



 Программирование
ПрограммированиеПохожие презентации:

")




 языка Java. (Лекция 16)")

")
")
Практика №1. Предварительная обработка чтений
1.
Основные Linux-командыpwd – показать текущую (рабочую) папку
cd XYZ – перейти в папку с именем XYZ
cd .. – перейти в папку на уровень выше, чем текущая
cd ~ - перейти в домашнюю папку (/data/Shared/ngs)
ls – посмотреть содержимое текущей папки
mkdir XYZ – создать папку с именем XYZ
Клавиша Tab – автодополнение
(нажать 1 или 2 раза)
Клавиши ↑ и ↓ - листать предыдущие команды:
↑ - предыдущая; ↓ - последующая
rm XYZ – удалить файл XYZ
rm -rf XYZ - удалить папку с именем XYZ и всё её содержимое
ln -s XYZ X1Y1Z1 – создать ссылку с именем X1Y1Z1 на объект XYZ
ln -s XYZ . – создать ссылку на объект XYZ в текущей папке с сохранением имени объекта
cp XYZ X1Y1Z1 – создать копию файла XYZ под именем X1Y1Z1 Ctrl+A – переход в начало строки
Ctrl+E – в конец
cp XYZ . – создать копию файла XYZ в текущей папке
cp -r XYZ X1Y1Z1 – создать копию папки XYZ под именем X1Y1Z1
cp -r XYZ . – создать копию папки XYZ в текущей папке
mv XYZ X1Y1Z1 – переименовать / переместить объект с именем XYZ в объект с именем X1Y1Z1
mv XYZ . - переименовать / переместить объект с именем XYZ в текущую папку
man XYZ – посмотреть справку по команде XYZ
2.
ЗАДАНИЕ1.
2.
Выполнить контроль качества ридов (fastqc) из папки ~/common/reads
Выполнить тримминг ридов (trimmomatic), включая удаление адаптеров,
фильтрацию по качеству с концов ридов и MINLEN. Обратить внимание на
статистику тримминга (каков процент ридов остался?)
http://www.usadellab.org/cms/?page=trimmomatic
http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomati
cManual_V0.32.pdf
3. Выполнить контроль качества оставшихся парных ридов
4. Объединить риды после тримминга в один файл и выполнить k-mer анализ
(jellyfish и kmergenie)
http://www.genome.umd.edu/docs/JellyfishUserGuide.pdf
http://kmergenie.bx.psu.edu/
3.
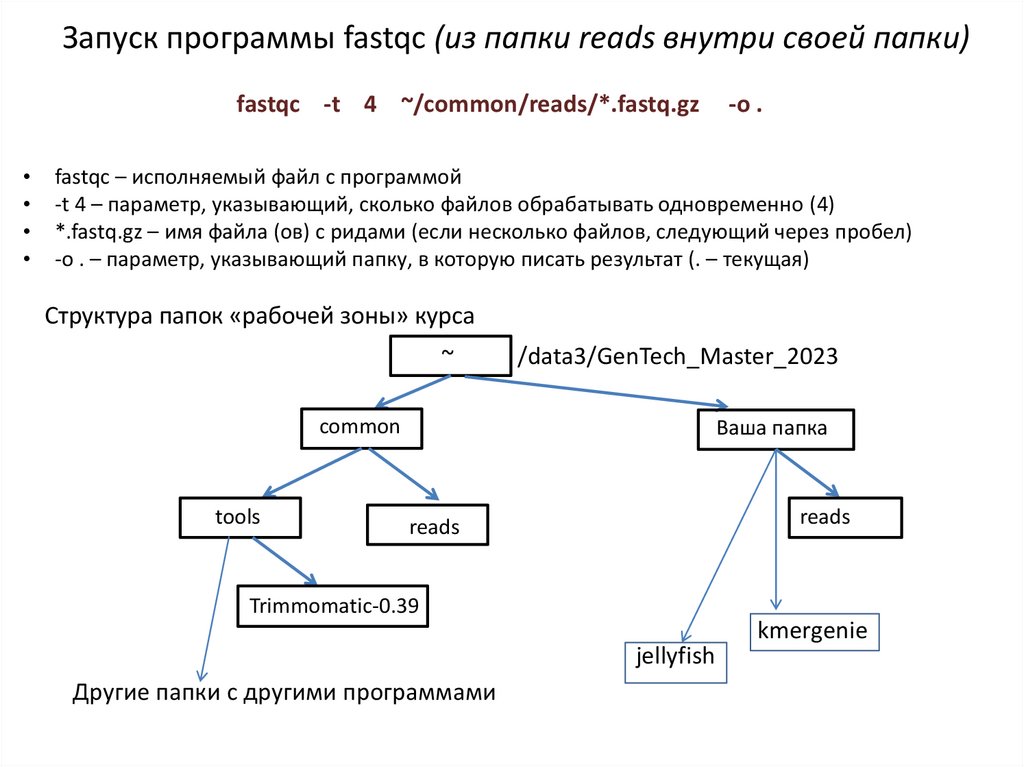
Запуск программы fastqc (из папки reads внутри своей папки)fastqc -t 4 ~/common/reads/*.fastq.gz
-o .
fastqc – исполняемый файл с программой
-t 4 – параметр, указывающий, сколько файлов обрабатывать одновременно (4)
*.fastq.gz – имя файла (ов) с ридами (если несколько файлов, следующий через пробел)
-o . – параметр, указывающий папку, в которую писать результат (. – текущая)
Структура папок «рабочей зоны» курса
~
↓
/data3/GenTech_Master_2023
common
↓
tools
↓
Ваша↓
папка
reads
↓
↓
reads
Trimmomatic-0.39
↓
jellyfish
Другие папки с другими программами
kmergenie
4.
Запуск программы Trimmomatic (для штамма 188)java -jar ~/common/tools/Trimmomatic-0.39/trimmomatic-0.39.jar PE -threads 2 -phred33
~/common/reads/188staph_S13_L001_R1_001.fastq.gz
~/common/reads/188staph_S13_L001_R2_001.fastq.gz -baseout 188.fastq.gz ТРИММЕРЫ
• java -jar ~/common/tools/Trimmomatic-0.36/trimmomatic-0.39.jar – запуск
исполняемого файла *.jar на Java
• PE – параметр, указывающий, что мы работаем с парными чтениями
• -threads 2 – параметр, указывающий, сколько CPU задействовать (2)
• -phred33 – параметр, указывающий, что качество чтений в кодировке phred+33
• 188_R1.fastq.gz 188_R2.fastq.gz – имена файлов с ридами
• -baseout 188.fastq.gz – общая часть имени выходных файлов:
188_1P.fastq.gz – выходной файл с левыми чтениями, у которых есть пара (paired)
188_2P.fastq.gz – выходной файл с правыми чтениями, у которых есть пара (paired)
188_1U.fastq.gz – выходной файл с левыми чтениями, оставшимися без пары (unpaired)
188_2U.fastq.gz – выходной файл с правыми чтениями, оставшимися без пары (unpaired)
ТРИММЕРЫ:
• ILLUMINACLIP – удаление адаптеров и праймеров из ридов
• LEADING:3 – удаление нуклеотидов с качеством прочтения Q менее заданного (3) с начала рида
• TRAILING:3 – удаление нуклеотидов с качеством прочтения Q менее заданного (3) с конца рида
• SLIDINGWINDOW:4:15 – удаление всех нуклеотидов в рамке длиной 4, если среднее качество Q
у них меньше заданного (15)
• CROP:100 – укорачивание всех ридов до заданной длины (100)
• HEADCROP:15 – удаление первых 15 нуклеотидов с начала рида
• MINLEN:100 – удаление всех ридов, длина которых менее заданной (100)
ПОРЯДОК ТРИММИНГА ВАЖЕН! ВАЖНО ПЕРВЫМ ДЕЛОМ ИЗБАВИТЬСЯ ОТ АДАПТЕРОВ!
5.
Запуск программы Trimmomatic: ILLUMINACLIPILLUMINACLIP:<fastaWithAdapters>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold>
• <fastaWithAdapters> - фаста-файл, содержащий адаптеры. Поставляются вместе с Триммоматиком,
лежат в папке adapters. HiSeq и MiSeq используют TruSeq-адаптеры версии 3 (TruSeq3-PE-2.fa).
• <seed mismatches> - максимальное число ошибок при выравнивании «куска» адаптера (max 16 bp)
Две стратегии тримминга адаптеров: simple и palindrome (только для парных ридов)
Palindrome:
• Имена последовательностей начинаются с Prefix и заканчиваются на /1 для левых ридов
и на /2 – для правых
• Адаптеры «пришиваются» к началу ридов и риды выравниваются друг с другом
3 признака «чтения сквозь»:
• Одинаковое кол-во «букв» образца
• Отсеквенированный адаптер с обоих
концов
• «Буквы» образца обратнокомплементарны
<palindrome clip threshold>
Точность выравнивания пары ридов
для палиндромной стратегии (30)
<simple clip threshold>
Точность выравнивания адаптера с ридом
для простой стратегии (10)
Точность: совпадение +0.6,
несовпадение –Q/10
Доп. параметры палиндромного режима - <minAdapterLength> (8) и <keepBothReads> (false)
6.
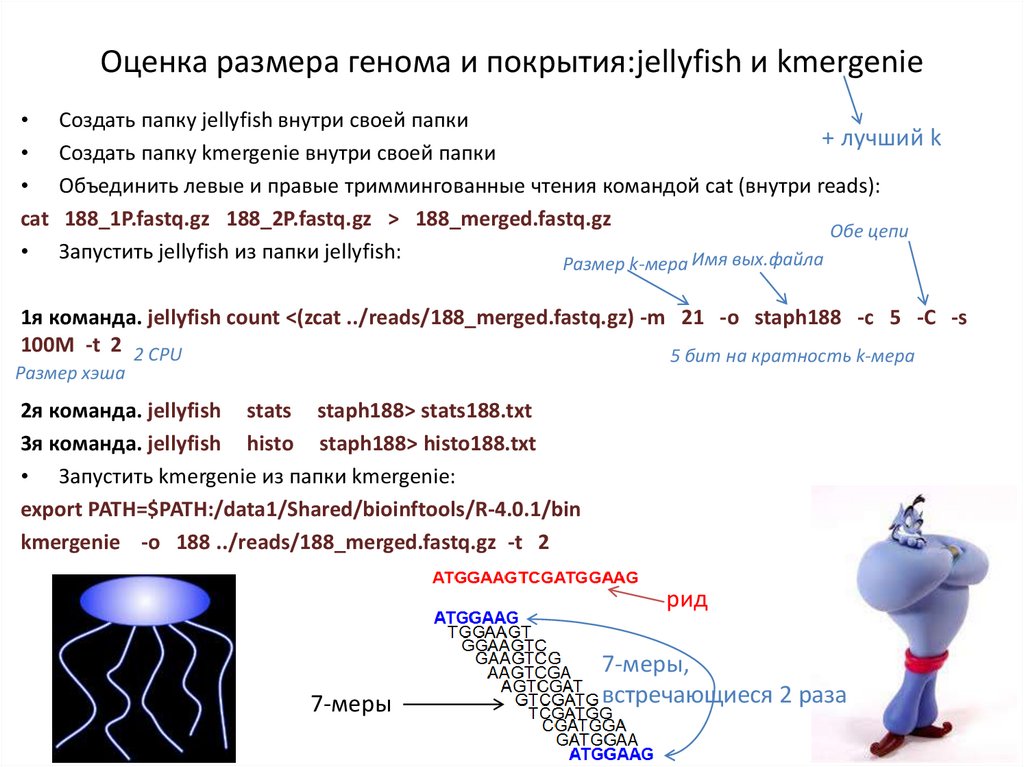
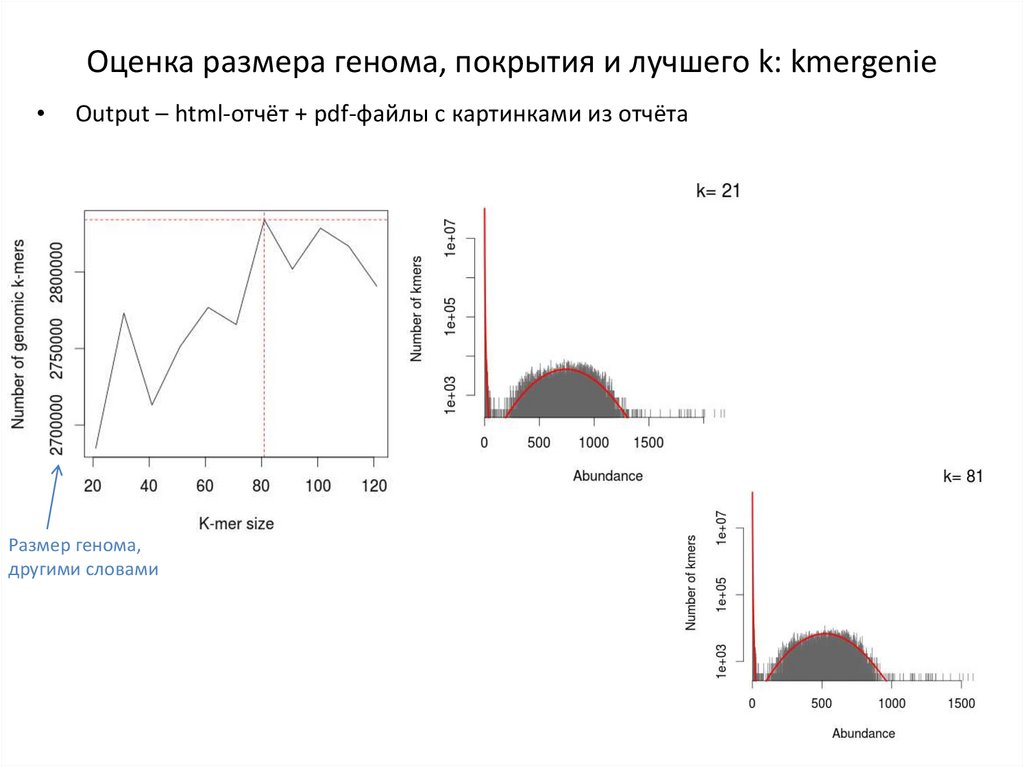
Оценка размера генома и покрытия:jellyfish и kmergenie• Создать папку jellyfish внутри своей папки
+ лучший k
• Создать папку kmergenie внутри своей папки
• Объединить левые и правые триммингованные чтения командой cat (внутри reads):
cat 188_1P.fastq.gz 188_2P.fastq.gz > 188_merged.fastq.gz
Обе цепи
• Запустить jellyfish из папки jellyfish:
Имя вых.файла
Размер k-мера
1я команда. jellyfish count <(zcat ../reads/188_merged.fastq.gz) -m 21 -o staph188 -c 5 -C -s
100M -t 2 2 CPU
5 бит на кратность k-мера
Размер хэша
2я команда. jellyfish stats staph188> stats188.txt
3я команда. jellyfish histo staph188> histo188.txt
• Запустить kmergenie из папки kmergenie:
export PATH=$PATH:/data1/Shared/bioinftools/R-4.0.1/bin
kmergenie -o 188 ../reads/188_merged.fastq.gz -t 2
рид
7-меры
7-меры,
встречающиеся 2 раза
7.
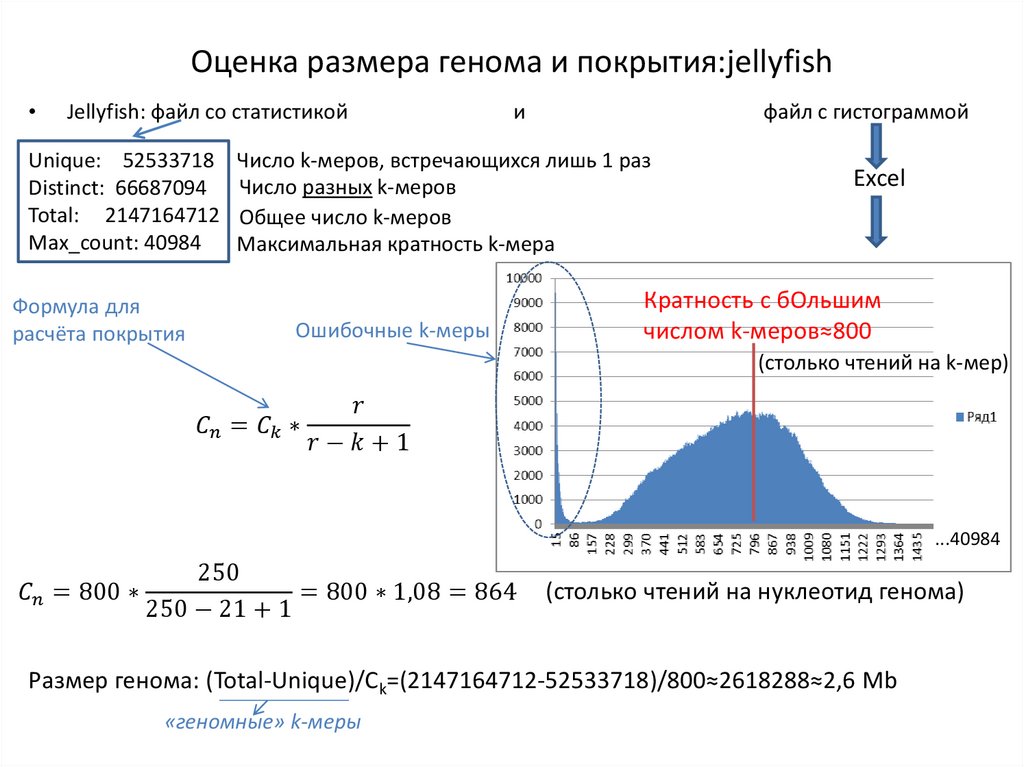
Оценка размера генома и покрытия:jellyfishJellyfish: файл со статистикой
Unique: 52533718
Distinct: 66687094
Total: 2147164712
Max_count: 40984
Формула для
расчёта покрытия
и
файл с гистограммой
Число k-меров, встречающихся лишь 1 раз
Число разных k-меров
Общее число k-меров
Максимальная кратность k-мера
Ошибочные k-меры
Excel
Кратность с бОльшим
числом k-меров≈800
(столько чтений на k-мер)