





























































 Медицина
МедицинаПохожие презентации:










Основы медико-генетического консультирования
1.
Основы медико-генетическогоконсультирования
Самойлова Людмила Руслановна
врач-лабораторный генетик, преподаватель кафедры
НППиМГ ИФМиБ
Казань, 2023
2.
«Наследственность — этоомнибус, в котором нас
сопровождают наши
предки, то и дело
кто-нибудь из них
высовывается оттуда,
ошеломляя нас своим
поведением».
Оливер Холмс
3.
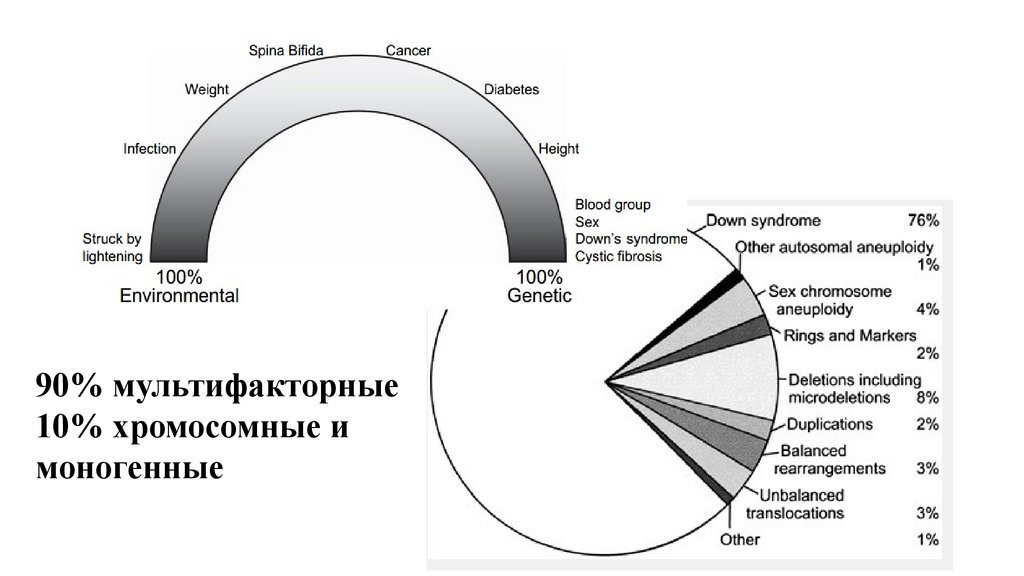
90% мультифакторные10% хромосомные и
моногенные
4.
• Первая в мире МГК была организованаДавиденковым С.Н. в 1929 г в Москве
• В 1932 г. открыта вторая в мире МГК в Ленинграде
• В США и Великобритании - после 1946 г.
• 1955г.-Шелдон Рид определяет МГК как социальную
службу
• 1979г- первая книга по практике МГК (Ш.Рид)
• 1979г – Кесслер –необходимость смены
евгенического курса на психосоциальный
5.
периоды в истории МГК• Евгенический. Цель - улучшение генофонда
• Профилактический (после второй мировой войны). Цельпредупреждение новых случаев в семье с отягощенным
анамнезом с помощью информирования о риске
• Период автономного принятия решений (с середины 60-х гг. 20
века). Эффективные методы диагностики и прогноза
способствуют утверждению недирективного консультирования
• Психотерапевтический
• Геномный
6.
Цель МГКУстановление степени генетического
риска в обследуемой семье и
разъяснение супругам в доступной
форме генетического заключения.
7.
Задачи МГК• Определение прогноза здоровья для будущего потомства в
семье, где был, есть или предполагается больной с
наследственной патологией
• Объяснение в доступной форме смысла генетического
прогноза и помощь в принятии решения
• Выбор наиболее эффективного способа профилактики
данного заболевания
• Пропаганда генетических знаний среди врачей и населения
8.
Показания для МГК• Рождение ребенка с врожденными пороками развития
• Установленная или подозреваемая наследственная патология в
семье
• Репродуктивные потери, мертворождения, бесплодие
• Близкородственные браки
• Неблагополучное протекание беременности
• Возраст беременной свыше 35 лет
• Отставание в развитии, наличие диспластических черт в
сочетании
9.
Этапы МГК1.Установление точного диагноза
2.Определение степени риска
математическими методами и прогноз
потомства
3.Выдача генетического заключения и
объяснения его сущности
консультирующимся
10.
Методы МГК• Клинико-генеалогический
• Близнецовый
• Дерматоглифический
• Популяционный
• Лабораторные (цитогенетический, биохимический,
молекулярный)
• Скрининговые программы (пренатальный, неонатальный)
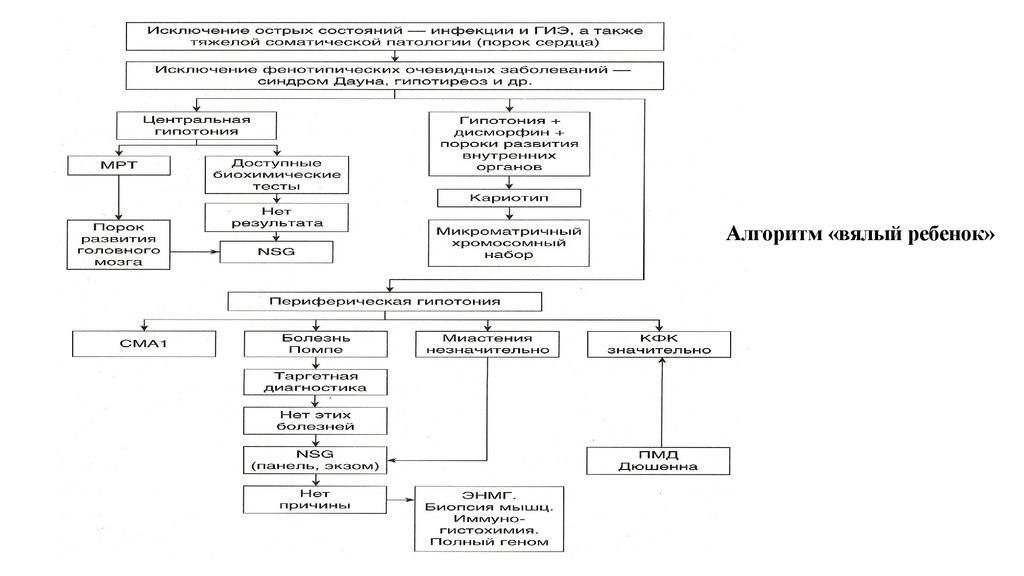
11.
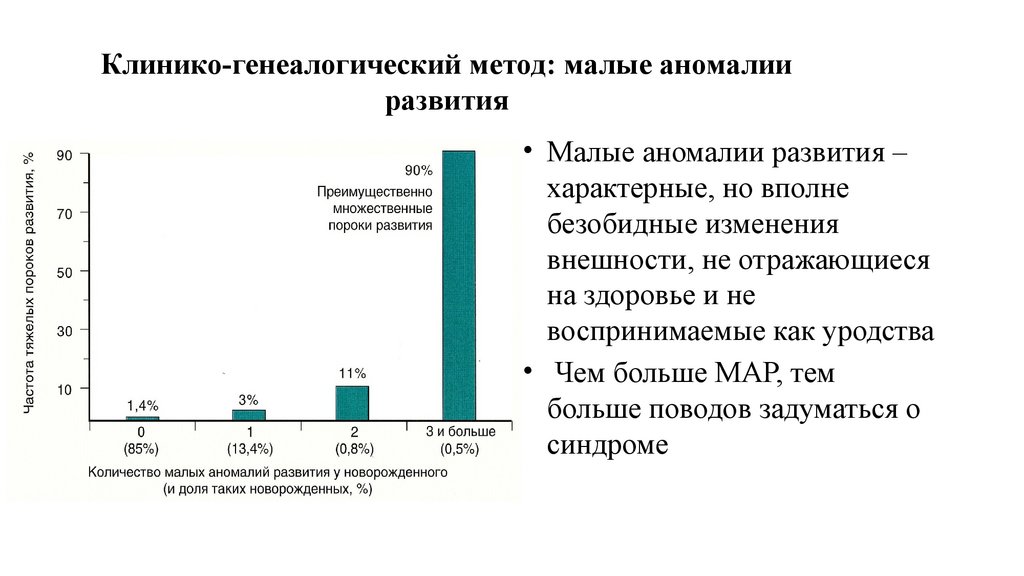
Клинико-генеалогический метод: малые аномалииразвития
• Малые аномалии развития –
характерные, но вполне
безобидные изменения
внешности, не отражающиеся
на здоровье и не
воспринимаемые как уродства
• Чем больше МАР, тем
больше поводов задуматься о
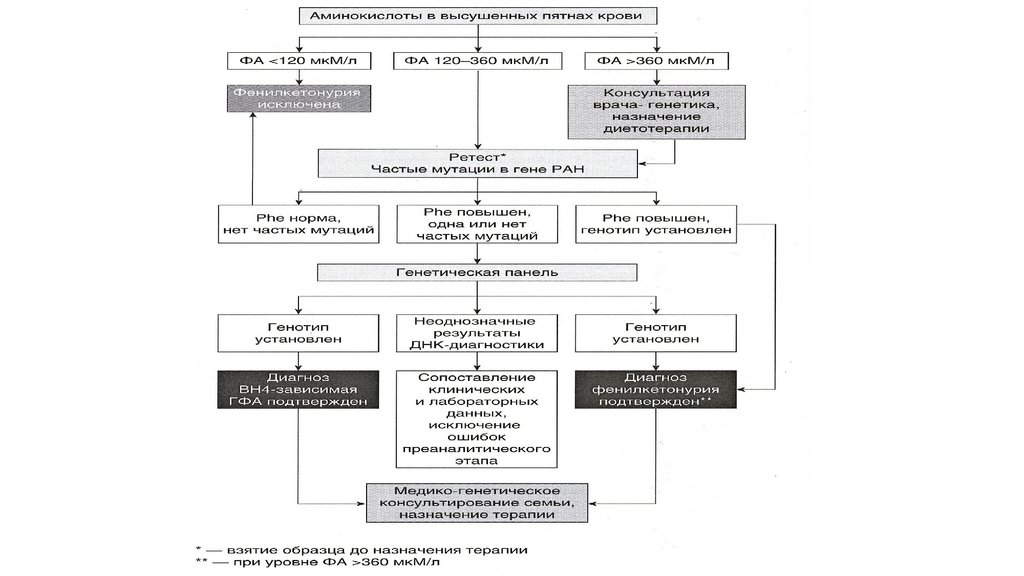
синдроме
12.
Малые аномалии развития глазной области• А и Б – эпикант
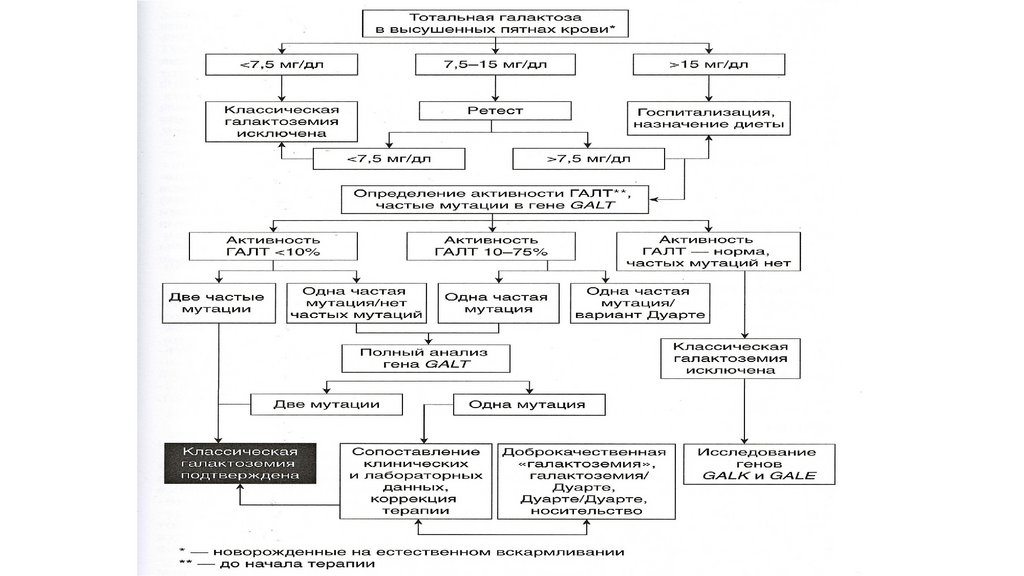
• В - односторонний эпикант
• Г- монголоидный разрез
• Д – антимонголоидный
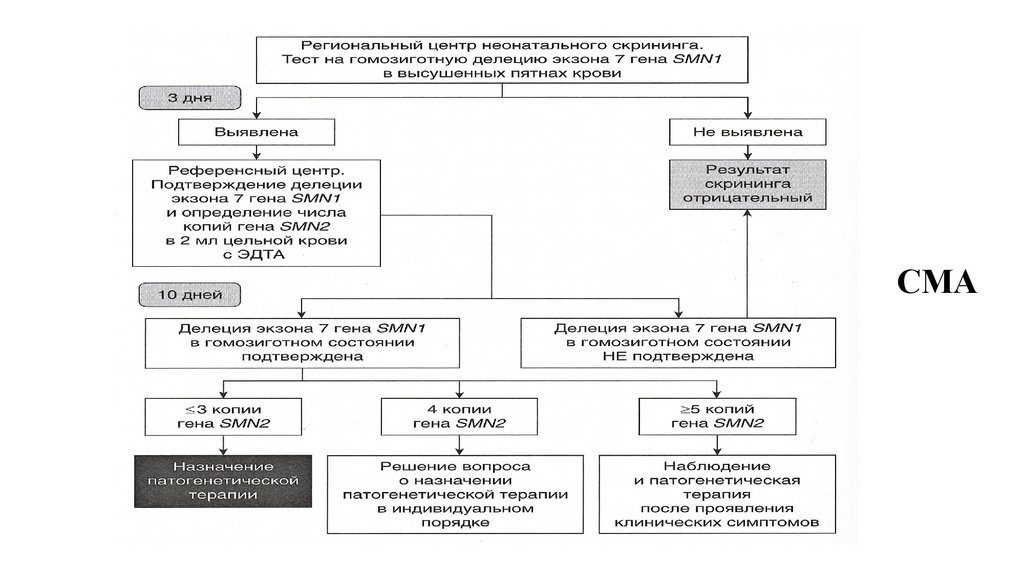
разрез
• Е – гипертелоризм глаз
• Ж – пятна Брашфилда
(Брушвильда) (в 80% при
синдроме Дауна)
13.
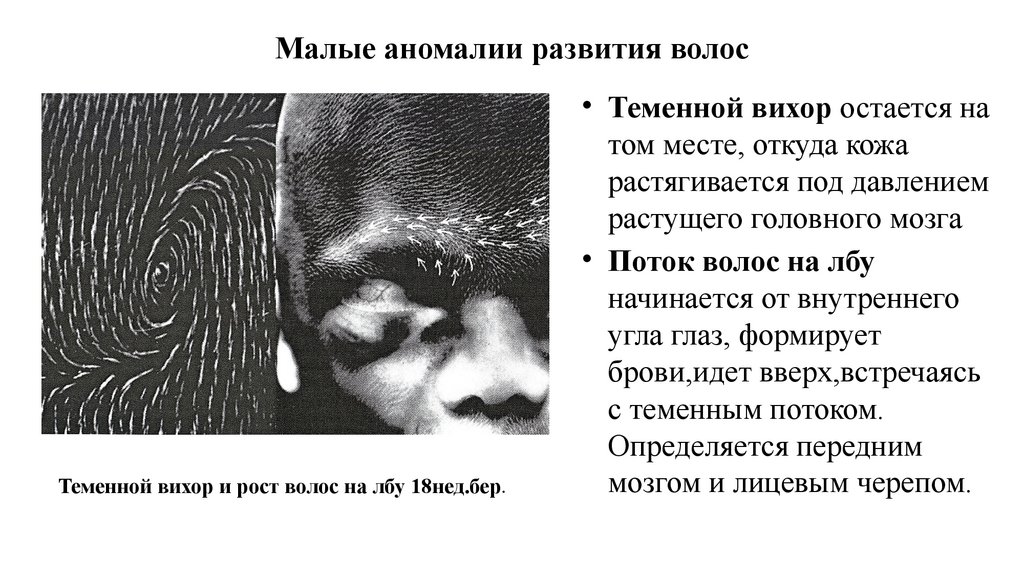
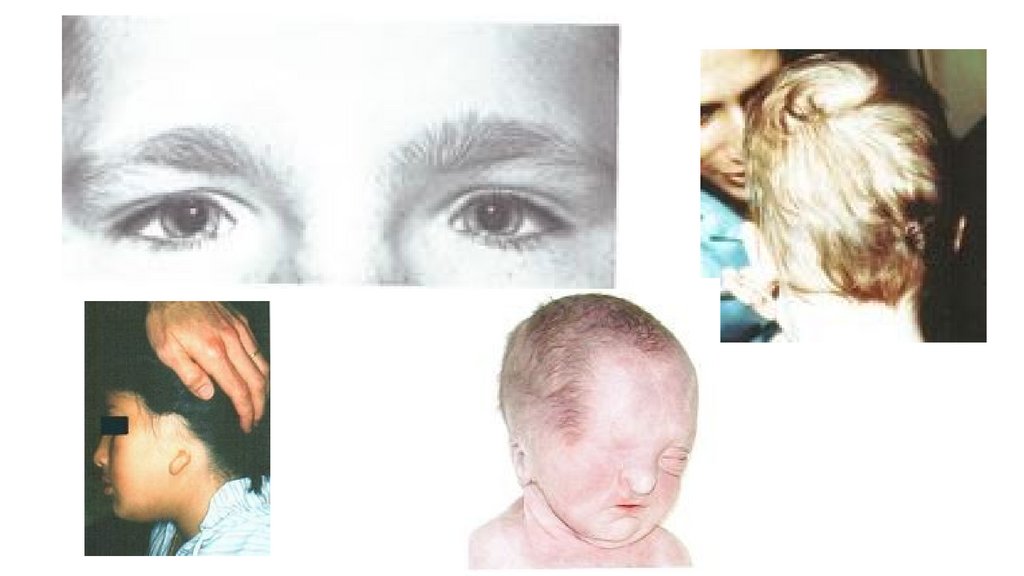
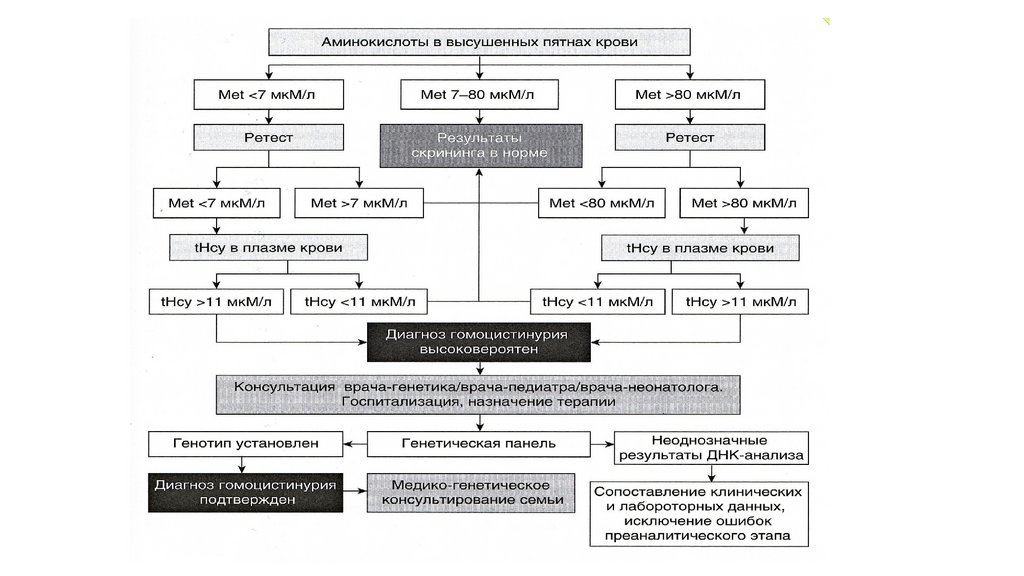
Малые аномалии развития волосТеменной вихор и рост волос на лбу 18нед.бер.
• Теменной вихор остается на
том месте, откуда кожа
растягивается под давлением
растущего головного мозга
• Поток волос на лбу
начинается от внутреннего
угла глаз, формирует
брови,идет вверх,встречаясь
с теменным потоком.
Определяется передним
мозгом и лицевым черепом.
14.
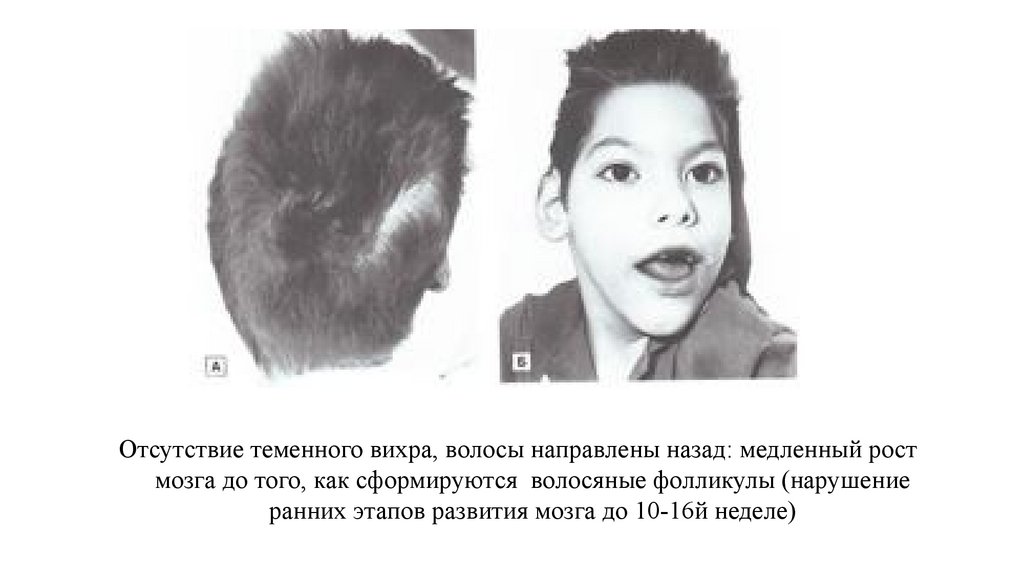
Отсутствие теменного вихра, волосы направлены назад: медленный ростмозга до того, как сформируются волосяные фолликулы (нарушение
ранних этапов развития мозга до 10-16й неделе)
15.
16.
Малые аномалии развития ушных раковин• А – ушные привески (аномалии 2
первых жаберных дуг, может быть
нарушение слуха)
• Б – предушные ямки, часто семейное ,
шире распространено у негров, у
девочек – вдвое чаще(может быть
нарушение слуха)
• В – макротия (результат в/у сдавления
плода, при маловодии)
• Г и Д – разный размер ушей (кривошея)
• Е-микротия (в 85% случаев глухота на
одно, в 15% - на оба уха)
• Ж – низкое и косое расположение
ушных раковин (нарушения
морфогенеза)
• З – бранхиогенные свищи
17.
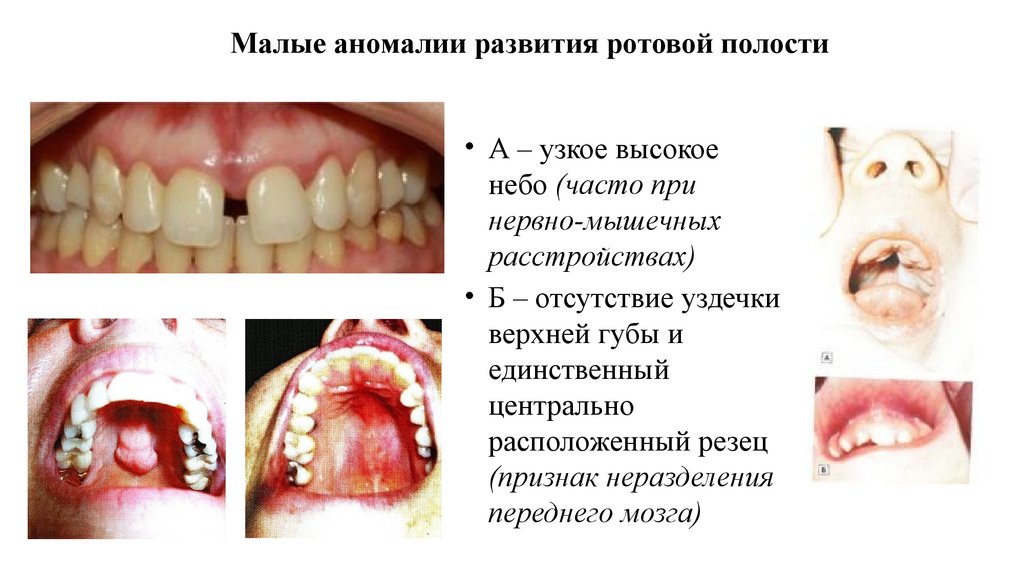
Малые аномалии развития ротовой полости• А – узкое высокое
небо (часто при
нервно-мышечных
расстройствах)
• Б – отсутствие уздечки
верхней губы и
единственный
центрально
расположенный резец
(признак неразделения
переднего мозга)
18.
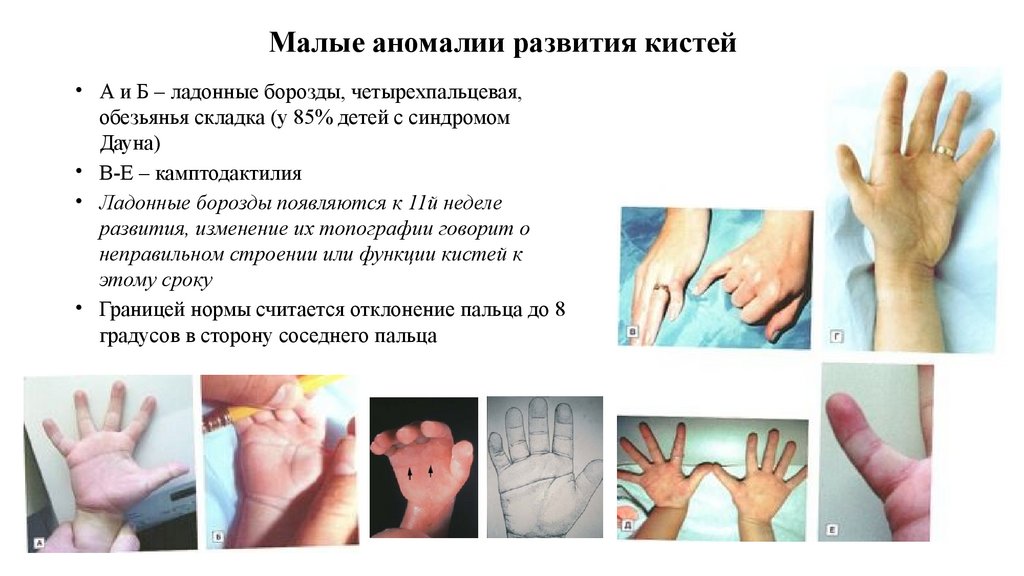
Малые аномалии развития кистей• А и Б – ладонные борозды, четырехпальцевая,
обезьянья складка (у 85% детей с синдромом
Дауна)
• В-Е – камптодактилия
• Ладонные борозды появляются к 11й неделе
развития, изменение их топографии говорит о
неправильном строении или функции кистей к
этому сроку
• Границей нормы считается отклонение пальца до 8
градусов в сторону соседнего пальца
19.
Дерматоглифический методдуга
завиток
петля
20.
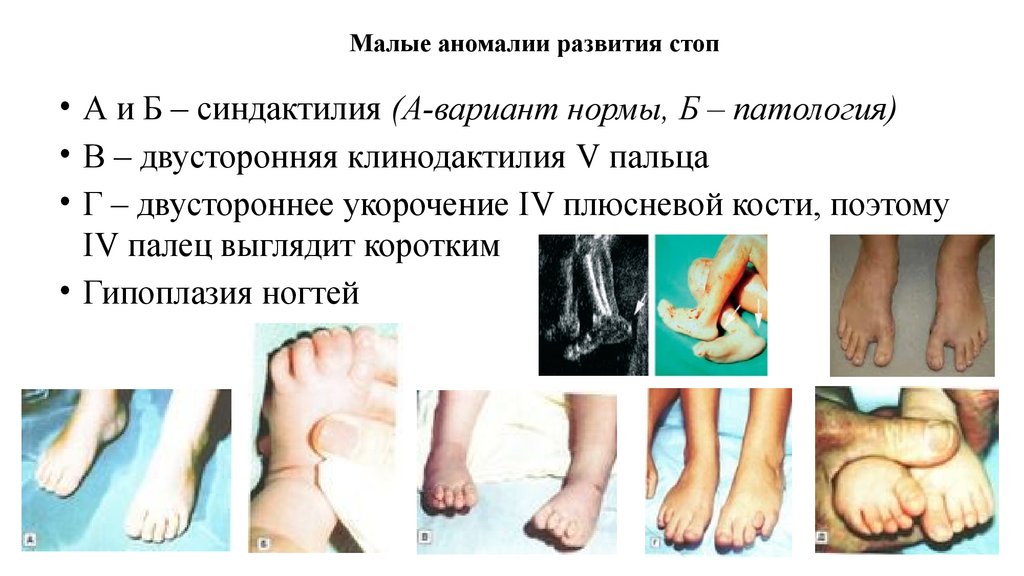
Малые аномалии развития стоп• А и Б – синдактилия (А-вариант нормы, Б – патология)
• В – двусторонняя клинодактилия V пальца
• Г – двустороннее укорочение IV плюсневой кости, поэтому
IV палец выглядит коротким
• Гипоплазия ногтей
21.
22.
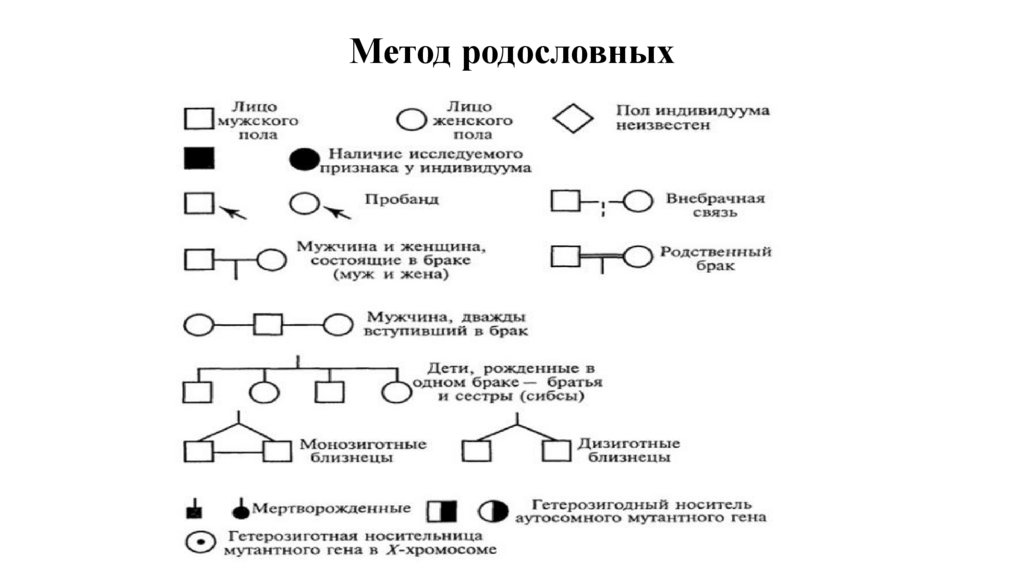
Метод родословных23.
Расчеты при медико-генетическом консультированииВсе расчеты при медико-генетическом консультировании основаны на
менделевских законах и элементах теории вероятностей. Есть 2
ключевых правила для расчетов:
• Если для того, чтобы произошло событие С необходимо, чтобы
произошли события А и В, то вероятность С будет равна
произведению вероятности событий А и В.
Р(С)=Р(А)хР(В), где Р- вероятность события.
• Чтобы вычислить вероятность данного события среди двух или более,
нужно вероятность этого события разделить на сумму вероятностей
всех событий
Р(А)=Р(А)/Р(А)+Р(В) и Р(В)=Р(В)/Р(А)+Р(В)
24.
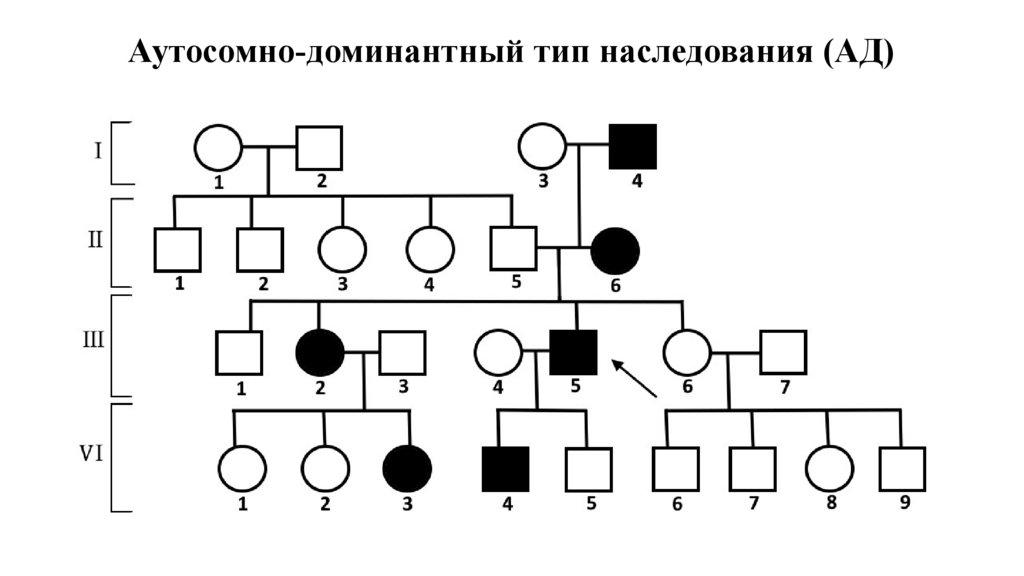
Аутосомно-доминантный тип наследования (АД)25.
Признаки АДЗ• Каждый больной член семьи имеет больного
родителя
• Больные встречаются в каждом поколении
(вертикальная передача)
• Одинаково болеют и мужчины, и женщины
• Если болен один из родителей, то риск
заболевания у детей 50%
26.
Особенности наследования АДЗ• Неполная пенетрантность-вероятность
проявления заболевания в фенотипе у носителей
патологического аллеля
• Доминантные аллели обладают и различной
экспрессивностью (тяжестью заболевания)
• Редко проявляются при рождении
27.
Прогноз потомства при АДЗ• Вероятность рождения больного ребенка ½ К
• Вероятность рождения здоровых гомозигот 50%
• Вероятность рождения здоровых гетерозигот
1/2(1-К)
• Доля гетерозиготных носителей среди здоровых
потомков (1-К)/(2-К)
• Доля детей НЕ носителей 1/(2-К)
28.
Пример: К=80% (или 0,8)• Вероятность рождения больного ребенка 1/2К = 0,4 (40%)
• Вероятность рождения здоровых гетерозигот ½(1-К) = 0,1 (10%)
• Доля гетерозиготных носителей среди здоровых потомков
(1-К)/(2-К)=0,166 (17%)
• Доля детей-НЕносителей (рецессивных гомозигот) 1/(2-К)=0,83
(83%)
Таким образом:
• 40% больных
• 60% здоровых, среди которых 17% - носители мутантного гена
29.
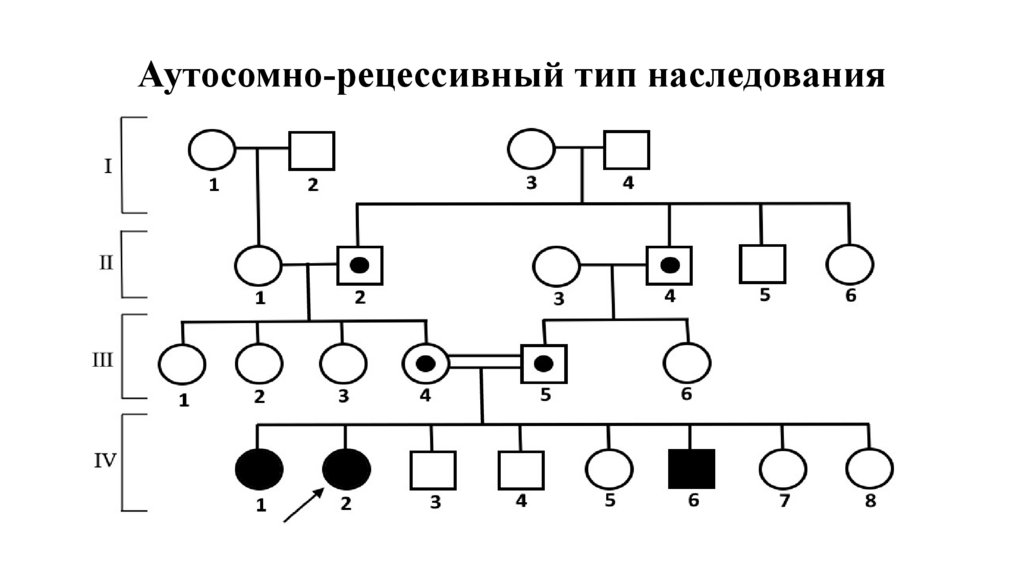
Аутосомно-рецессивный тип наследования30.
Признаки АРЗ• Частота встречаемости АР заболеваний в популяции невысока
• Заболевание проявляется в раннем возрасте
• Больные присутствуют в одном поколении (горизонтальный тип)
• Родители больных клинически здоровы, оба родителя являются
носителями патологичного аллеля
• У больного родителя в браке со здоровым партнером дети будут
клинически здоровы, но носителями мутантного аллеля
• При близкородственных браках вероятность рождения больного
возрастает по сравнению с популяцией.
• Мужчины и женщины болеют с одинаковой частотой
31.
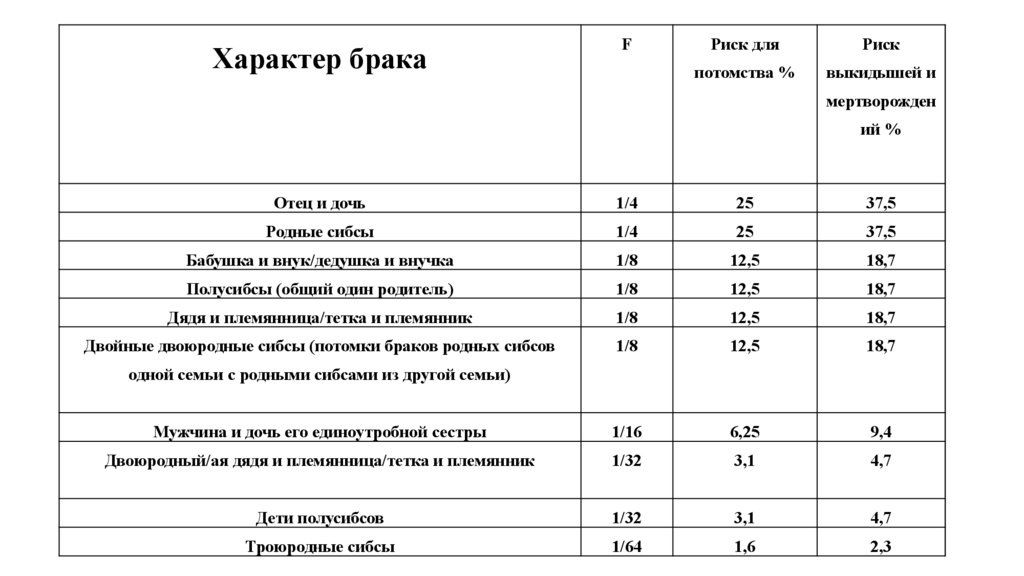
Близкородственные браки. Коэффициент инбридингаМера вероятности патогенетических эффектов, возникающих при
близкородственных браках, необходим для оценки генетического
риска для потомков при близкородственных браках
где А - удвоенное количество общих предков, В - число ступеней
передачи через отца, С - ступеней передачи через мать
32.
Характер бракаF
Риск для
Риск
потомства %
выкидышей и
мертворожден
ий %
Отец и дочь
1/4
25
37,5
Родные сибсы
1/4
25
37,5
Бабушка и внук/дедушка и внучка
1/8
12,5
18,7
Полусибсы (общий один родитель)
1/8
12,5
18,7
Дядя и племянница/тетка и племянник
1/8
12,5
18,7
Двойные двоюродные сибсы (потомки браков родных сибсов
1/8
12,5
18,7
Мужчина и дочь его единоутробной сестры
1/16
6,25
9,4
Двоюродный/ая дядя и племянница/тетка и племянник
1/32
3,1
4,7
Дети полусибсов
1/32
3,1
4,7
Троюродные сибсы
1/64
1,6
2,3
одной семьи с родными сибсами из другой семьи)
33.
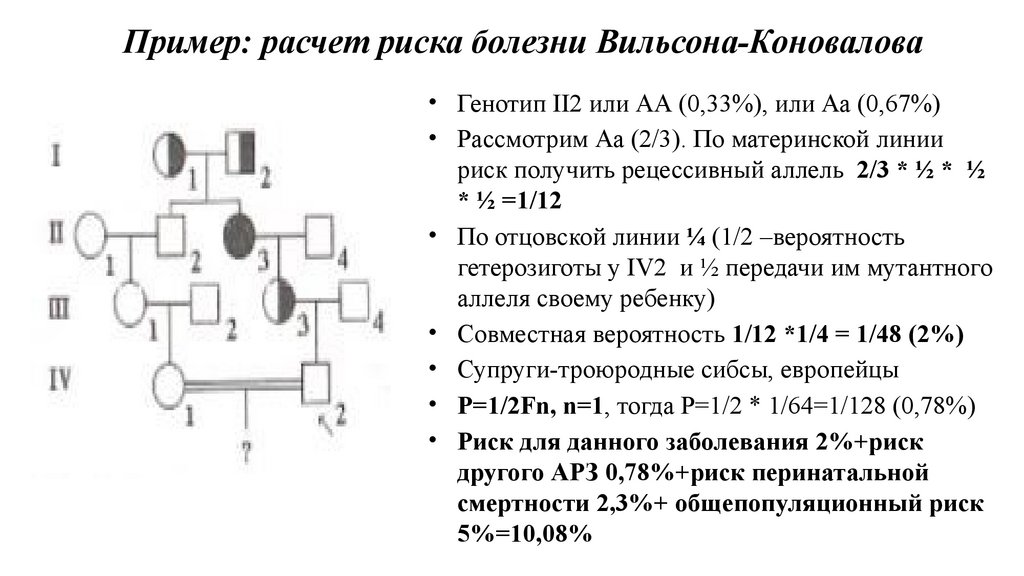
Пример: расчет риска болезни Вильсона-Коновалова• Генотип II2 или АА (0,33%), или Аа (0,67%)
• Рассмотрим Аа (2/3). По материнской линии
риск получить рецессивный аллель 2/3 * ½ * ½
* ½ =1/12
• По отцовской линии ¼ (1/2 –вероятность
гетерозиготы у IV2 и ½ передачи им мутантного
аллеля своему ребенку)
• Совместная вероятность 1/12 *1/4 = 1/48 (2%)
• Супруги-троюродные сибсы, европейцы
• P=1/2Fn, n=1, тогда Р=1/2 * 1/64=1/128 (0,78%)
• Риск для данного заболевания 2%+риск
другого АРЗ 0,78%+риск перинатальной
смертности 2,3%+ общепопуляционный риск
5%=10,08%
34.
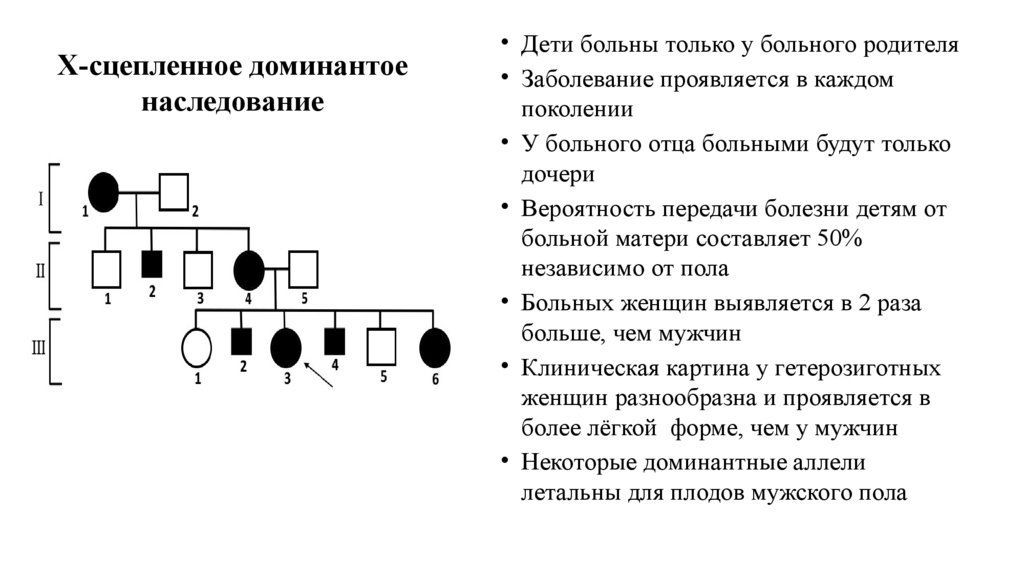
Х-сцепленное доминантоенаследование
• Дети больны только у больного родителя
• Заболевание проявляется в каждом
поколении
• У больного отца больными будут только
дочери
• Вероятность передачи болезни детям от
больной матери составляет 50%
независимо от пола
• Больных женщин выявляется в 2 раза
больше, чем мужчин
• Клиническая картина у гетерозиготных
женщин разнообразна и проявляется в
более лёгкой форме, чем у мужчин
• Некоторые доминантные аллели
летальны для плодов мужского пола
35.
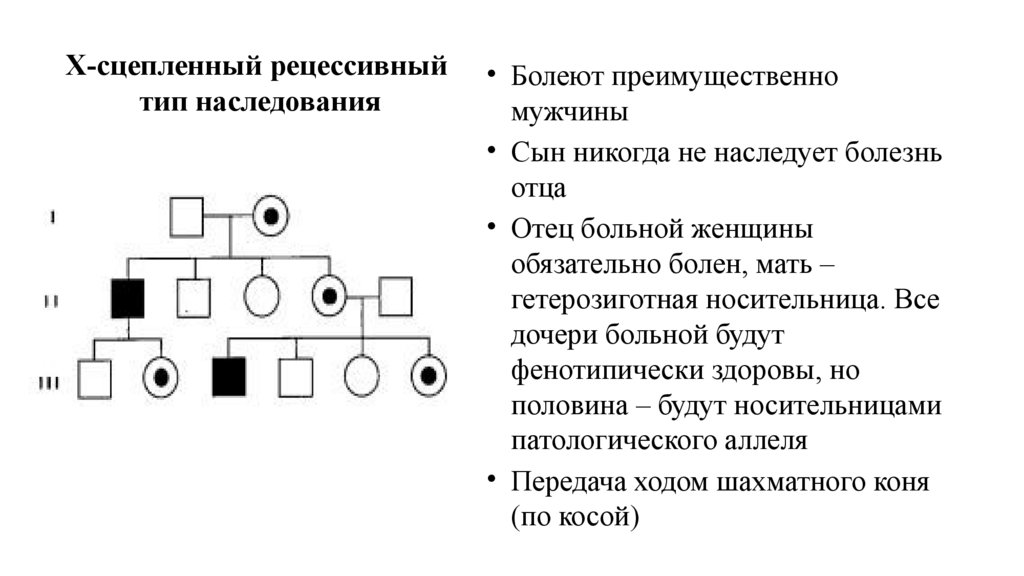
Х-сцепленный рецессивныйтип наследования
• Болеют преимущественно
мужчины
• Сын никогда не наследует болезнь
отца
• Отец больной женщины
обязательно болен, мать –
гетерозиготная носительница. Все
дочери больной будут
фенотипически здоровы, но
половина – будут носительницами
патологического аллеля
• Передача ходом шахматного коня
(по косой)
36.
Y-сцепленный типнаследования
• В Y-хромосоме
расположено около 40 генов,
в основном,
контролирующих процессы
детерминации пола и
сперматогенеза
• Признаки и заболевания,
связанные с этой гоносомой
передаются от отца к сыну
37.
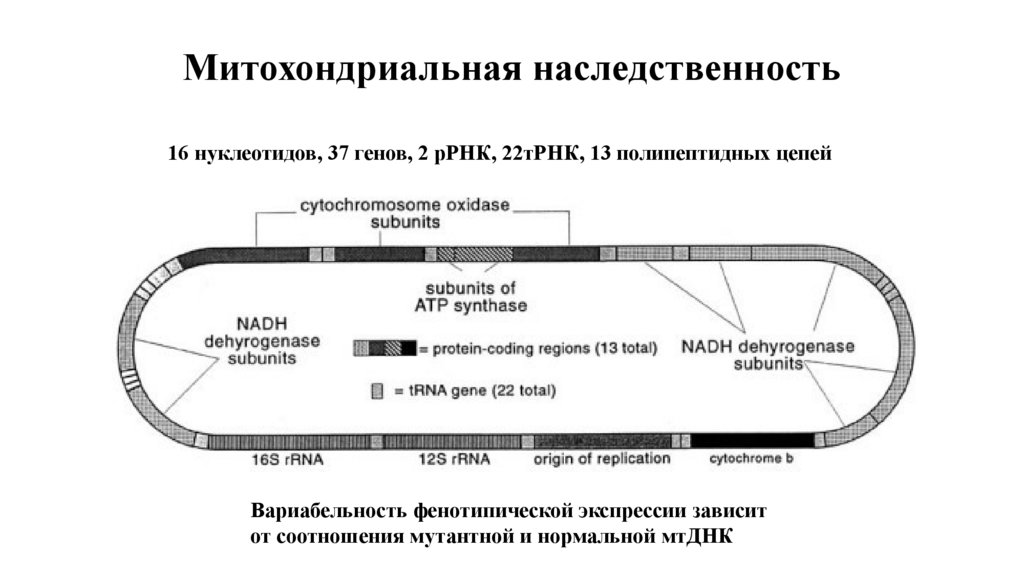
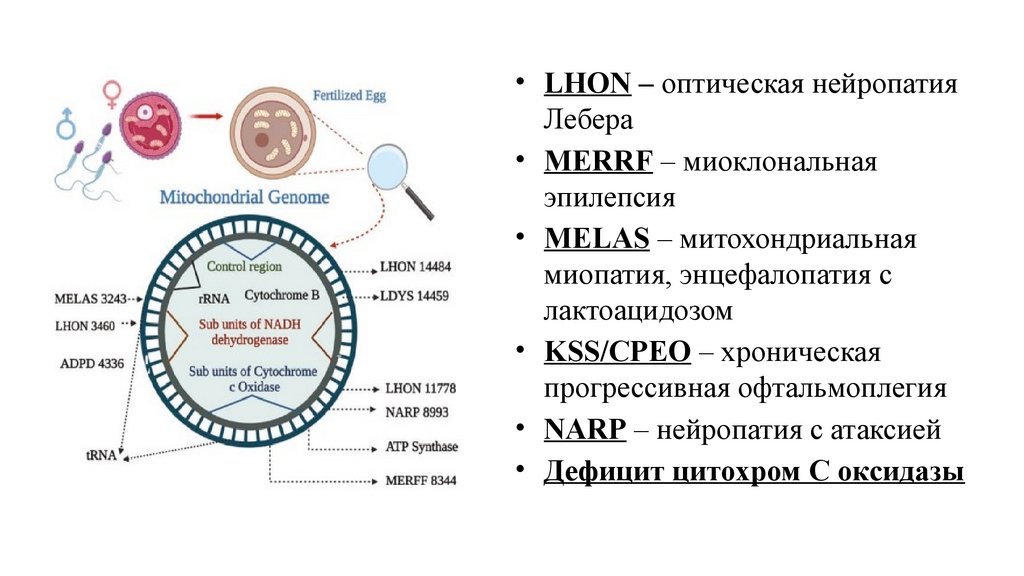
Митохондриальная наследственность16 нуклеотидов, 37 генов, 2 рРНК, 22тРНК, 13 полипептидных цепей
Вариабельность фенотипической экспрессии зависит
от соотношения мутантной и нормальной мтДНК
38.
• LHON – оптическая нейропатияЛебера
• MERRF – миоклональная
эпилепсия
• MELAS – митохондриальная
миопатия, энцефалопатия с
лактоацидозом
• KSS/CPEO – хроническая
прогрессивная офтальмоплегия
• NARP – нейропатия с атаксией
• Дефицит цитохром С оксидазы
39.
Митохондриальная патология• Передача патологии только по материнской
линии
• Поражаются оба пола в равной степени
• Гетероплазмия (наличие в одной клетке
различных вариантов митохондриальной ДНК)
40.
Особенности прогноза при мультифакторных заболеваниях• Вероятность проявления заболевания зависит от степени родства с
пораженным членом семьи (играет роль количество общих генов)
• Количество больных в семье определяет прогноз для пробанда
• Прогноз зависит от степени тяжести заболевания
• Генетический прогноз зависит от пола пробанда, если имеется
преимущественное поражение пола
• Риск увеличивается при ассортативных браках (подбор партнеров
по какому-либо признаку)
• Риск для потомства возрастает в кровнородственных браках
41.
Расчет риска при хромосомной патологииОпределение повторного риска проводится в случаях:
• Повторное рождение ребенка с анеуплоидией при нормальных
кариотипах родителей
• Повторное рождение больного при мозаицизме у родителя
• Прогноз при носительстве сбалансированной перестройки у родителя
При мозаицизме у одного из родителей риск определяется по формуле
[Х/(2-Х)] × К,
где Х-доля аномального клона, К-коэффициент элиминации
несбалансированной зиготы (например, для трисомии 21 К=0,5)
42.
Немного цифр…• Более 16000 генов отвечают за фенотипические моногенные
признаки
• Для 11000 установлен тип наследования
• Картировано более 8000 генов
• Около 4500 генов связано с моногенными заболеваниями
• Для 4000 установлен тип наследования
• Более 3500 аутосомных болезней
• Более 300 Х-сцепленных
• Более 20-митохондриальных
• 10 – Y-сцепленные
43.
1. АД тип наследования2. АР тип наследования
3. Х-сцепленный тип наследования
4. Ген локализован в Y-хромосоме
5. Ген локализован в ДНК митохондрий
6. Ген локализован в аутосомах после 15 мая 1994 года
* Перед MIM-номером – тип наследования установлен
# Перед MIM-номером - заболевание м.б. вызвано мутациями в 2 или более генах
44.
От 306900.0001 до 306900.0101 – аллельные варианты45.
Синдром Элерса-Данлоса (кожно-суставной тип)• Описан в 1682 г, подробно в 1901г
• Разболтанность суставов,
чрезмерная растяжимость кожи,
плохое заживление ран с
образованием атрофических
рубцов
• Мутации генов COL5A1, COL5A2
и COL1A1 – синтез коллагена V и
I типов
• Предполагается, что Паганини
страдал этим заболеванием
OMIM #130000
АДз
46.
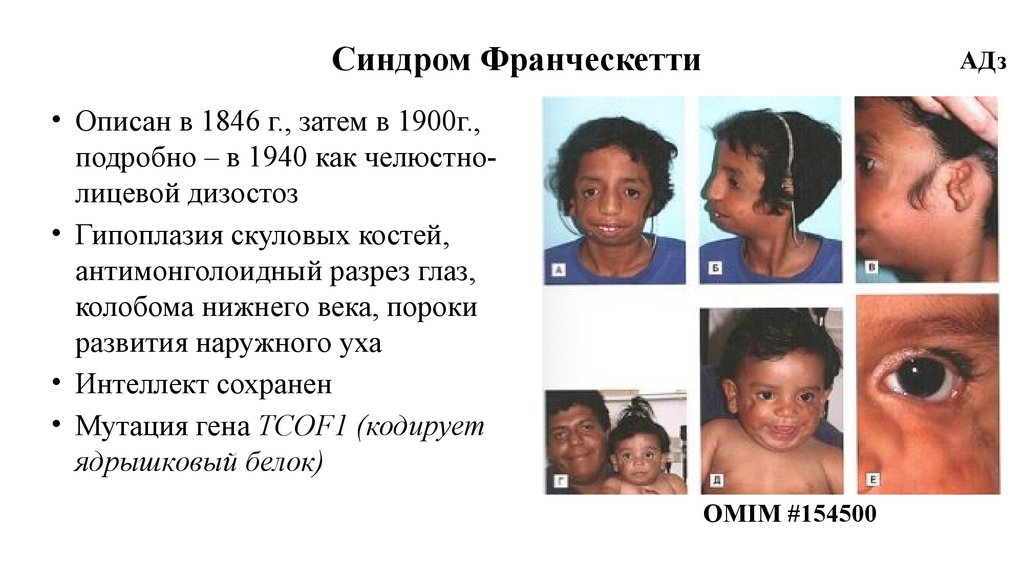
Синдром ФранческеттиАДз
• Описан в 1846 г., затем в 1900г.,
подробно – в 1940 как челюстнолицевой дизостоз
• Гипоплазия скуловых костей,
антимонголоидный разрез глаз,
колобома нижнего века, пороки
развития наружного уха
• Интеллект сохранен
• Мутация гена TCOF1 (кодирует
ядрышковый белок)
OMIM #154500
47.
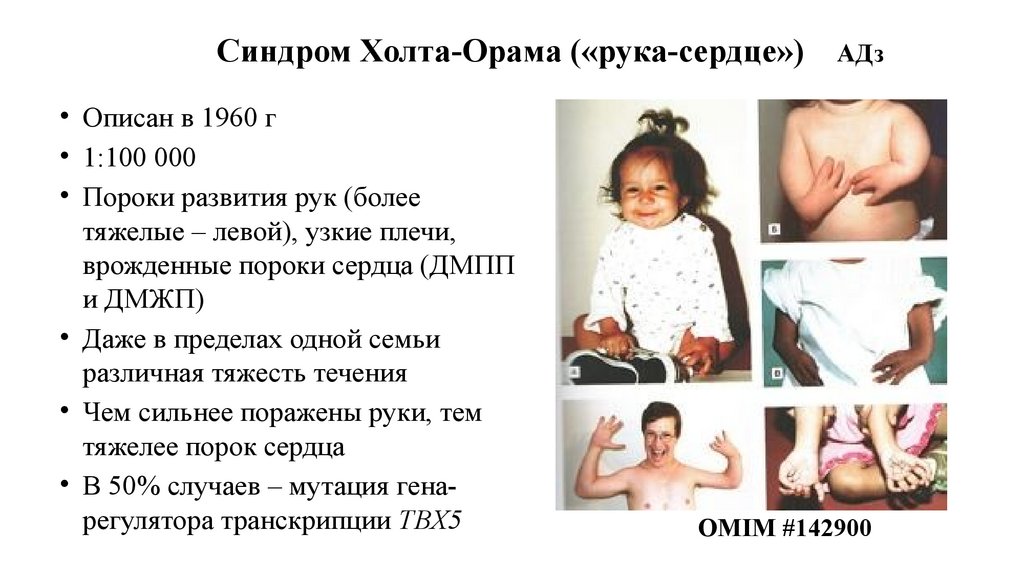
Синдром Холта-Орама («рука-сердце»)• Описан в 1960 г
• 1:100 000
• Пороки развития рук (более
тяжелые – левой), узкие плечи,
врожденные пороки сердца (ДМПП
и ДМЖП)
• Даже в пределах одной семьи
различная тяжесть течения
• Чем сильнее поражены руки, тем
тяжелее порок сердца
• В 50% случаев – мутация генарегулятора транскрипции ТВХ5
АДз
OMIM #142900
48.
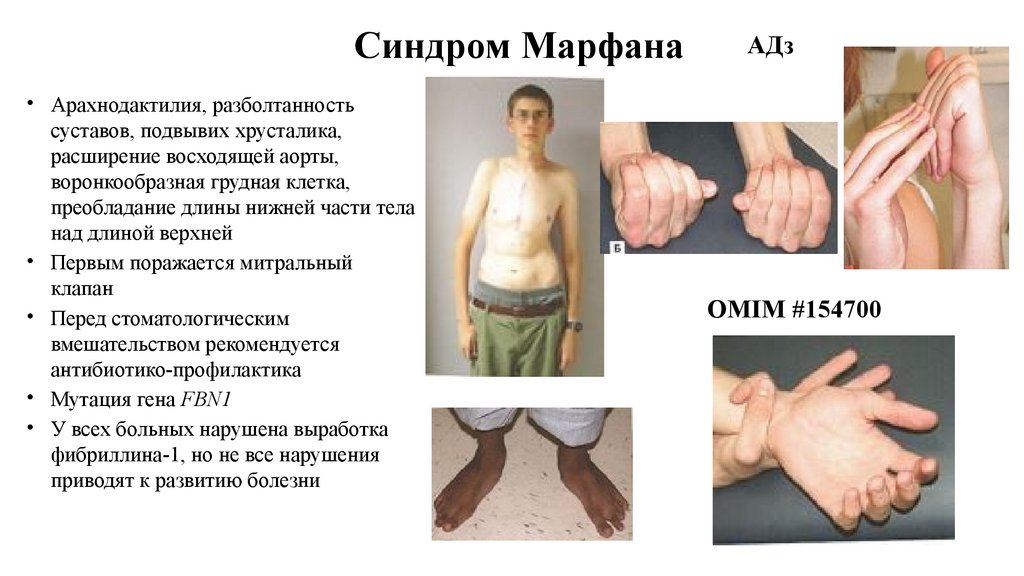
Синдром Марфана• Арахнодактилия, разболтанность
суставов, подвывих хрусталика,
расширение восходящей аорты,
воронкообразная грудная клетка,
преобладание длины нижней части тела
над длиной верхней
• Первым поражается митральный
клапан
• Перед стоматологическим
вмешательством рекомендуется
антибиотико-профилактика
• Мутация гена FBN1
• У всех больных нарушена выработка
фибриллина-1, но не все нарушения
приводят к развитию болезни
АДз
OMIM #154700
49.
АРзСиндром Миллера
• Гипоплазия скуловых
костей, лицо, как у с-ма
Франческетти, колобомы
и выворот век, расщелина
верхней губы и неба,
отсутствие V пальца на
обеих руках и ногах,
гипоплазия лучевой и
локтевой костей,
добавочные соски
• Интеллект сохранен
OMIM#263750
50.
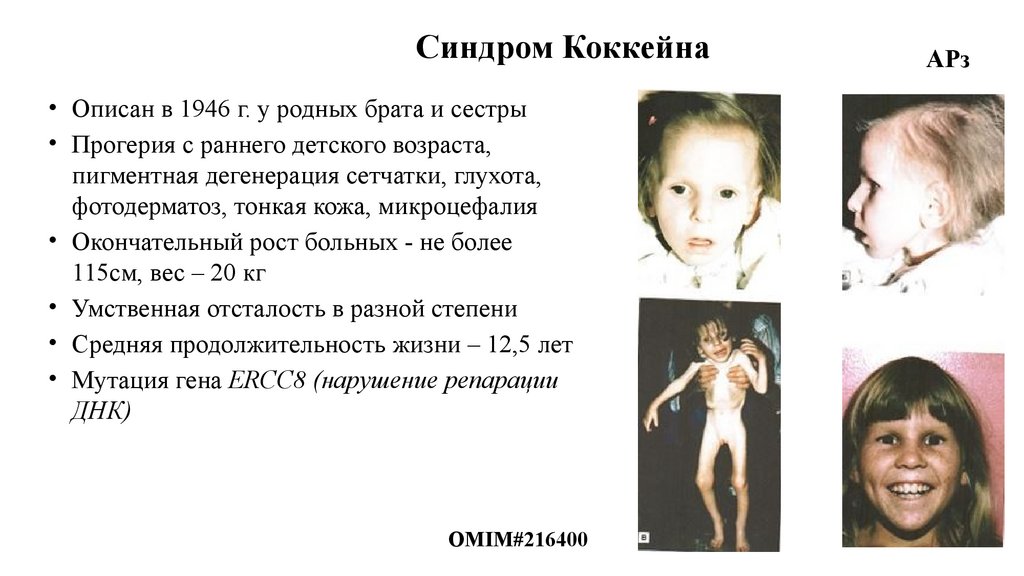
Синдром Коккейна• Описан в 1946 г. у родных брата и сестры
• Прогерия с раннего детского возраста,
пигментная дегенерация сетчатки, глухота,
фотодерматоз, тонкая кожа, микроцефалия
• Окончательный рост больных - не более
115см, вес – 20 кг
• Умственная отсталость в разной степени
• Средняя продолжительность жизни – 12,5 лет
• Мутация гена ERCC8 (нарушение репарации
ДНК)
OMIM#216400
АРз
51.
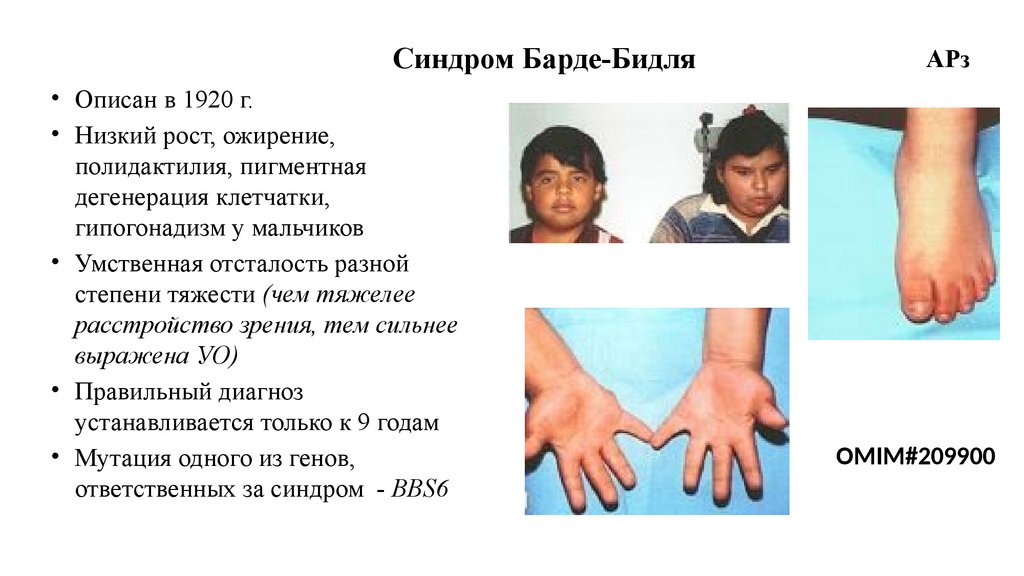
Синдром Барде-Бидля• Описан в 1920 г.
• Низкий рост, ожирение,
полидактилия, пигментная
дегенерация клетчатки,
гипогонадизм у мальчиков
• Умственная отсталость разной
степени тяжести (чем тяжелее
расстройство зрения, тем сильнее
выражена УО)
• Правильный диагноз
устанавливается только к 9 годам
• Мутация одного из генов,
ответственных за синдром - BBS6
АРз
OMIM#209900
52.
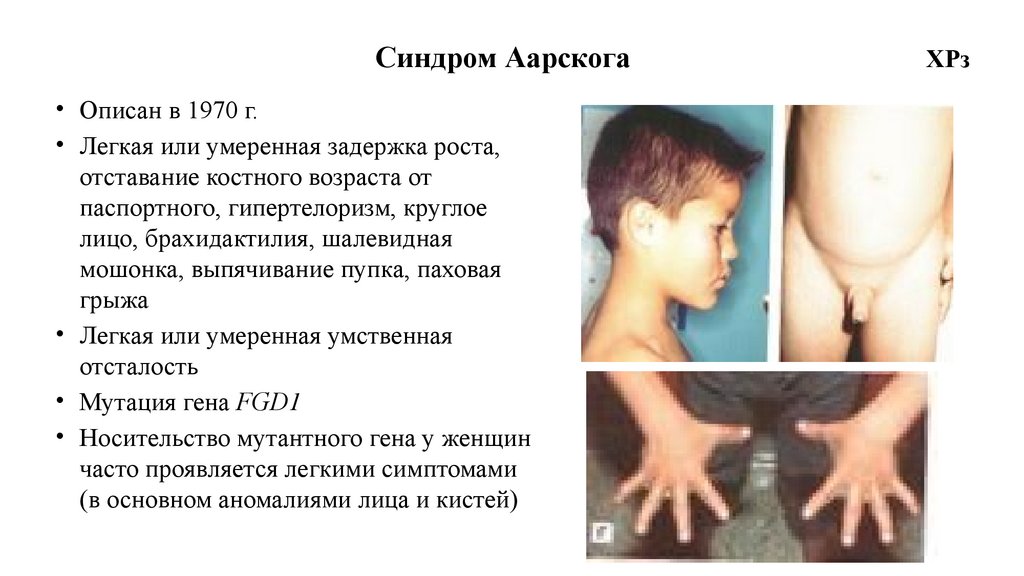
Синдром Аарскога• Описан в 1970 г.
• Легкая или умеренная задержка роста,
отставание костного возраста от
паспортного, гипертелоризм, круглое
лицо, брахидактилия, шалевидная
мошонка, выпячивание пупка, паховая
грыжа
• Легкая или умеренная умственная
отсталость
• Мутация гена FGD1
• Носительство мутантного гена у женщин
часто проявляется легкими симптомами
(в основном аномалиями лица и кистей)
ХРз
53.
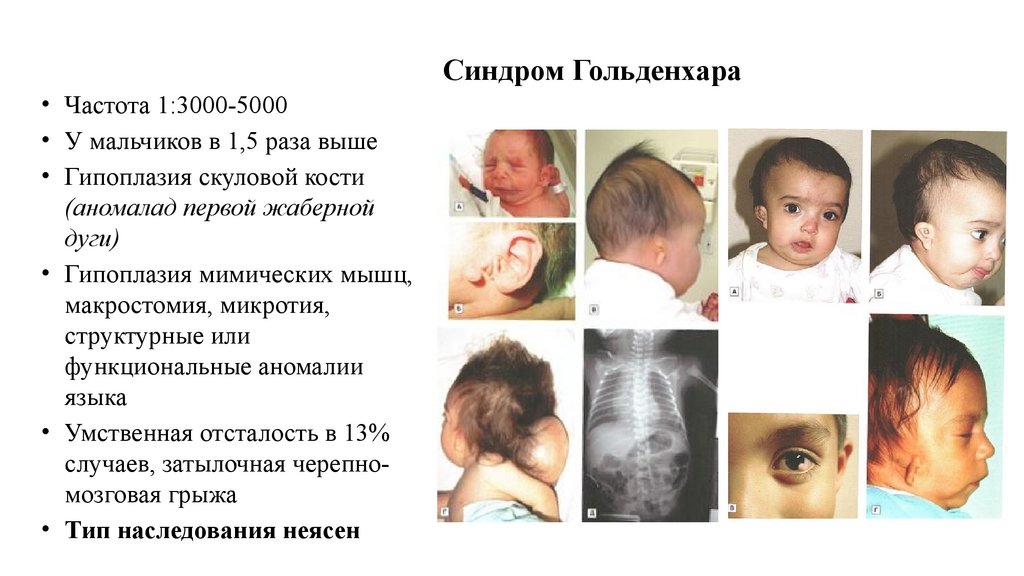
• Частота 1:3000-5000• У мальчиков в 1,5 раза выше
• Гипоплазия скуловой кости
(аномалад первой жаберной
дуги)
• Гипоплазия мимических мышц,
макростомия, микротия,
структурные или
функциональные аномалии
языка
• Умственная отсталость в 13%
случаев, затылочная черепномозговая грыжа
• Тип наследования неясен
Синдром Гольденхара
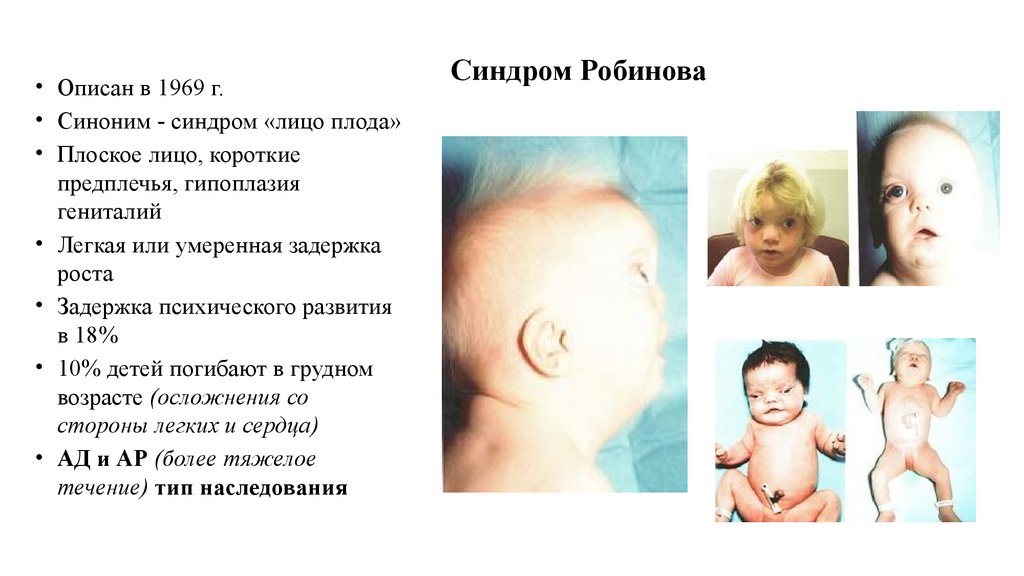
54.
• Описан в 1969 г.• Синоним - синдром «лицо плода»
• Плоское лицо, короткие
предплечья, гипоплазия
гениталий
• Легкая или умеренная задержка
роста
• Задержка психического развития
в 18%
• 10% детей погибают в грудном
возрасте (осложнения со
стороны легких и сердца)
• АД и АР (более тяжелое
течение) тип наследования
Синдром Робинова
55.
«Никто из нас генетически несовершенен, и по мере
прогресса генетических
исследований каждый
оказывается
предрасположенным к тому
или иному заболеванию»
Фрэнсиз Коллинз, директор
программы «Геном человека»
56.
Предиктивная медицина• Цель – раннее выявление наследственной
предрасположенности к хроническим заболеваниям,
их предупреждение на досимптоматическом этапе,
разработка индивидуальных методов профилактики и
лечения
• Генетическое тестирование (генетический паспорт) в
досимтоматический период позволяет выявить геныкандидаты и наметить пути мониторинга и ранней
профилактики МФЗ
57.
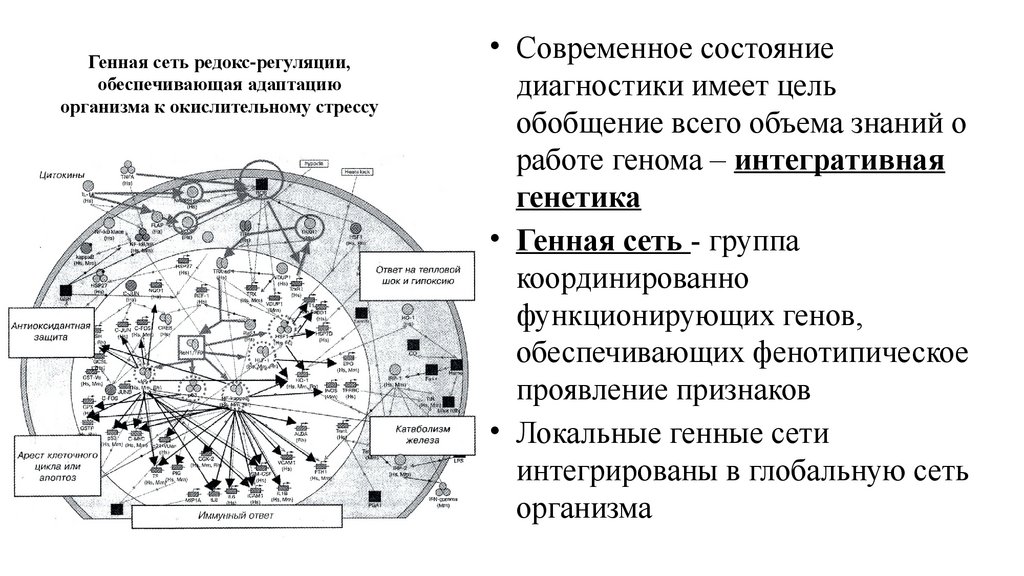
Генная сеть редокс-регуляции,обеспечивающая адаптацию
организма к окислительному стрессу
• Современное состояние
диагностики имеет цель
обобщение всего объема знаний о
работе генома – интегративная
генетика
• Генная сеть - группа
координированно
функционирующих генов,
обеспечивающих фенотипическое
проявление признаков
• Локальные генные сети
интегрированы в глобальную сеть
организма
58.
Иерархия локальных генных сетей59.
• Качество работы локальных генных сетей зависит от генгенных (внутри сети), интегральных (между геннымисетями) и адаптивных (между генными сетями и факторами
внешней среды) взаимодействий
• Гены, образующие структурный остов каждой генной сети,
полиморфны. В определенных условиях этот полиморфизм
может предрасполагать или препятствовать проявлению
заболевания – гены предрасположенности
• Гены предрасположенности – мутантные гены (аллели),
совместимые с рождением и жизнью и при определенных
условиях способствую развитию заболевания
60.
Гены предрасположенности• Гены внешней среды
• Гены-триггеры (метаболические шунты)
• Гены клеточных рецепторов
• Гены иммунной защиты
• Гены старения
• Гены предрасположенности к МФЗ (более 1500)
61.
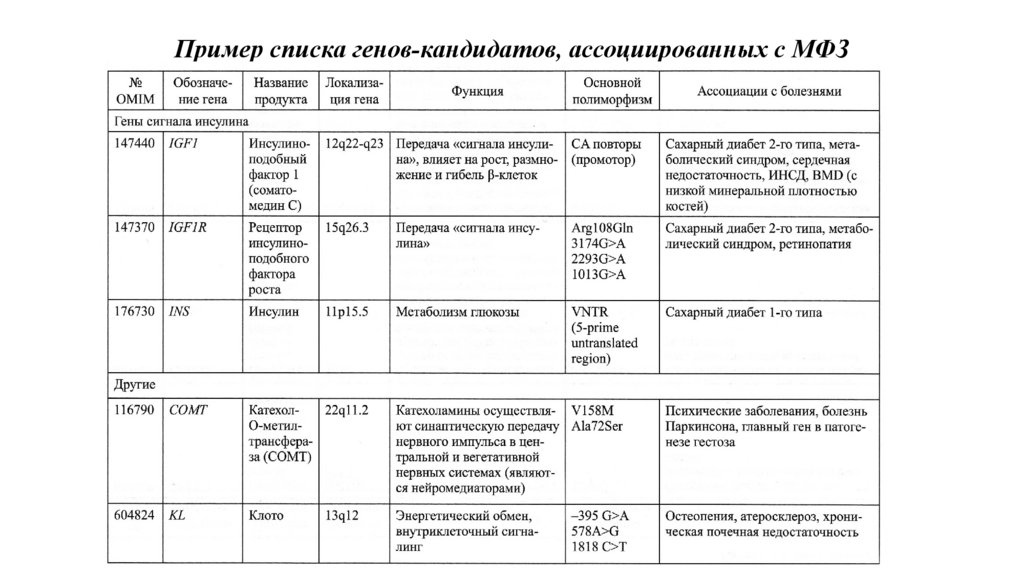
Пример списка генов-кандидатов, ассоциированных с МФЗ62.
63.
64.
65.
Организация помощи и структура медикогенетической консультации• Медико-генетическая служба (МГС) создана в РФ в
соответствие с приказом Министерства
здравоохранения РФ № 316 от 30.12.1993 «О
дальнейшем развитии медико-генетической службы»
• Порядок организации медико-генетической службы
(МГС) изложен в Приложении к приказу «Положение
о медико-генетической службе Минздрава России»
66.
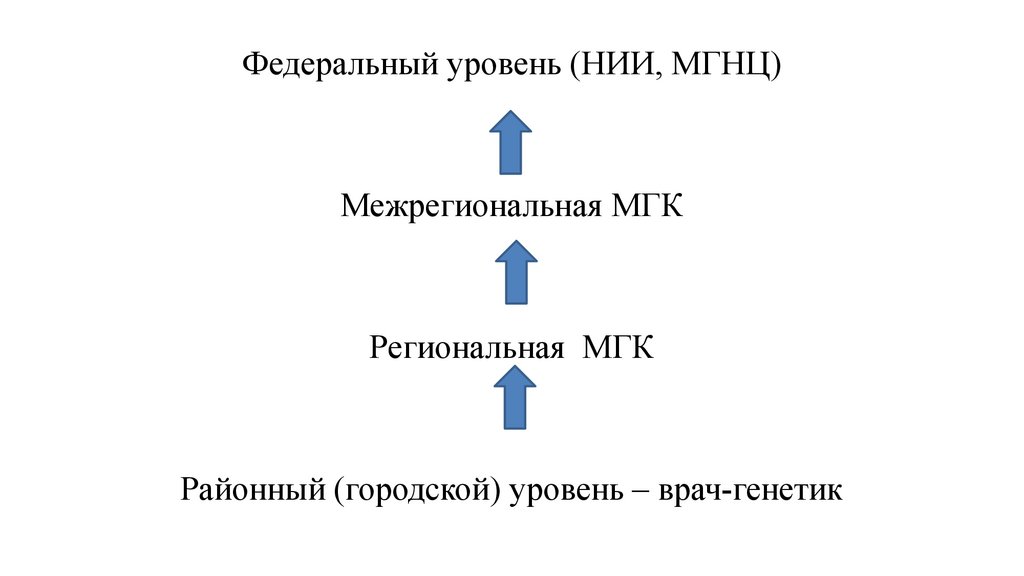
Федеральный уровень (НИИ, МГНЦ)Межрегиональная МГК
Региональная МГК
Районный (городской) уровень – врач-генетик
67.
Наследственные ошибкиметаболизма
и методы их диагностики
Самойлова Людмила Руслановна, врач-лабораторный генетик,
преподаватель кафедры НППиМГ ИФМиБ КФУ
Казань
2023
68.
• Наследственные ошибки метаболизма (НОМ)-обширная группазаболеваний, полиморфных и гетерогенных по генетической
сути
• НОМ в большинстве своем подчиняются менделевским
правилам наследования: значительная часть наследуется по АР и
Х-сцепленному типу, существенно меньшая – АД
• Подавляющее большинство НОМ - результат нарушения работы
ферментов или транспортных белков
• НОМ включают в себя группу митохондриальных болезней и
болезней экспансии
• В группу НОМ объединяются в основном моногенные болезни
69.
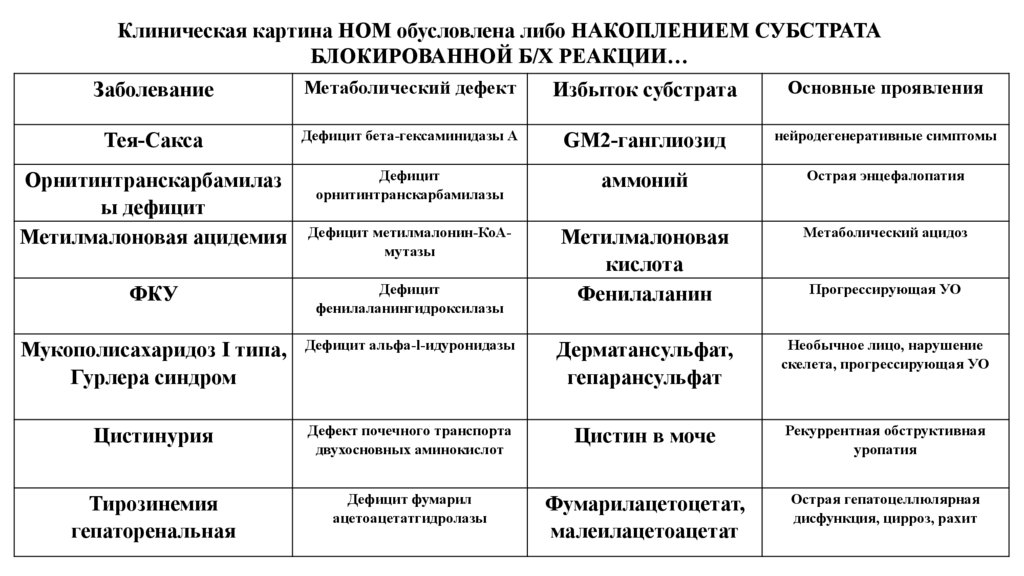
Клиническая картина НОМ обусловлена либо НАКОПЛЕНИЕМ СУБСТРАТАБЛОКИРОВАННОЙ Б/Х РЕАКЦИИ…
Заболевание
Метаболический дефект
Избыток субстрата
Основные проявления
Тея-Сакса
Дефицит бета-гексаминидазы А
GM2-ганглиозид
нейродегенеративные симптомы
Орнитинтранскарбамилаз
ы дефицит
Метилмалоновая ацидемия
Дефицит
орнитинтранскарбамилазы
аммоний
Острая энцефалопатия
Дефицит метилмалонин-КоАмутазы
Метилмалоновая
кислота
Фенилаланин
Метаболический ацидоз
ФКУ
Дефицит
фенилаланингидроксилазы
Прогрессирующая УО
Мукополисахаридоз I типа,
Гурлера синдром
Дефицит альфа-l-идуронидазы
Дерматансульфат,
гепарансульфат
Необычное лицо, нарушение
скелета, прогрессирующая УО
Цистинурия
Дефект почечного транспорта
двухосновных аминокислот
Цистин в моче
Рекуррентная обструктивная
уропатия
Тирозинемия
гепаторенальная
Дефицит фумарил
ацетоацетатгидролазы
Фумарилацетоцетат,
малеилацетоацетат
Острая гепатоцеллюлярная
дисфункция, цирроз, рахит
70.
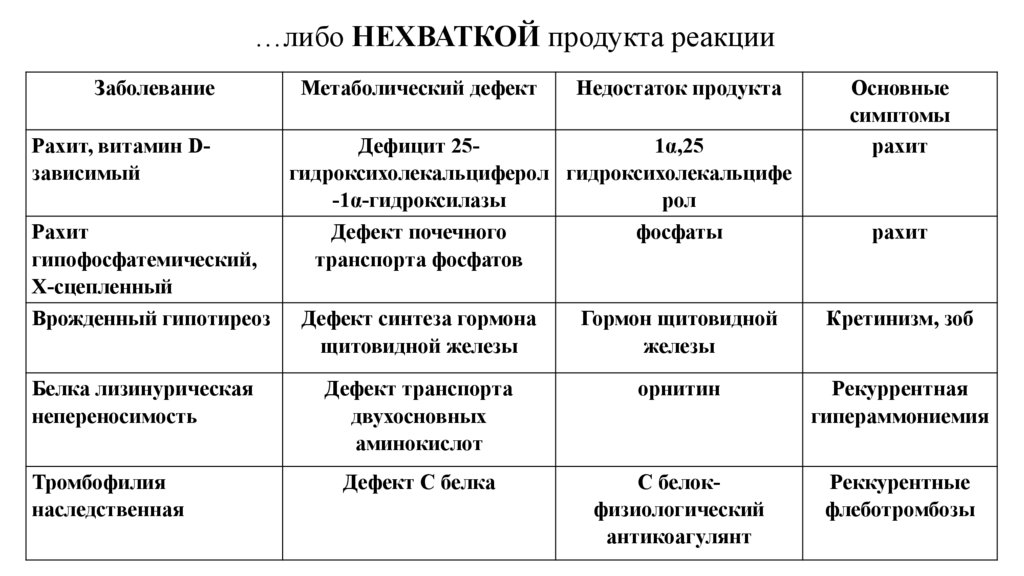
…либо НЕХВАТКОЙ продукта реакцииЗаболевание
Рахит, витамин Dзависимый
Рахит
гипофосфатемический,
Х-сцепленный
Метаболический дефект
Недостаток продукта
Дефицит 251α,25
гидроксихолекальциферол гидроксихолекальцифе
-1α-гидроксилазы
рол
Дефект почечного
фосфаты
транспорта фосфатов
Основные
симптомы
рахит
рахит
Врожденный гипотиреоз
Дефект синтеза гормона
щитовидной железы
Гормон щитовидной
железы
Кретинизм, зоб
Белка лизинурическая
непереносимость
Дефект транспорта
двухосновных
аминокислот
орнитин
Рекуррентная
гипераммониемия
Дефект С белка
С белокфизиологический
антикоагулянт
Реккурентные
флеботромбозы
Тромбофилия
наследственная
71.
• На практике нередко встречаются состояния, в основекоторых НЕ лежат нарушения обмена, но симптоматически
напоминают НОМ
Диагностические ошибки могут могут быть обусловлены:
Наличием интеркуррентных заболеваний
Генетической гетерогенностью НОМ
ВПР
• Начальные проявления НОМ нередко ошибочно
расцениваются как инфекционное заболевание,
интоксикация неясной природы и пр.
72.
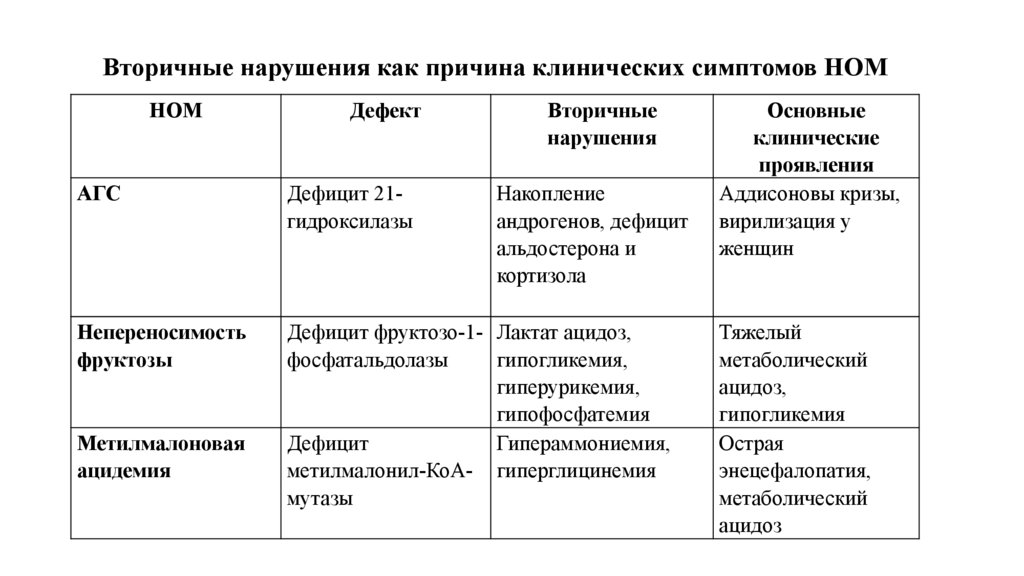
Вторичные нарушения как причина клинических симптомов НОМНОМ
Дефект
Вторичные
нарушения
АГС
Дефицит 21гидроксилазы
Непереносимость
фруктозы
Дефицит фруктозо-1- Лактат ацидоз,
фосфатальдолазы
гипогликемия,
гиперурикемия,
гипофосфатемия
Дефицит
Гипераммониемия,
метилмалонил-КоА- гиперглицинемия
мутазы
Метилмалоновая
ацидемия
Накопление
андрогенов, дефицит
альдостерона и
кортизола
Основные
клинические
проявления
Аддисоновы кризы,
вирилизация у
женщин
Тяжелый
метаболический
ацидоз,
гипогликемия
Острая
энецефалопатия,
метаболический
ацидоз
73.
Генетическая гетерогенность НОМ являетсяпричиной диагностических ошибок, если не
используются адекватные методы
параклинического исследования пробанда
Например, диетоустойчивые формы ФКУ
становятся курабельными после внедрения методов
исследования неоптерина и биоптерина
74.
Ряд синдромов ВПР обоснованно рассматриваютсякак проявления моногенных НОМ
Микроцефалия, УО
Пороки сердца, лёгких, почек,
пищеварительного тракта и половых органов.
Пониженный мышечный тонус
Задержка в развитии, нарушения поведения и
способности к обучению
Птоз (опущение) верхнего века, катаракта
Нарушение слуха
Нарушения иммунной системы
Сколиоз, остеопороз
Синдактилия (сращение) второго и третьего
пальцев стопы, полидактилия
Смита-Лемли-Опитца
дефицит 7-дегидрохолестерол редуктазы
75.
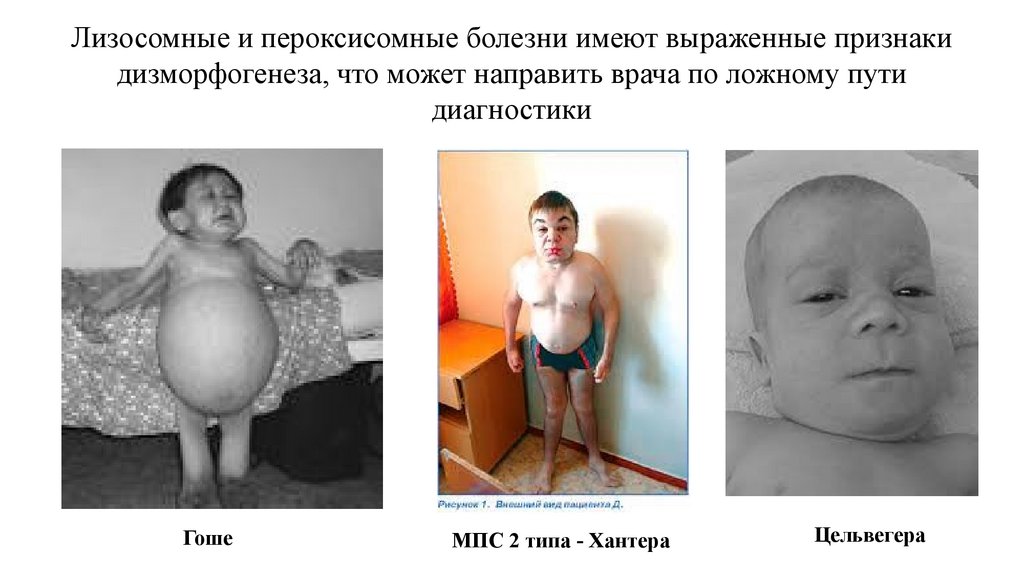
Лизосомные и пероксисомные болезни имеют выраженные признакидизморфогенеза, что может направить врача по ложному пути
диагностики
Гоше
МПС 2 типа - Хантера
Цельвегера
76.
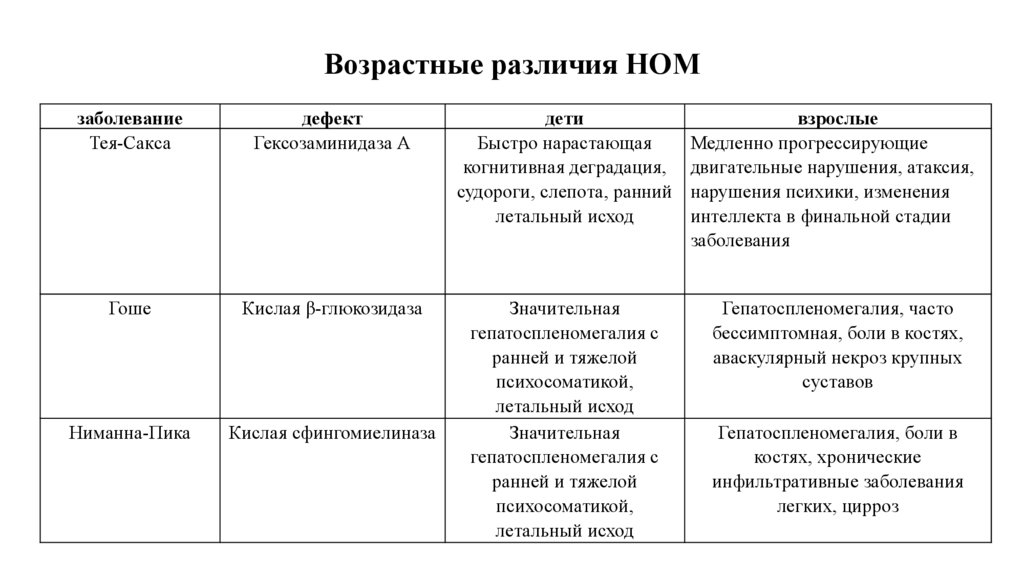
Возрастные различия НОМзаболевание
Тея-Сакса
дефект
Гексозаминидаза А
дети
Быстро нарастающая
когнитивная деградация,
судороги, слепота, ранний
летальный исход
взрослые
Медленно прогрессирующие
двигательные нарушения, атаксия,
нарушения психики, изменения
интеллекта в финальной стадии
заболевания
Гоше
Кислая β-глюкозидаза
Гепатоспленомегалия, часто
бессимптомная, боли в костях,
аваскулярный некроз крупных
суставов
Ниманна-Пика
Кислая сфингомиелиназа
Значительная
гепатоспленомегалия с
ранней и тяжелой
психосоматикой,
летальный исход
Значительная
гепатоспленомегалия с
ранней и тяжелой
психосоматикой,
летальный исход
Гепатоспленомегалия, боли в
костях, хронические
инфильтративные заболевания
легких, цирроз
77.
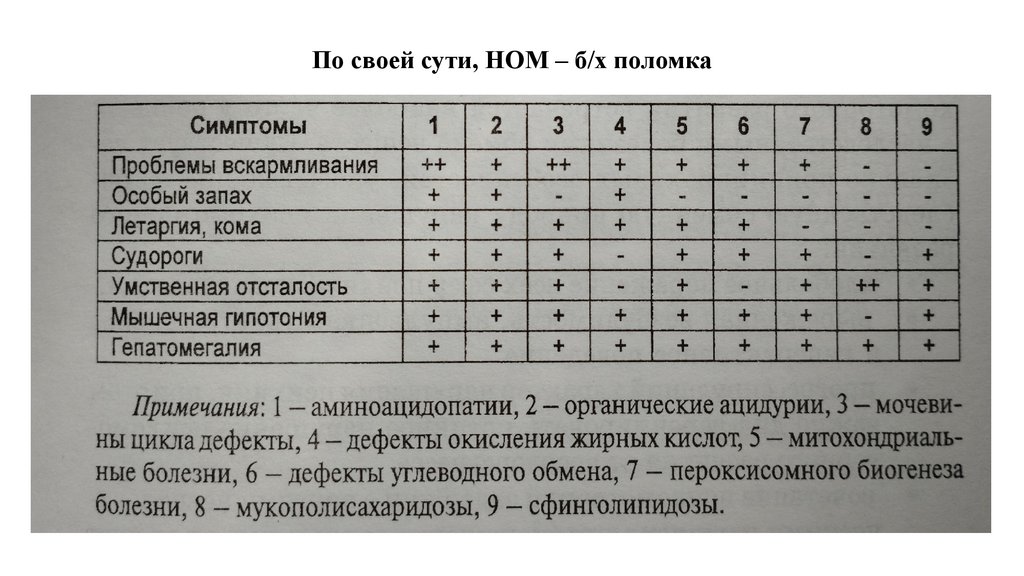
По своей сути, НОМ – б/х поломка78.
НОМ в соответствии с фенотипическими проявлениямиС признаками поражения нервной системы и психики
С признаками поражения функции печени
С признаками поражения сердца
С признаками дизморфогенеза
С признаками метаболического ацидоза
НОМ как острая угрожающая жизни патология
новорожденного
79.
Принципиальные особенности, указывающие на наследственнуюобменную природу нарушения психомоторного развития:
глобальное поражение всех сфер психического развития
выраженная возбудимость, импульсивность, гиперактивное
поведение
прогрессирующий характер нарушения психики
Сочетание психомоторной задержки с другими
неврологическими изменениями (нарушение тонуса, потеря
зрения и слуха, судороги, симптомы поражения
экстрапирамидальной системы и пр.)
80.
НОМ как моногенные болезниТипы мутаций, меняющих трансляцию:
• Нонсенс – останавливает трансляцию
• Миссенс – изменение в кодоне замена аминокислоты
• Регуляторные – нарушения в промоторе
• Сплайсинг – на стыке экзон-интрон, нарушение сплайсинга
• Фреймшифт нарушение структуры полипептида
• Динамические увеличение количества триплетных
повторов в промоторной или кодирующей области гена
81.
Лечение моногенных болезней (патогенетическое)• Восполнение дефицита фермента (гемофилия, наследственный
панкреатит,болезнь Фабри, болезнь Гоше)
• Восполнение дефицита кофактора - витамины и коэнзимы
(пиридоксинзависимые судороги, атаксия с дефицитом вит.Е)
• Восполнение дефицита конечного продукта ( АГС, ВГ, гипофизарный нанизм)
• Ограничение субстрата в пище (ФКУ, тирозинемия, галактоземия)
• Коррекция выведения продукта (Вильсона-Коновалова)
• Ограничение применения лекарственной терапии (дефицит глюкозо-6фосфатдегидрогеназы)
• Замещение поврежденных тканей (поликистоз почек)
• Удаление поврежденных тканей (семейный аденоматозный полипоз,
сфероцитарная анемия, нейрофиброматоз)
82.
муковисцидоз• Описан в 1938 г. Андерсоном
• Частота в западной Европе 1:2000, в России 1: 4900. В странах
Азии и Африки почти не встречается
• Тип наследования АР
• Ген CFTR картирован в 1985 г. расположен в 7q31.2
• Мажорная мутация-delF508 – утрата 3 нуклеотидов в 10м
экзоне гена, сопровождается утратой фенилаланина в 508
положении
• В настоящее время картировано более 2000 миссенс-мутаций
83.
• delF508 нарушает процессинг белка недостигает апикальной мембраны
хлорный канал не формируется
тяжелое течение
• R117H не затрагивает процессинг
канал формируется, но работает менее
интенсивно более легкое течение
84.
• В 70-80% молекулярная диагностика успешна• Когда НЕ удается провести идентификацию
мутаций – на сроке 17 нед. беременности
проводится диагностика по анализу активности
ферментов кишечного происхождения (снижены
при болезни)
85.
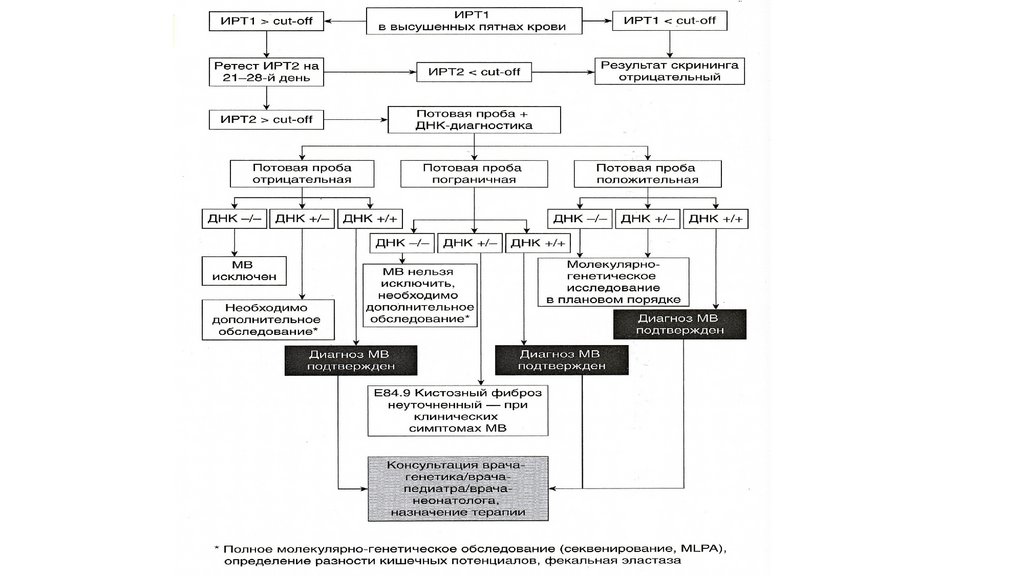
Диагностика МВПри скрининге иммунореактивный трипсин (ИРТ) > 70 нг/мл
Повторное определение ИТР на 4й неделе жизни (не позднее 8й
недели)> 40нг/мл
Потовая проба в Центре муковисцидоза > 40ммоль/л (до 1 года), >
60ммоль/л (старше 1 года)
При пограничном значении потовой пробы – ДНК диагностика на 1030 мутаций CFTR
86.
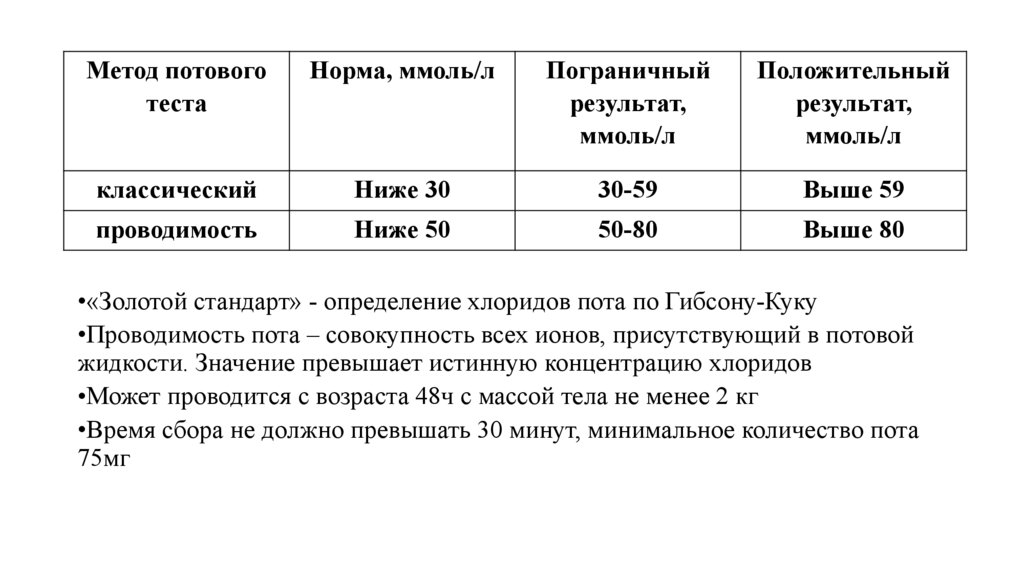
Метод потовоготеста
Норма, ммоль/л
Пограничный
результат,
ммоль/л
Положительный
результат,
ммоль/л
классический
Ниже 30
30-59
Выше 59
проводимость
Ниже 50
50-80
Выше 80
•«Золотой стандарт» - определение хлоридов пота по Гибсону-Куку
•Проводимость пота – совокупность всех ионов, присутствующий в потовой
жидкости. Значение превышает истинную концентрацию хлоридов
•Может проводится с возраста 48ч с массой тела не менее 2 кг
•Время сбора не должно превышать 30 минут, минимальное количество пота
75мг
87.
Причины пограничных результатов ПП• Индивидуальные особенности у людей
без МВ, особенно у взрослых
• Неправильная подготовка к пробе
• Носительство «мягких» мутаций при
МВ
88.
89.
фенилкетонурия• Описана в 1934 г Феллингом
• В нашей стране в 1970 г Блюминой
• Частота 1: 8-10 тыс.
• Блок на этапе превращения Phe в Tyr
• С первых недель жизни – повышенная возбудимость, мышечная
ригидность, судорожный синдром, повторяющаяся рвота
• Дефект пигментации (дефицит меланина)
• Больные-гомозиготы по мутации в гене PAH (12q22-24)
• В России, Польше, Беларуси мажорные мутации R408W
90.
• Лечение диетотерапия• Фенил-Фри, Лофеналак, Тетрафен
• Первые – институт экспериментальной медицины
РАМН
• Предпочтительно придерживаться диеты всю жизнь
• При наступлении беременности –
бесфенилаланиновая диета, иначе – фенилаланиновая
эмбриопатия у плода (микроцефалия, пороками
сердца, пренатальной гипоплазией и УО)
91.
92.
галактоземия• Описана в 1908 г. Ресом
• Частота 1:30 тыс в Европе, в России 1:32 800 новорожденных
• Мутации в гене GALT (9q13)
• Недостаток/отсутствие галакотозо-1-фосфатуридил трансферазы
• Токсическое влияние на ткани мозга, печени, почек, кишечника
ингибирование бактерицидной активности лейкоцитов
• При очень высоком накоплении галактозы образование
галактиола патология глаз
• Рвота, диарея, желтуха (увеличение пр Bi), гепатомегалия,
диспепсия, асцит, гипотрофия, катаракта, УО, галактозурия,
протеинурия,аминоацидурия
93.
94.
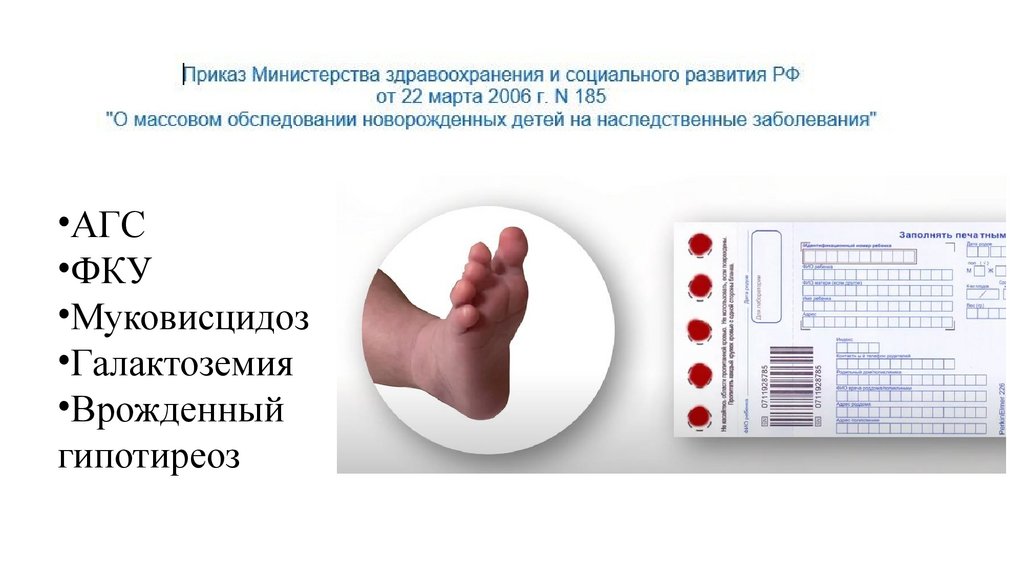
•АГС•ФКУ
•Муковисцидоз
•Галактоземия
•Врожденный
гипотиреоз
95.
Приказ МЗ РФ от 21.04.2022 №274н «Об утверждении Порядка оказаниямедицинской помощи пациентам с врожденными и (или) наследственными
заболеванями» (вступил в силу 1.01.2023)
• 2 вида скрининга (неонатальный 5 заболеваний и
расширенный неонатальный 36 заболеваний)
• Расширенный выполняется методом тандемной массспектрометрии (29 нозологий группы нарушения обмена) и
ПЦР реал-тайм (СМА и группу первичных иммунодефицитов)
• Взятие крови из пятки в возрасте 24-48ч жизни у
доношенного, на 7е сутки у недоношенного
• На 2 бланка (5 капель для НС на один, 3 капли для РНС на
другой)
96.
• Тест-бланки доставляются в МГК 1 раз в 2 дня• Время проведения скрининга не более 72 ч
• В течение 24ч новорожденный повышенного
риска приглашается для ретеста
• Время проведения ретеста не более 72 ч
• Общее проведение подтверждающих
исследований составляет не более 10 дней
97.
Врожденный гипотиреоз• Частота сильно варьирует от популяции: Перу
1:1638, Европа и Северная Америка 1:4000, Япония
1:7000, Россия 1:3617
• У девочек ВГ возникает в 2 раза чаще, чем у
мальчиков
• Типичная картина возникает у детей первого месяца
жизни в 10-15%
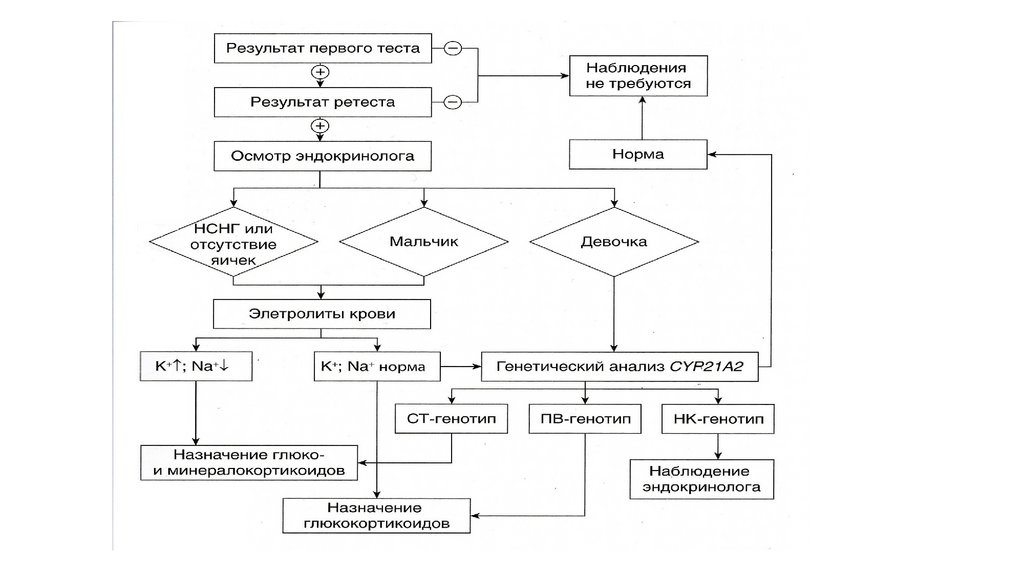
98.
99.
Адрено-генитальный синдром• Врожденная вирилизирующая гиперплазия
коры надпочечников – нарушение биосинтеза
глюко- и минералкортикоидов при повышенной
секреции андрогенов в коре надпочечников
• Не менее 5 генов, мутации которых приводят к
АГС
• Частота 1:12000
100.
101.
гомоцистинурия• Классическая гомоцистинурия – это редкое аутосомно-рецессивное
заболевание, связанное с нарушением обмена метионина.
Причиной заболевания являются мутации гена CBS, кодирующего
цистатион-β-синтазу (CbS), которая участвует в транcсульфурации
гомоцистеина, образующегося из метионина
• Проявление - эктопия хрусталиков и/или тяжелая миопия,
поражение скелета (марфаноподобная внешность, остеопороз и
деформация костей), нарушение умственного развития, судороги,
психические и поведенческие расстройства, венозные и
артериальные тромбозы.
102.
• Частота в популяции разнообразна: в Катаре1:1800, в США 1:100 000, в Ирландии 1:65000
• Лечение – диетотерапия, содержание
метионина в суточном рационе не должно
превышать 1-15 мг на 1 кг массы тела
• Диагностика – методом тандемной массспектрометрии в рамках расширенного
неонатального скрининга