working days after the submission or
upon the request of the CMS. The CMS shall approve the variation within 15 working days
The CMS is entitled t o refuse to amend the country-specific d o c u m e n t s within 10 days
from t h e date of gaining the access to the dossier
SCHEME: 20 working days for the RMS + 5 working days for the RMS (registry)
3. Variations to the country-specific documents M1 only in the CMS
SCHEME: (20 + 5 working days for the register)
Type-IB
variations
Type-II variations
To be submitted to all CMS and RMS including the country-specific documents
The access to the dossier is granted in 5 working days or upon the request of the CMS
SCHEME: 5 working days (with the request) + 30 working days for the expertise in the RMS
(with the request) + 15 working days for the CMS to decide whether to refuse + 10 working
days to obtain the opinion of the RMS or to consult with the CMS + 5 working days for a
final decision of the RMS and 10 working days for the registry; the CMS to approve within
10 working days.
A final consistent decision is taken via the consultations or at the EC
Submission of full dossiers to all CMS and RMS The access is granted for the RMS within
5 working days. Written or other types of consultations between the countries are
stipulated.
SCHEME: 10 working days (with the request) + 60 working days for the expertise by the
RMS (with the request) + 20 working days for the CMS + 15 working days for
consultations and report updating + 10 working days for the RMS to take a decision + 5
working days for the RMS to use the register; 10 working days for the CMS to take a
decision + 10 working days for the CMS to use the register.
The part related to the Expert Committee has been deleted. In different countries, the result
may be associated with a various decision!
21.
AIPMIntroducing
amendments
1. without a r e v i s i o n of t h e data in clause 1.6 and the data in the registry —
to be submitted and published only in the RMS
SCHEME: 20 working days
Notifications of type-IA
and -IAin
2. with a revision of the data i n c l a u s e 1 . 6 and the d a t a in the registry —
t o be submitted and published i n t h e R M S a n d i n t h e C M S Realization
is possible only after the information is introduced into the registry.
An access to the dossier for the CMS is granted within 5 working days after the submission or
upon the request of the CMS. The CMS shall approve the variation within 15 working days
The CMS is entitled t o refuse to amend the country-specific d o c u m e n t s within 10 days
from t h e date of gaining the access to the dossier
SCHEME: 20 working days for the RMS + 5 working days for the RMS (registry)
3. Variations to the country-specific documents M1 only in the CMS
SCHEME: (20 + 5 working days for the register)
Type-IB
variations
Type-II variations
To be submitted to all CMS and RMS including the country-specific documents
The access to the dossier is granted in 5 working days or upon the request of the CMS
SCHEME: 5 working days (with the request) + 30 working days for the expertise in the RMS
(with the request) + 15 working days for the CMS to decide whether to refuse + 10 working
days to obtain the opinion of the RMS or to consult with the CMS + 5 working days for a
final decision of the RMS and 10 working days for the registry; the CMS to approve within
10 working days.
A final consistent decision is taken via the consultations or at the EC
Submission of full dossiers to all CMS and RMS The access is granted for the RMS within
5 working days. Written or other types of consultations between the countries are
stipulated.
SCHEME: 10 working days (with the request) + 60 working days for the expertise by the
RMS (with the request) + 20 working days for the CMS + 15 working days for
consultations and report updating + 10 working days for the RMS to take a decision + 5
working days for the RMS to use the register; 10 working days for the CMS to take a
decision + 10 working days for the CMS to use the register.
The part related to the Expert Committee has been deleted. In different countries, the result
may be associated with a various decision!
22.
Deadlines for approval of the changes to the EECCouncil Decision No. 78
related to the procedure of registration dossier
amending and bringing into compliance are expected
not earlier than in Q4/2023
At the moment, there is an active coordination of
the procedures between the authorized bodies of
the EAEU Member States
Not for distribution. For internal use
23.
Association ofInternational
Pharmaceutical
Manufacturers
ISSUES RELATED TO THE
GMP INSPECTIONS
Deadlines for obtaining certificates — a quiz ?
Requirements for certificates — introduction of licenses and certificate numbers
Requirementswhileplanning
the inspection –
all
sites within a production chain
a list of
Potential risk of refusal to issue a GMP certificate with absent non-compliances in
the inspection report
Receipt of the requests months onward after the inspections
Use of the term "intermediate product" instead of "active pharmaceutical
ingredient" ("active pharmaceutical substance") in manufacturing of biological
medicinal products
Requirement to manufacture such products as bisphosphonates, cytostatic agents,
cytotoxic agents, and products that induce apoptosis, in the dedicated area
(dedicated premises and equipment)
Issues related to the inspections of biotechnology-derived products, vaccines, etc.
— sterile substances, manufacturing in grade A areas, etc.