

















































 Медицина
МедицинаПохожие презентации:










Хромосомные болезни
1.
2.
ХРОМОСОМНЫЕ БОЛЕЗНИ-
Наследственные заболевания обусловленные изменением числа
или структуры хромосом
-
Возникают в результате мутаций в половых клетках одного из
родителей
-
Хромосомными нарушениями обусловлены 50% спонтанных
абортов и 7% всех мертворождений
3.
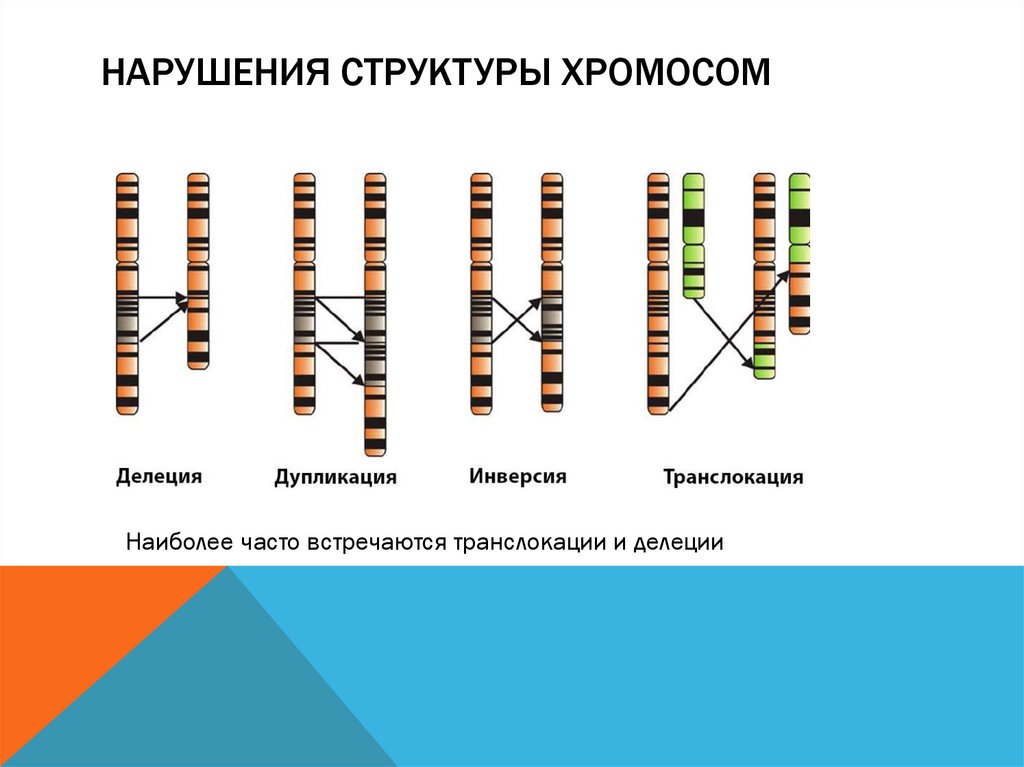
НАРУШЕНИЯ СТРУКТУРЫ ХРОМОСОМНаиболее часто встречаются транслокации и делеции
4.
НАРУШЕНИЯ СТРУКТУРЫ ХРОМОСОМ-
Изохромосомия – хромосомы с повторяющимся генетическим
материалом в обоих плечах
-
Возникновение кольцевых хромомосом – соединение двух
концевых делеций в обоих плечах хромосомы
-
утрата – отрыв части хромосомы
5.
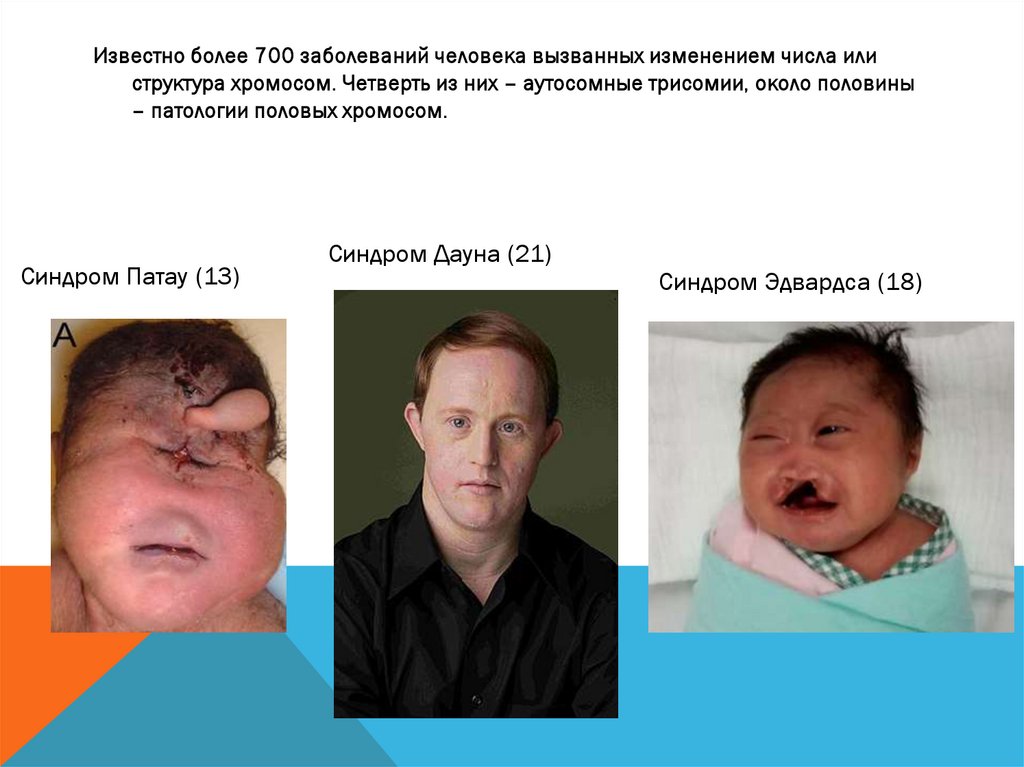
Известно более 700 заболеваний человека вызванных изменением числа илиструктура хромосом. Четверть из них – аутосомные трисомии, около половины
– патологии половых хромосом.
Синдром Патау (13)
Синдром Дауна (21)
Синдром Эдвардса (18)
6.
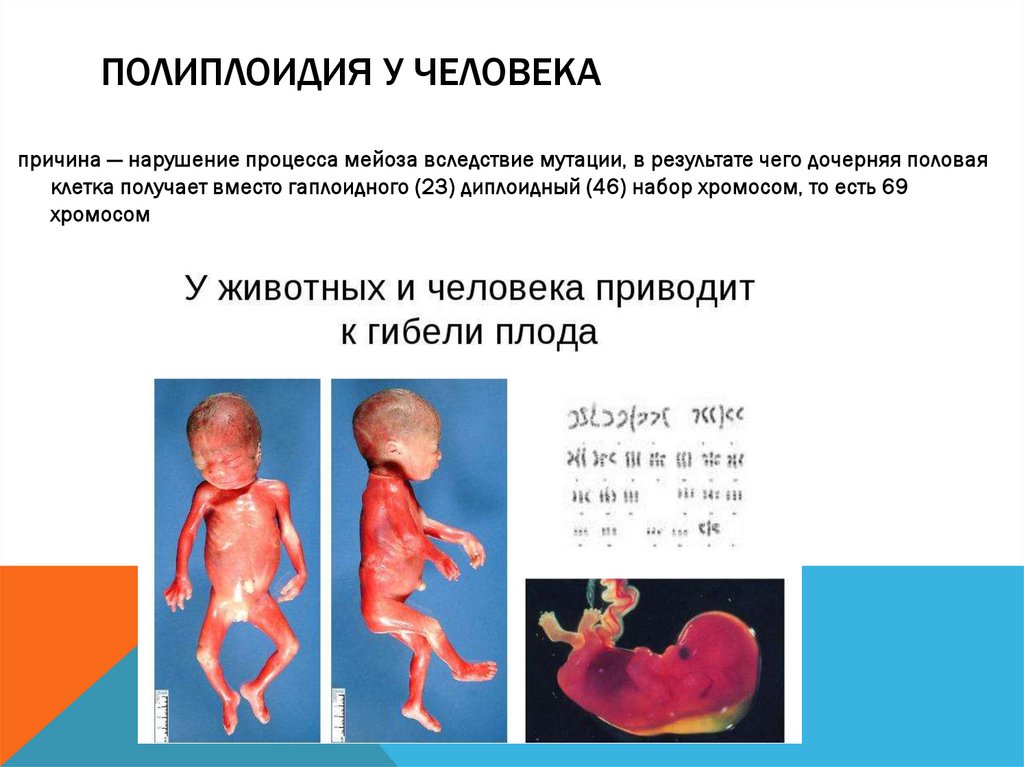
ПОЛИПЛОИДИЯ У ЧЕЛОВЕКАпричина — нарушение процесса мейоза вследствие мутации, в результате чего дочерняя половая
клетка получает вместо гаплоидного (23) диплоидный (46) набор хромосом, то есть 69
хромосом
7.
8.
АНЕУПЛОИДИЯАбсолютное большинство эмбрионов с
анеуплоидией погибает на ранних
сроках беременности.
Чем меньше генов в хромосоме, тем
вероятнее, что плод с
анеуплоидией доживет до
рождения.
Нарушения развития всегда
затрагивают многие органы и
ткани
9.
10.
11.
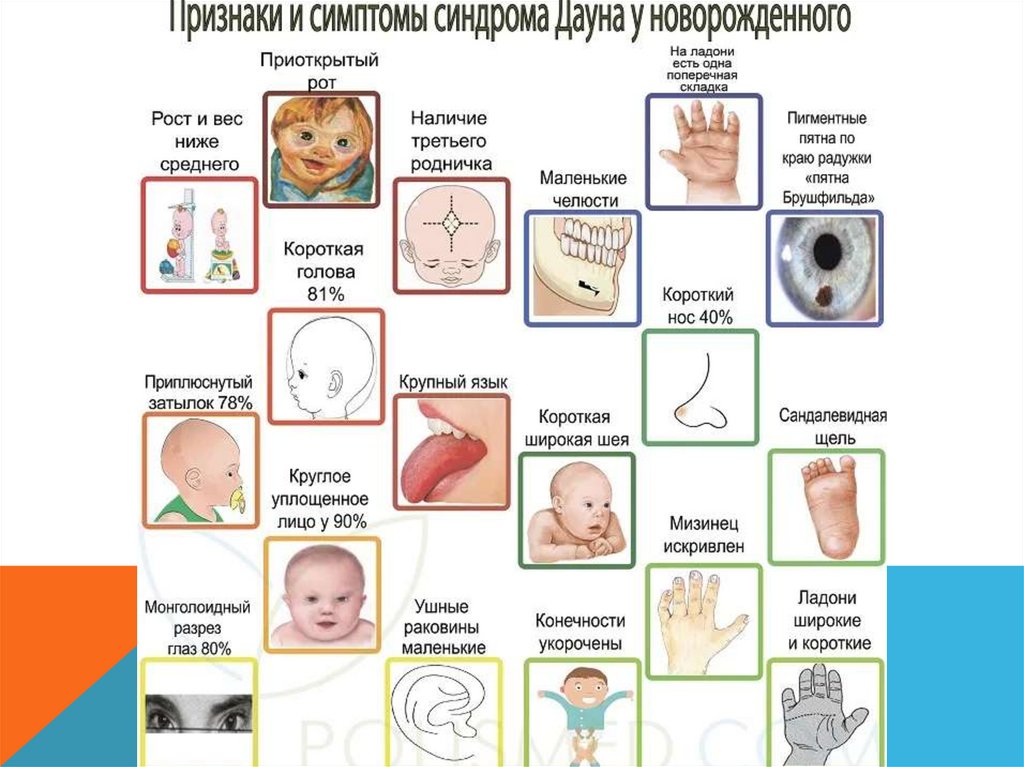
СИНДРОМ ДАУНАОписан в 1866 году
Частота в популяции 1: 625
Среди всех умственно отсталых 10 –
12% дауны
Причина: нерасхождение хромосом в
мейозе (95% - трисомия по 21
хромосоме, 4% - 21 хромосома
транслоцирована на 13 -15
хромосомы, 1% - мозаичные формы)
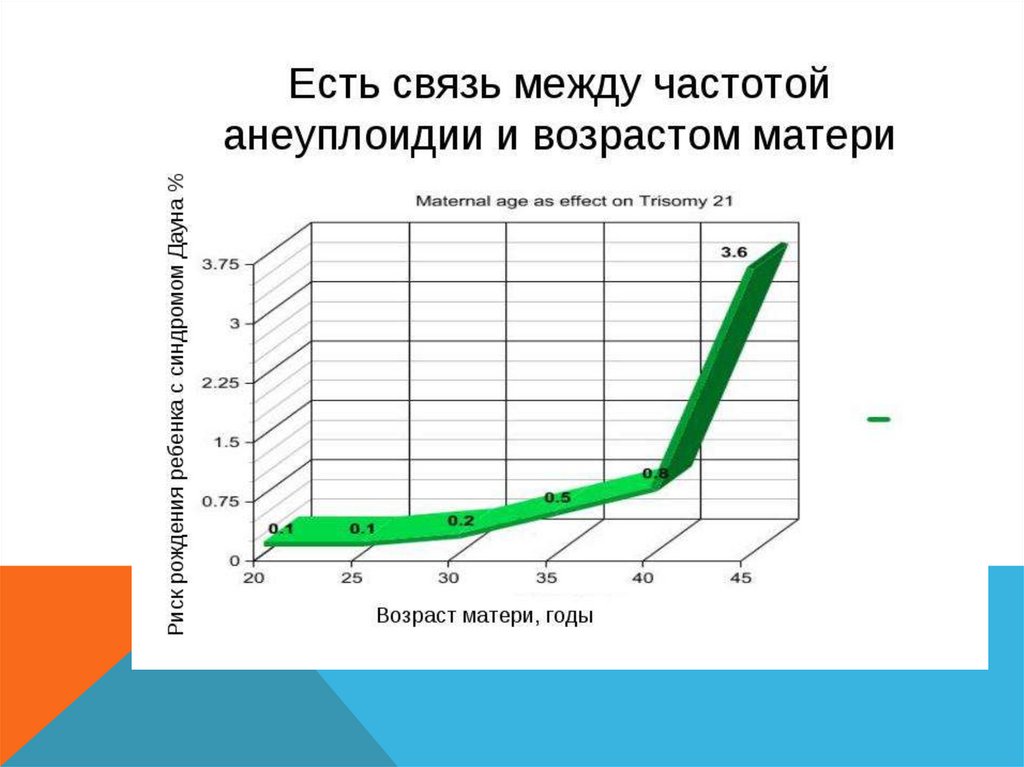
Сильная зависимость вероятности
рождения такого ребенка от
возраста матери (повышена до 19 и
после 35)
12.
13.
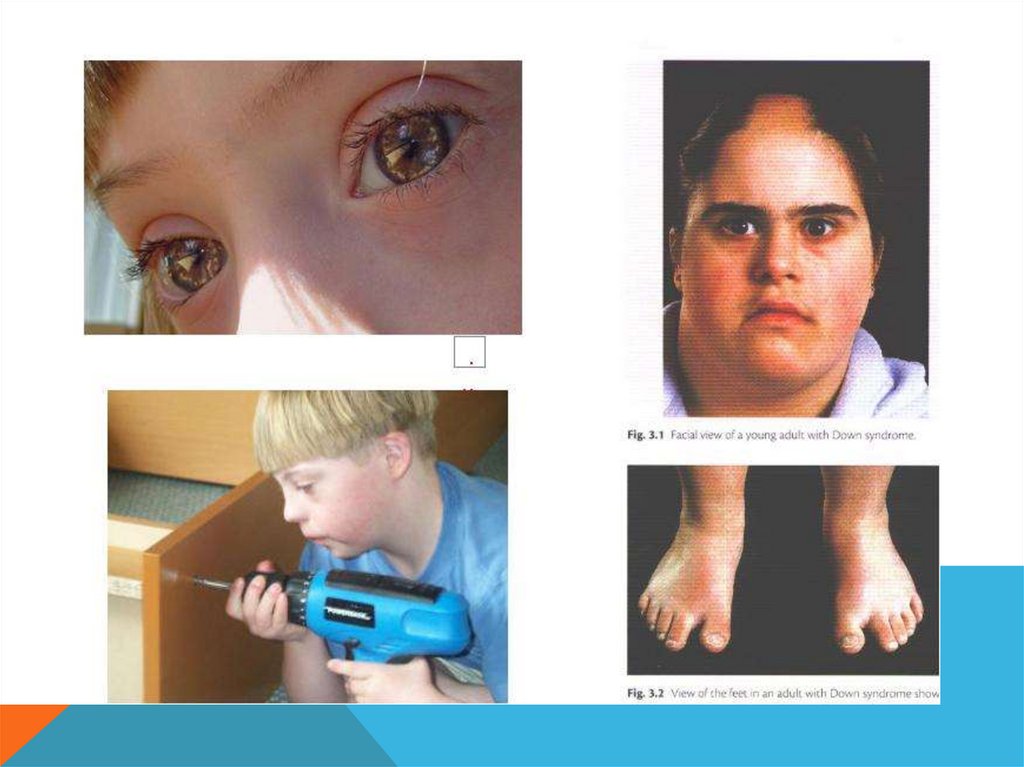
СИНДРОМ ДАУНА-
Дети отстают в умственном развитии
-
очень ласковые и привязываются к
тем, кто их обслуживает
-
Возможна адаптация к жизни ( могут
обслуживать себя, выполнять
неквалифицированную работу)
-
Могут иметь детей
-
Средняя продолжительность жизни 25
– 30 лет
14.
15.
16.
17.
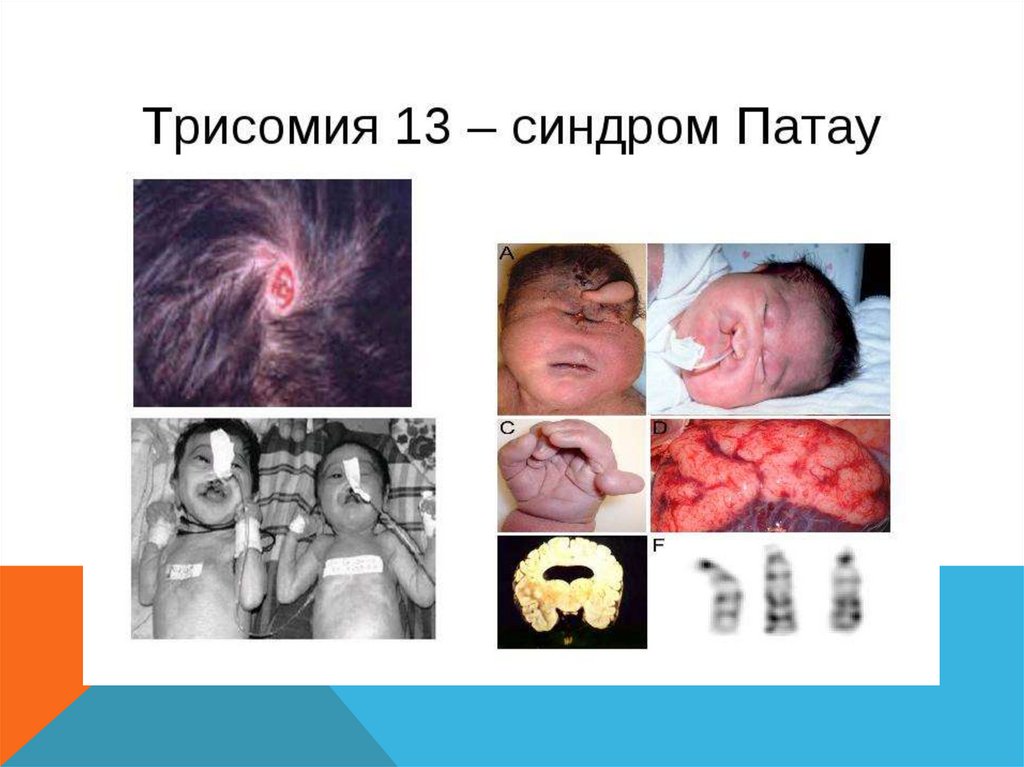
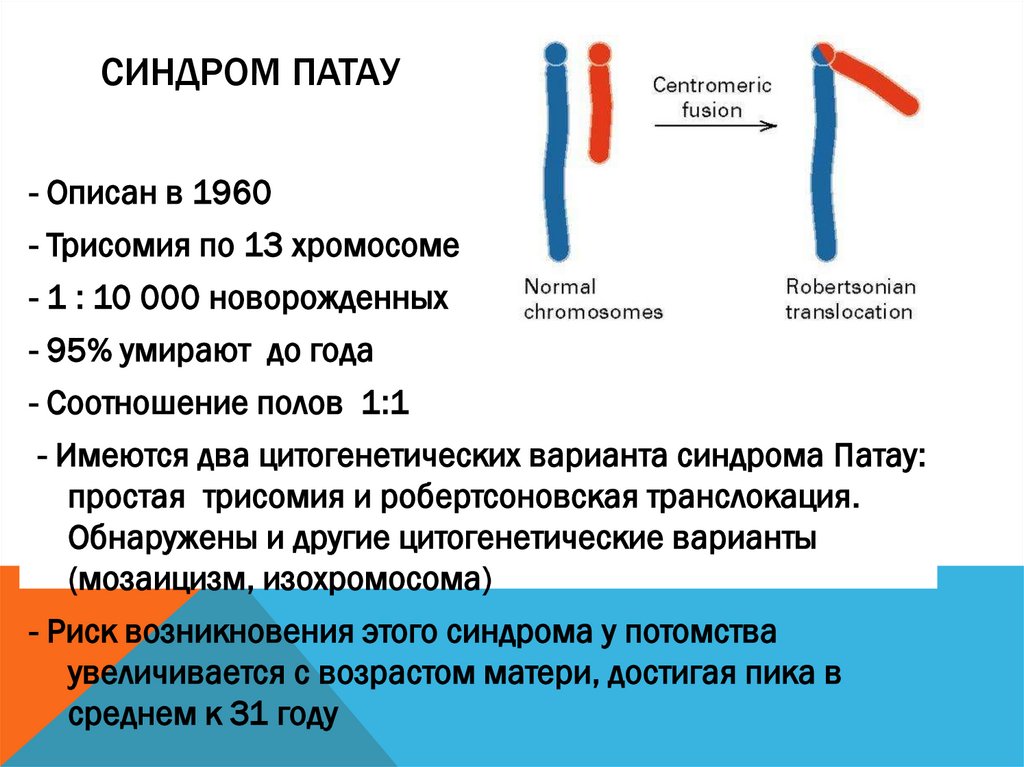
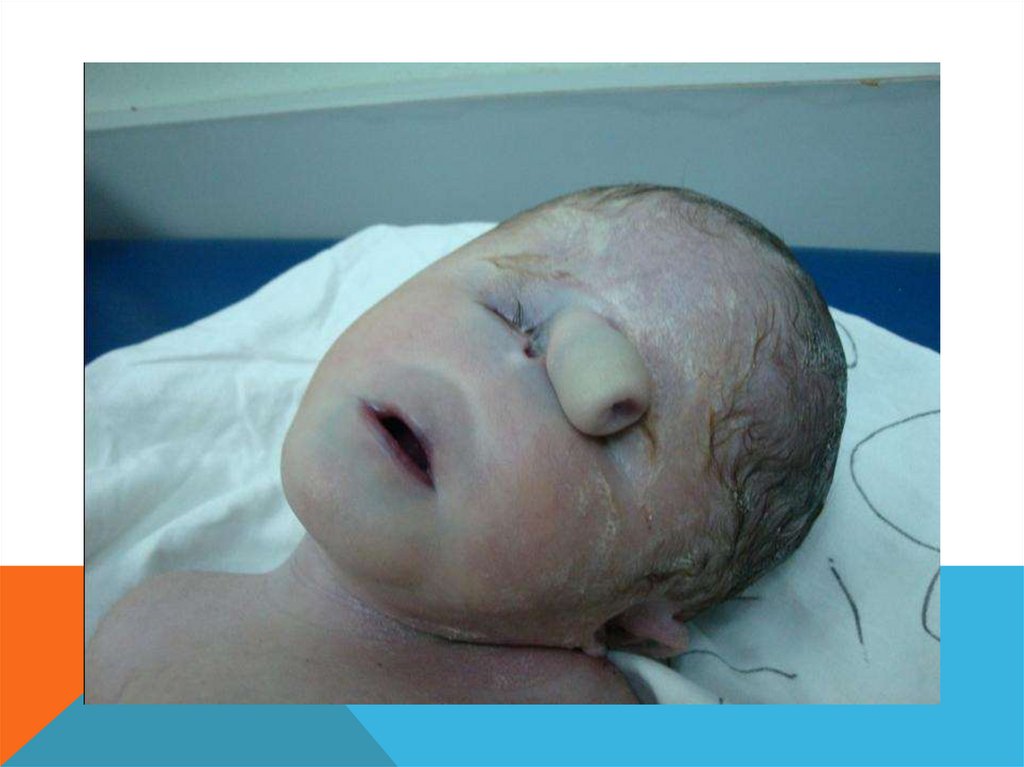
СИНДРОМ ПАТАУ- Описан в 1960
- Трисомия по 13 хромосоме
- 1 : 10 000 новорожденных
- 95% умирают до года
- Соотношение полов 1:1
- Имеются два цитогенетических варианта синдрома Патау:
простая трисомия и робертсоновская транслокация.
Обнаружены и другие цитогенетические варианты
(мозаицизм, изохромосома)
- Риск возникновения этого синдрома у потомства
увеличивается с возрастом матери, достигая пика в
среднем к 31 году
18.
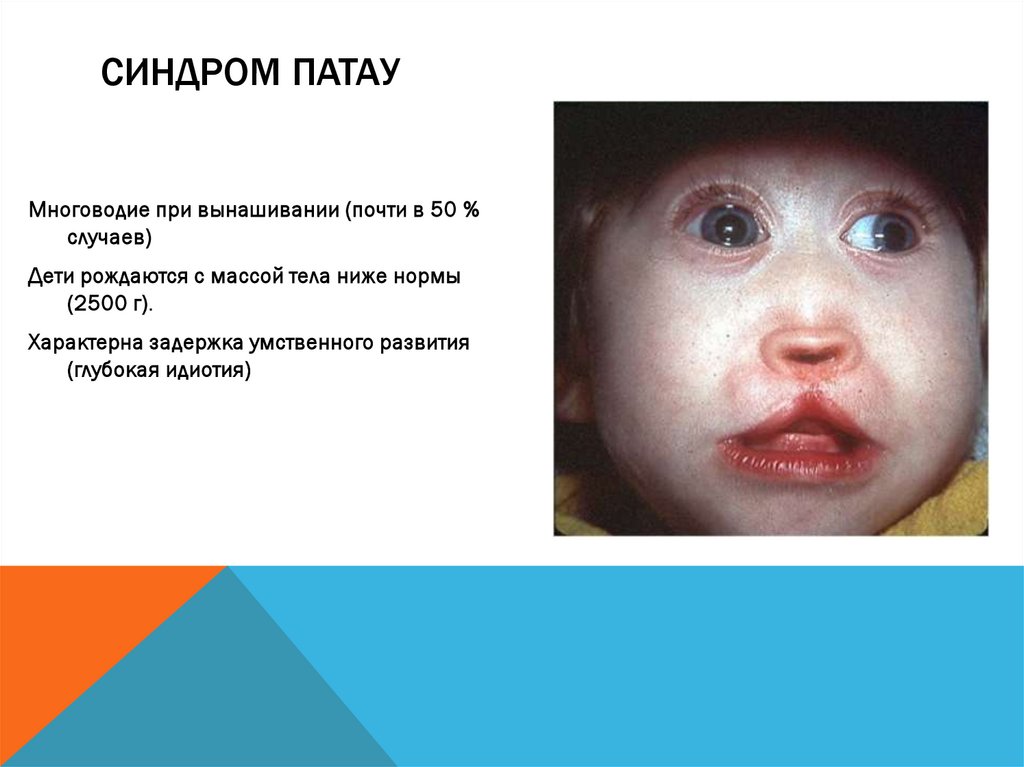
СИНДРОМ ПАТАУМноговодие при вынашивании (почти в 50 %
случаев)
Дети рождаются с массой тела ниже нормы
(2500 г).
Характерна задержка умственного развития
(глубокая идиотия)
19.
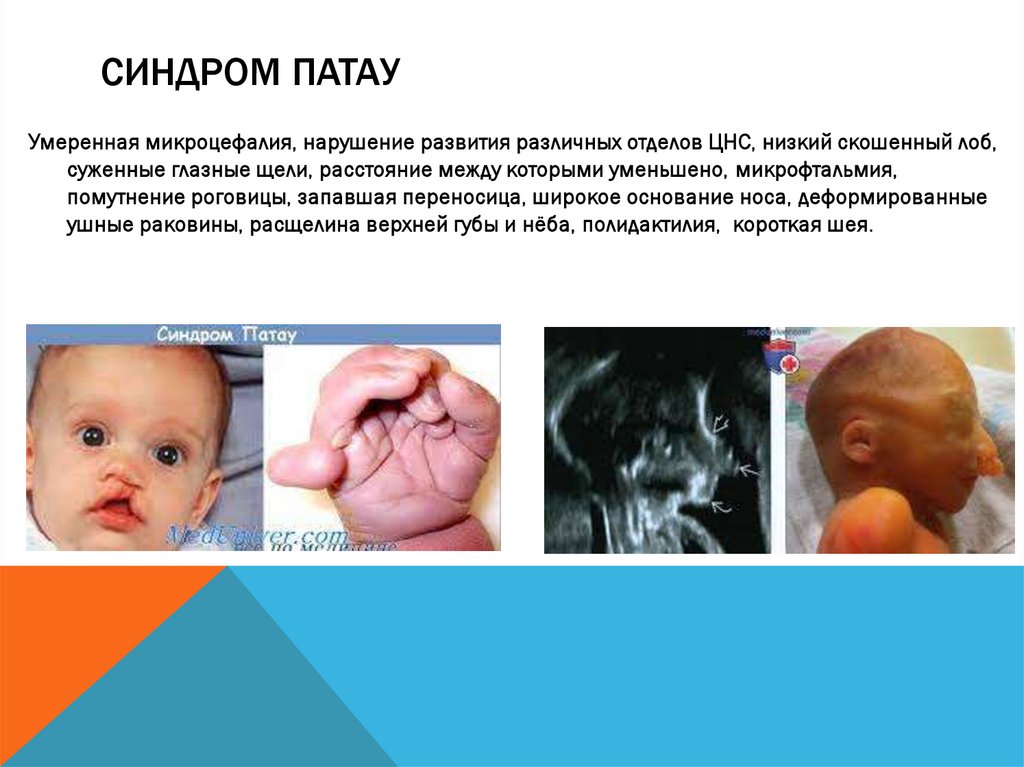
СИНДРОМ ПАТАУУмеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб,
суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия,
помутнение роговицы, запавшая переносица, широкое основание носа, деформированные
ушные раковины, расщелина верхней губы и нёба, полидактилия, короткая шея.
20.
СИНДРОМ ПАТАУУ 80 % новорождённых встречаются пороки развития сердца: дефекты межжелудочковой и
межпредсердной перегородок, транспозиции сосудов.
Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки,
эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и
кисты в корковом слое, выявляются пороки развития половых органов.
21.
СИНДРОМ ПАТАУНа данном этапе развития медицины возможность исправить хромосомные нарушения
отсутствует.
Комплексная работа группы различных специалистов заключается в постоянном контроле за
состоянием здоровья больного и поддержке семьи.
22.
23.
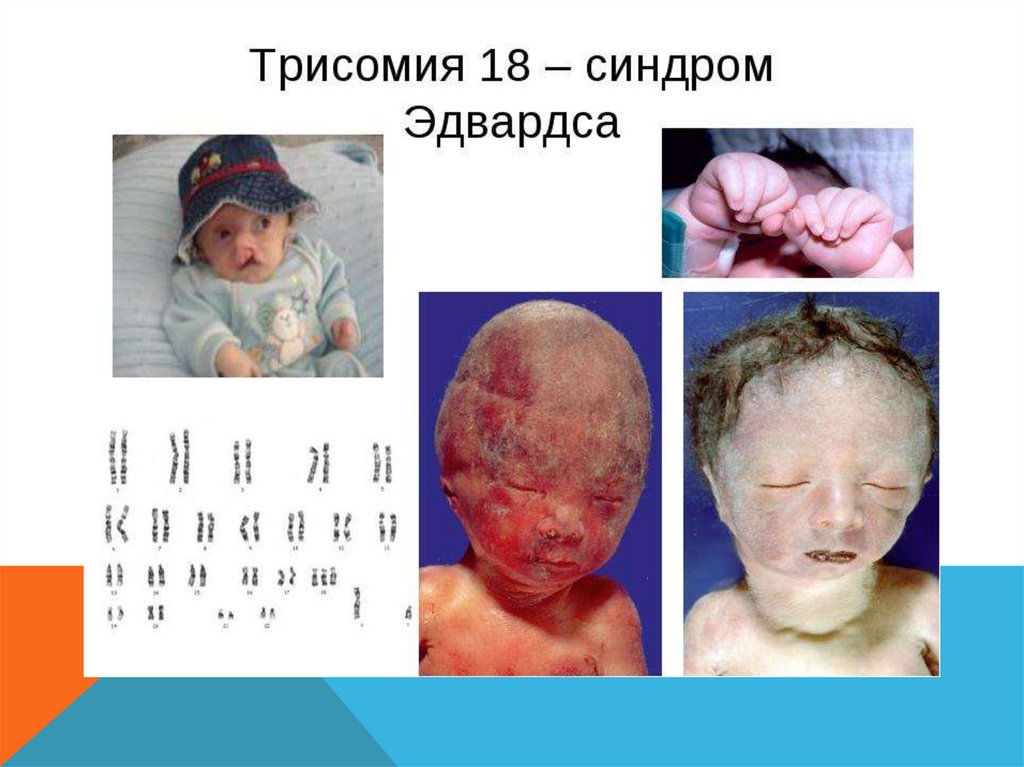
СИНДРОМ ЭДВАРДСА- Описан в 1960 году
- Трисомия по 18 хромосоме
-
1 : 4000 новорожденных
-
Девочки рождаются втрое чаще
- дети чаще рождаются у пожилых матерей, (для женщин старше 45 лет риск
родить больного ребёнка составляет 0,7 %)
- Риск увеличивается если мать больна диабетом
- Выживание после года жизни составляет около 5-10%
24.
25.
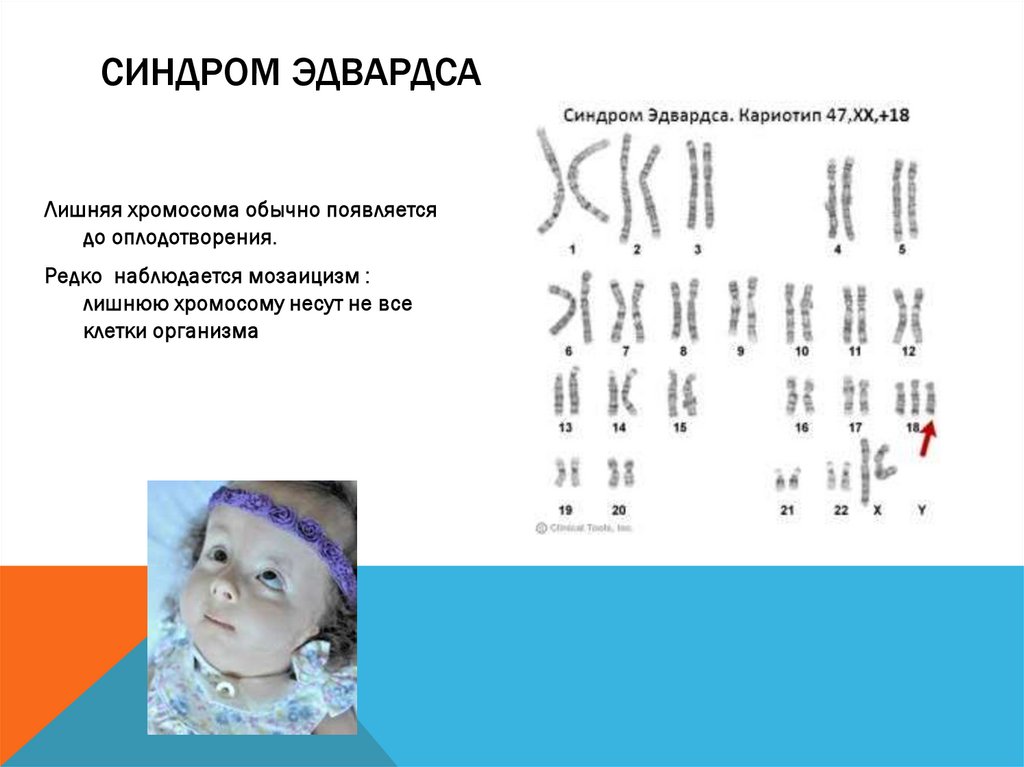
СИНДРОМ ЭДВАРДСАЛишняя хромосома обычно появляется
до оплодотворения.
Редко наблюдается мозаицизм :
лишнюю хромосому несут не все
клетки организма
26.
27.
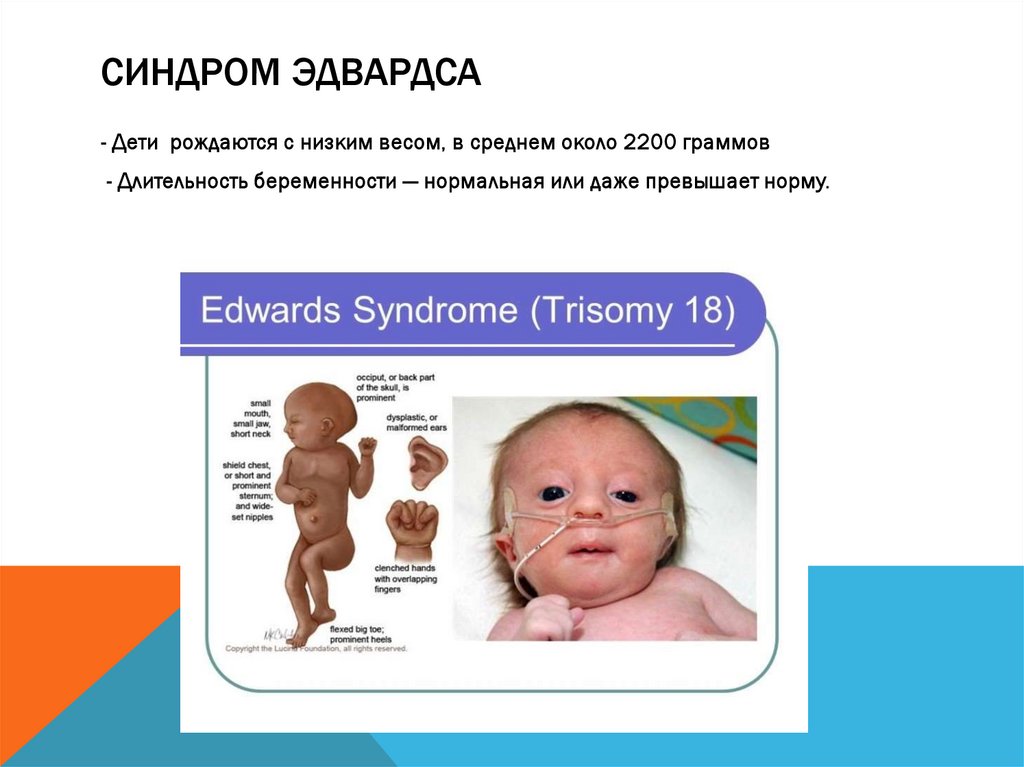
СИНДРОМ ЭДВАРДСА- Дети рождаются с низким весом, в среднем около 2200 граммов
- Длительность беременности — нормальная или даже превышает норму.
28.
СИНДРОМ ЭДВАРДСАНижняя челюсть и ротовое отверстие маленькие.
Глазные щели узкие и короткие.
Ушные раковины деформированы и
расположены низко. Мочка часто отсутствует.
Наружный слуховой проход сужен, иногда
отсутствует.
Грудина короткая, из-за чего межреберные
промежутки уменьшены и грудная клетка
шире и короче нормальной.
29.
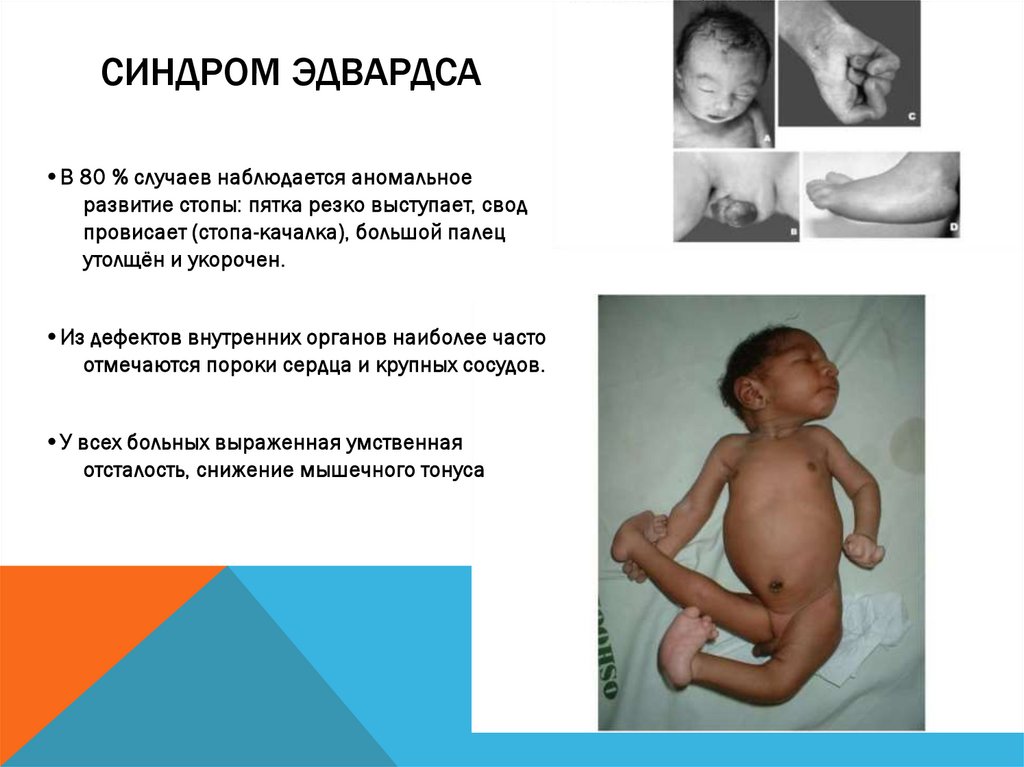
СИНДРОМ ЭДВАРДСА•В 80 % случаев наблюдается аномальное
развитие стопы: пятка резко выступает, свод
провисает (стопа-качалка), большой палец
утолщён и укорочен.
•Из дефектов внутренних органов наиболее часто
отмечаются пороки сердца и крупных сосудов.
•У всех больных выраженная умственная
отсталость, снижение мышечного тонуса
30.
СИНДРОМ ЭДВАРДСА31.
32.
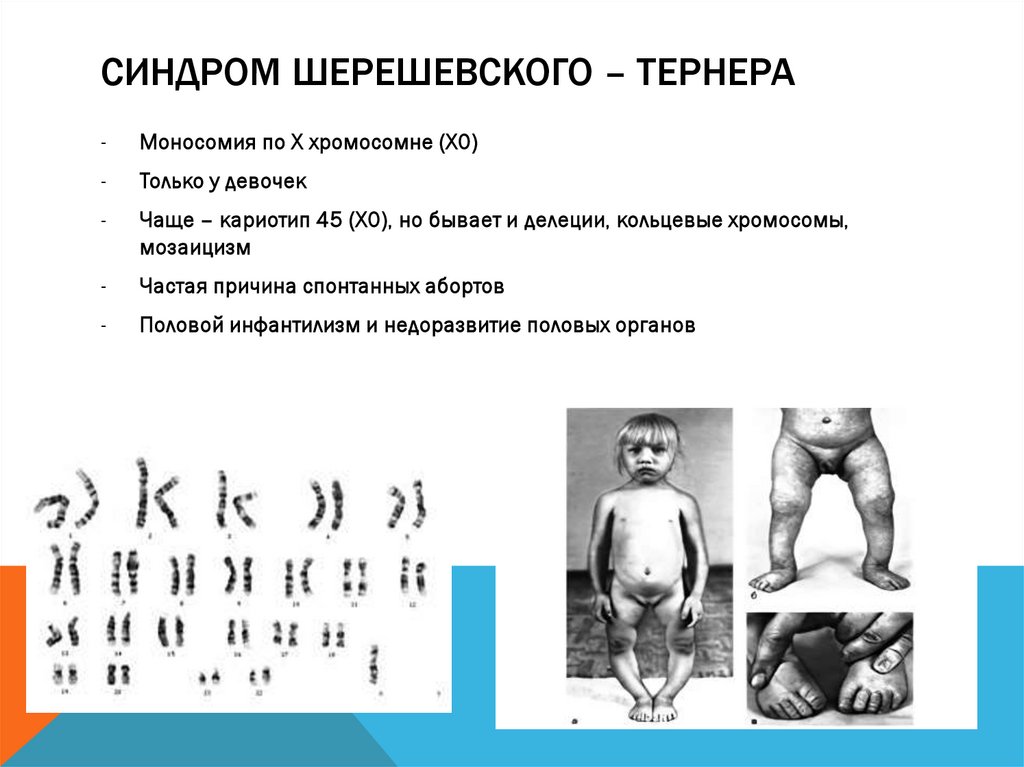
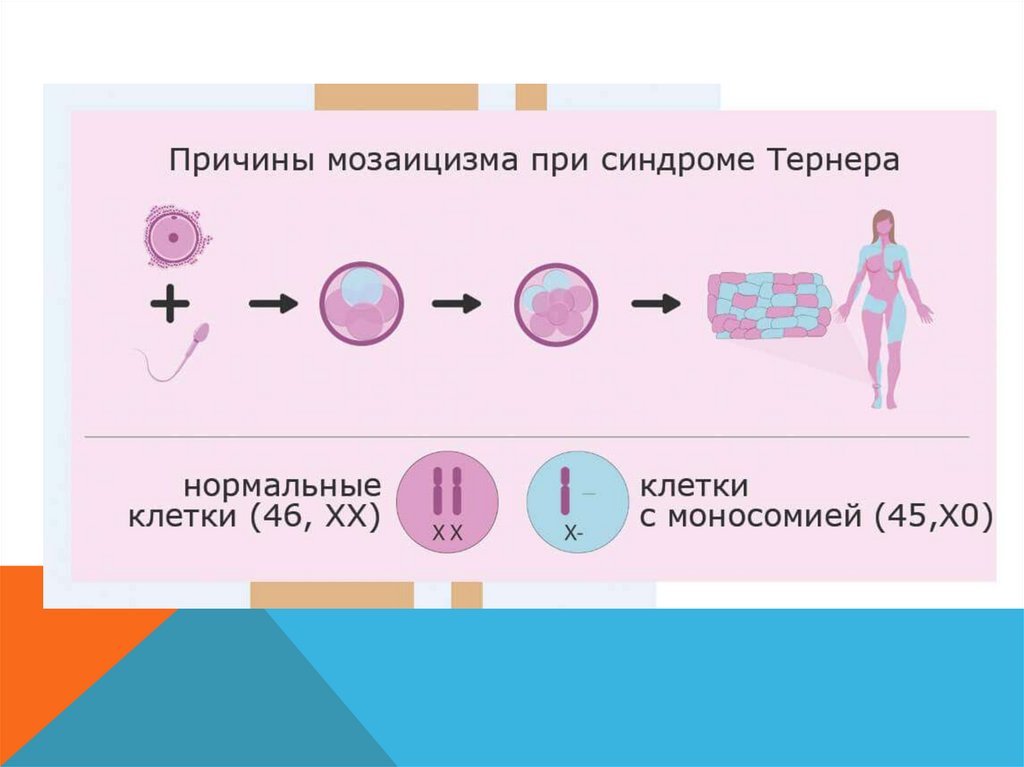
СИНДРОМ ШЕРЕШЕВСКОГО – ТЕРНЕРА-
Моносомия по Х хромосомне (Х0)
-
Только у девочек
-
Чаще – кариотип 45 (Х0), но бывает и делеции, кольцевые хромосомы,
мозаицизм
-
Частая причина спонтанных абортов
-
Половой инфантилизм и недоразвитие половых органов
33.
34.
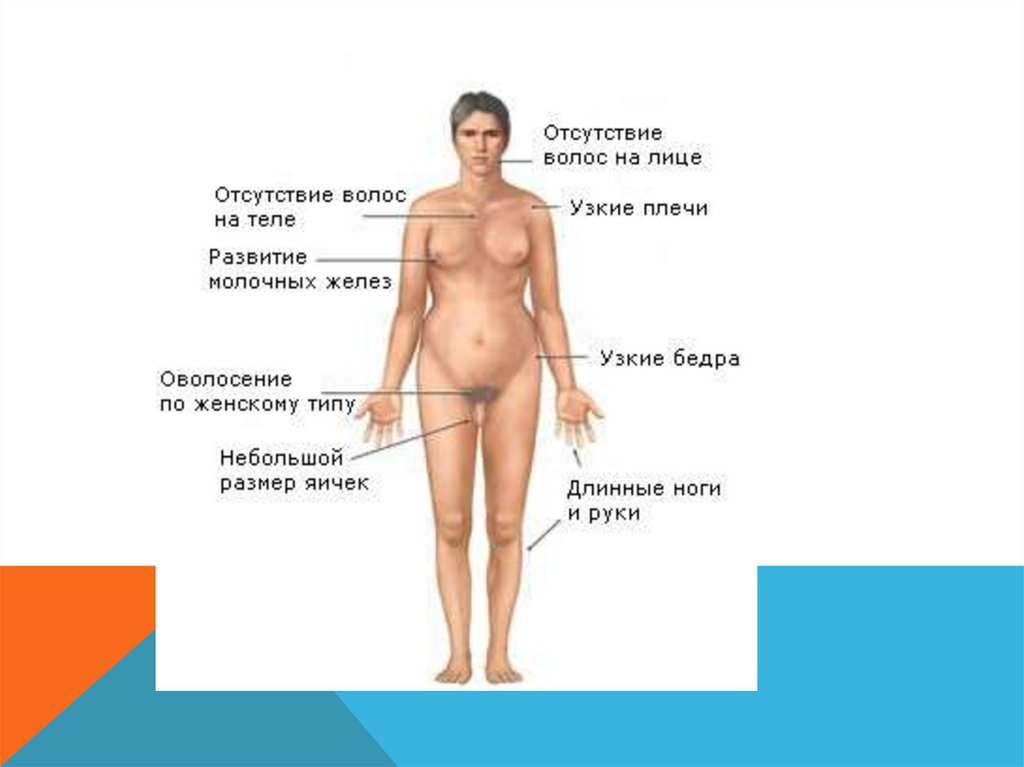
СИНДРОМ ШЕРЕШЕВСКОГО – ТЕРНЕРА-
Характерная крыловидная складка на
шее
-
Множественные малые аномалии
развития (лимфотические отеки
кистей и стоп)
-
Поражения сердца, аномалии почек
*полная или частичная моносомия по Ххромосоме.
35.
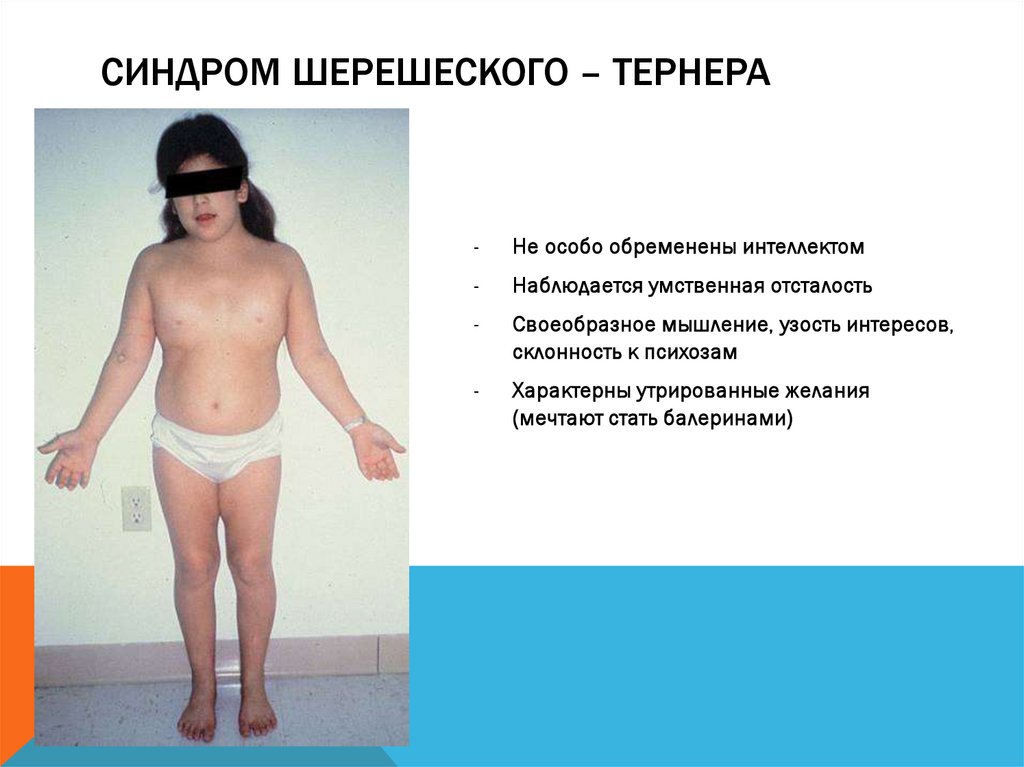
СИНДРОМ ШЕРЕШЕСКОГО – ТЕРНЕРА-
Не особо обременены интеллектом
-
Наблюдается умственная отсталость
-
Своеобразное мышление, узость интересов,
склонность к психозам
-
Характерны утрированные желания
(мечтают стать балеринами)
36.
СИНДРОМ ШЕРЕШЕВСКОГО ТЕРНЕРА (ЛЕЧЕНИЕ)На первом этапе терапия заключается в стимуляции роста тела анаболическими
препаратами.
Главным видом терапии больных является эстрогенизация, которую следует
проводить с 14—16 лет. Лечение приводит к феминизации телосложения,
развитию женских вторичных половых признаков, улучшает трофику половых
путей. Лечение следует проводить в течение всего детородного возраста.
Если с помощью гормональной терапии удаётся вырастить до нормальных
размеров матку, то беременность у таких больных возможна с помощью
ЭКО. (ибо случаи, где сохранились свои яйцеклетки, единичны)
В последнее время для увеличения показателей окончательного роста
проводится терапия соматотропином.
37.
СИНДРОМ ШЕРЕШЕВСКОГО ТЕРНЕРАПрогноз для жизни при синдроме Шерешевского — Тёрнера
благоприятный
исключение составляют больные с тяжёлыми врождёнными пороками
сердца и крупных сосудов и почечной гипертензией.
38.
- трисомия ХХХ– частота возникновения 1:700, не резкие отклонения в физическомразвитии, нарушения функции яичников, преждевременный климакс. Больные с
такой мутацией даже не догадываются о своем кариотипе;
- тетросомия ХХХХ приводит к умственной недостаточности разной степени;
- пентосомия ХХХХХ сопровождается тяжелыми повреждениями органов и сознания;
- моносомия Х0 (синдром Тернера) – единственная совместимая с жизнью, частота
возникновения 1:4000, недоразвитые яичники и матка, физическая и умственная
отсталость.
39.
40.
41.
42.
- синдром Клайнфельтера встречается в двух формах – полисомия по Ххромосоме и полисомия по Y-хромосоме, больные с кариотипом ХХ Y – этомужчины женоподобного типа, у них развита грудь, женский голос, длинные
ноги, недоразвиты семенники. Они бесплодны, но психически совершенно
нормальны;
- больные с кариотипом Х Y Y – нормальные высокие мужчины, умственно и
психически здоровы, но склонны к агрессии, асоциальны.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
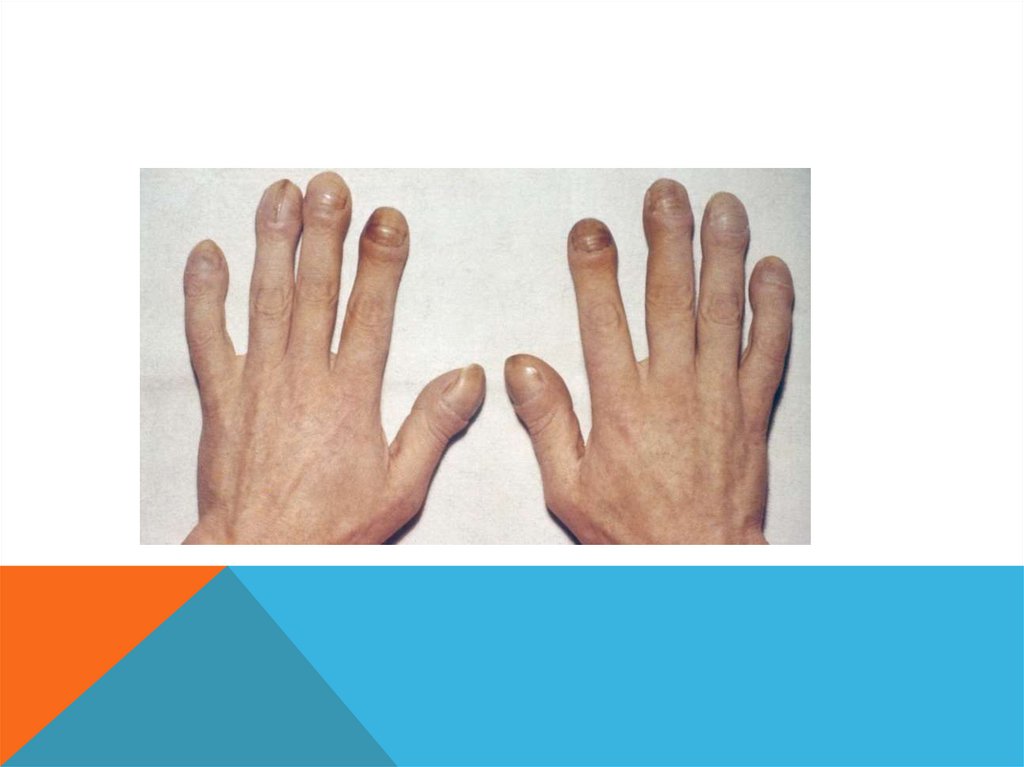
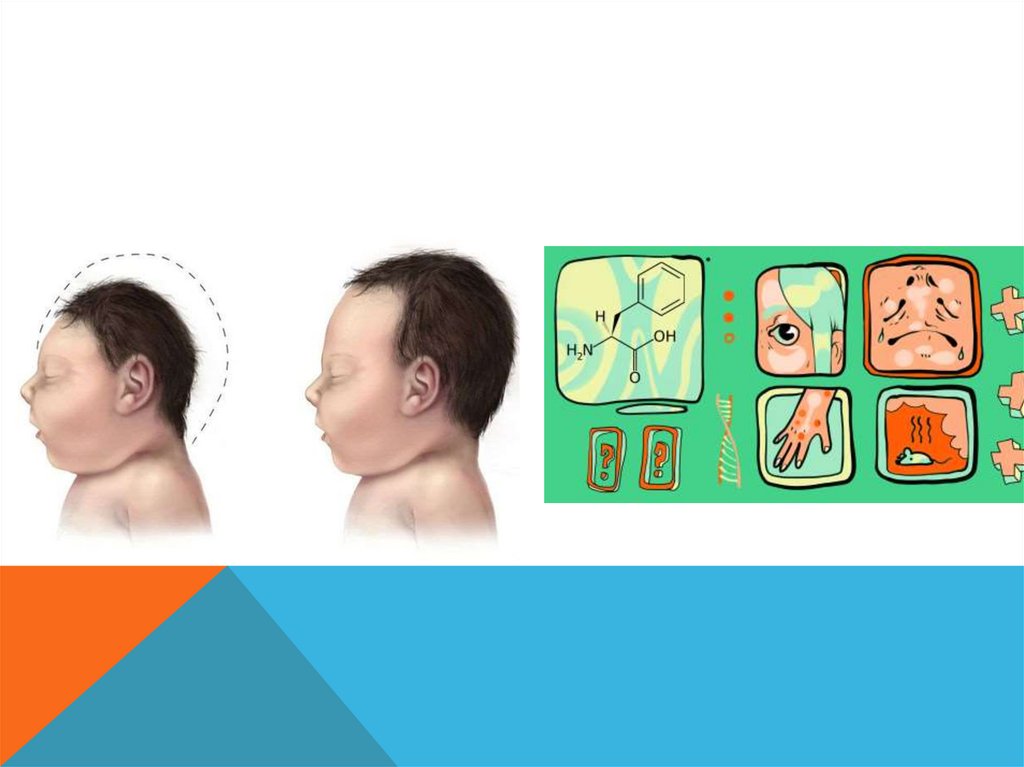
СИНДРОМ ПРАДЕРА-ВИЛЛИ.Описан в 1956 году.
Популяционная частота 1 на 15 000. Встречается в основном спорадически, хотя
описаны и семейные случаи с аутосомно-доминантным типом наследования.
55.
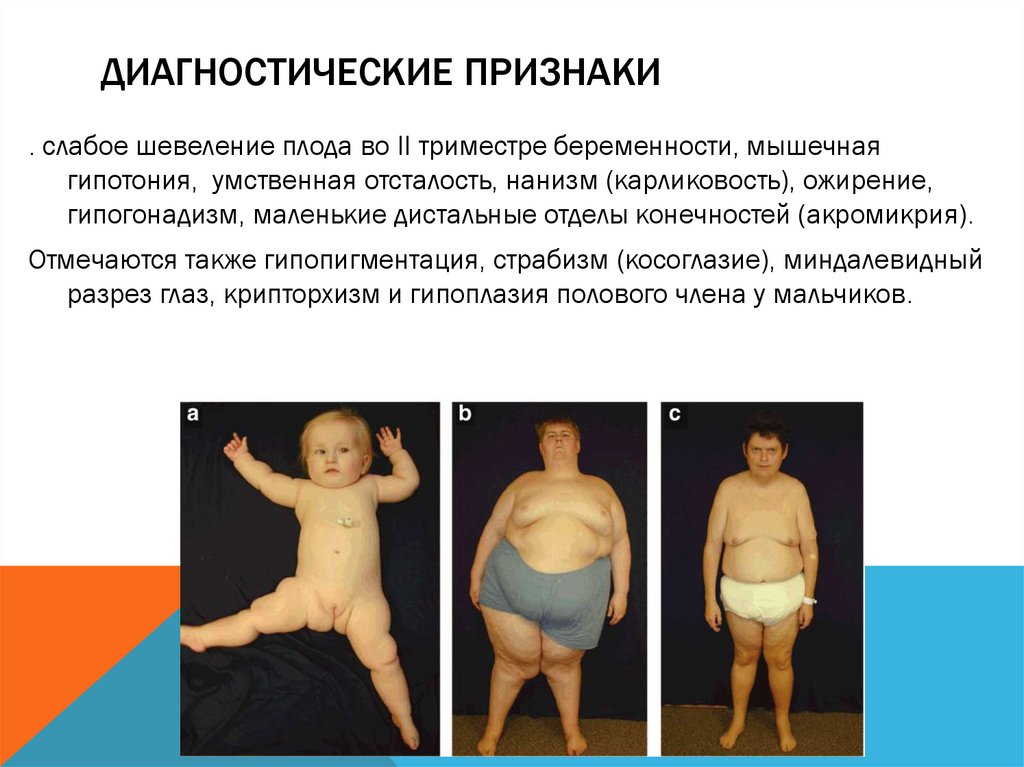
ДИАГНОСТИЧЕСКИЕ ПРИЗНАКИ. слабое шевеление плода во II триместре беременности, мышечная
гипотония, умственная отсталость, нанизм (карликовость), ожирение,
гипогонадизм, маленькие дистальные отделы конечностей (акромикрия).
Отмечаются также гипопигментация, страбизм (косоглазие), миндалевидный
разрез глаз, крипторхизм и гипоплазия полового члена у мальчиков.
56.
57.
58.
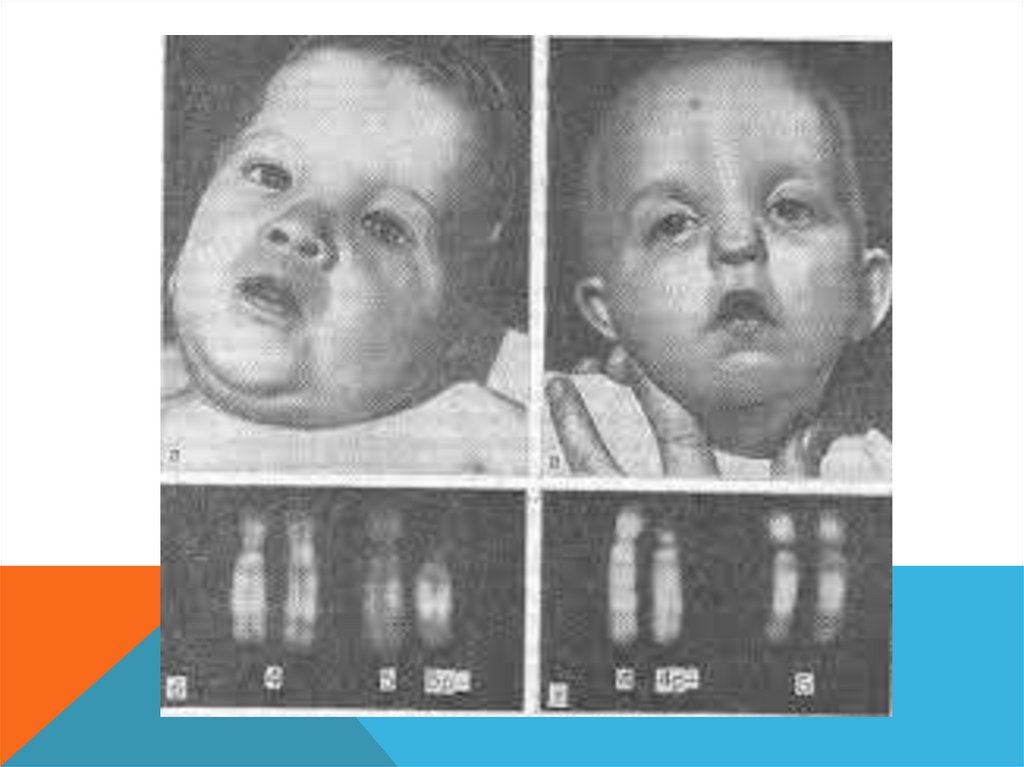
ТРИСОМИЯ ПО 8 ХРОМОСОМЕВпервые описана в 1962 году.
Частота 1 на 50 000.
Результат нерасхождения хромосом на ранних стадиях бластулы (кроме редких
случаев мутаций de novo в гаметогенезе).
Цитогенетически выявляются как полные, так и мозаичные формы, которые
составляют до 90% всех случаев. Различий по клинической картине таких
форм нет.
59.
Основные диагностические признакиМакроцефалия, микрогнатия, вывернутая нижняя губа, массивный
выступающий лоб, широкая спинка носа, высокое акровидное небо,
большие оттопыренные уши с выступающим противозавитком.
Аномалии скелета (добавочные ребра и позвонки, закрытые спинномозговые
грыжи в шейном и грудном отделах позвоночника, аплазия и гипоплазия
надколенника, короткая шея)
Имеются множественные контрактуры суставов, клинодактилия,
камптодактилия.
Распространены пороки мочевой системы, особенно гидронефроз.
Диагностируют пороки сердца, особенно дефекты перегородок и крупных
сосудов.
60.
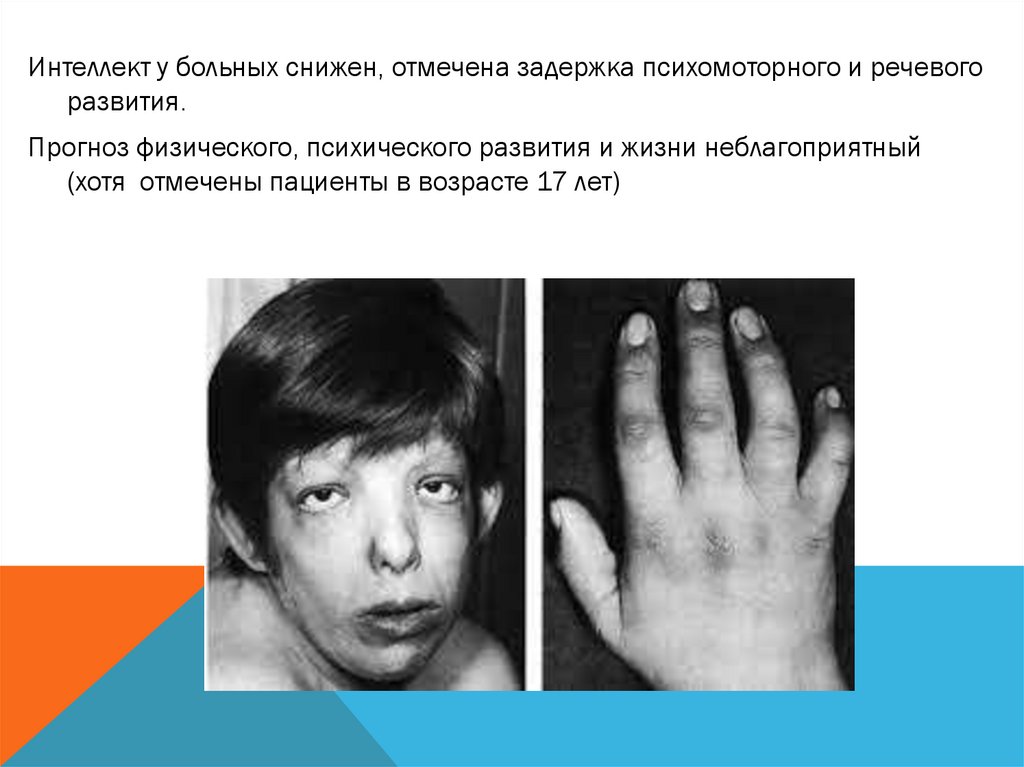
Интеллект у больных снижен, отмечена задержка психомоторного и речевогоразвития.
Прогноз физического, психического развития и жизни неблагоприятный
(хотя отмечены пациенты в возрасте 17 лет)
61.
62.
ТРИСОМИЯ ПО 9 ХРОМОСОМЕОписан в 1970 году.
В основе этиологии лежит нерасхождение хромосом на ранних стадиях
бластулы, кроме редких случаев мутаций de novo в гаметогенезе.
Цитогенетически отмечены полные формы трисомии и мозаики (50%).
Мутации de novo составляют 1/3 всех описанных случаев. Кроме полных
аутосомных трисомий хромосомы 9 выявлены также делеции ее длинного
плеча. Транслокационные варианты встречаются редко.
63.
64.
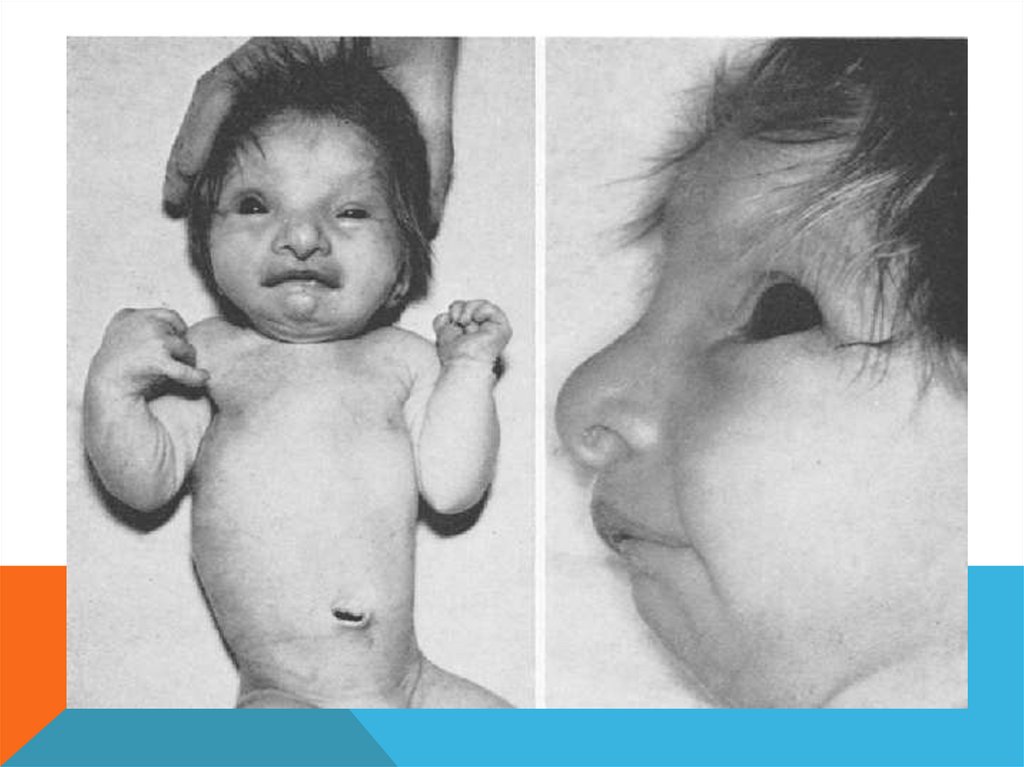
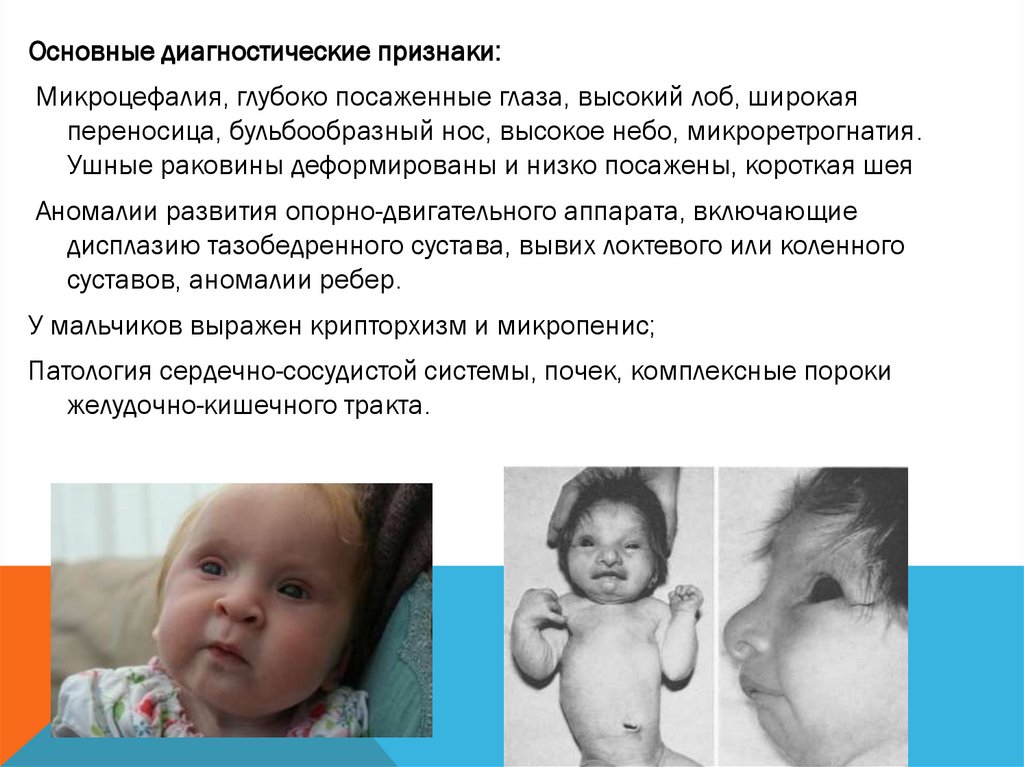
Основные диагностические признаки:Микроцефалия, глубоко посаженные глаза, высокий лоб, широкая
переносица, бульбообразный нос, высокое небо, микроретрогнатия.
Ушные раковины деформированы и низко посажены, короткая шея
Аномалии развития опорно-двигательного аппарата, включающие
дисплазию тазобедренного сустава, вывих локтевого или коленного
суставов, аномалии ребер.
У мальчиков выражен крипторхизм и микропенис;
Патология сердечно-сосудистой системы, почек, комплексные пороки
желудочно-кишечного тракта.
65.
Прогноз жизни неблагоприятный.Большинство больных погибают в первые 4 месяца жизни,
особенно от респираторных инфекций.
66.
67.
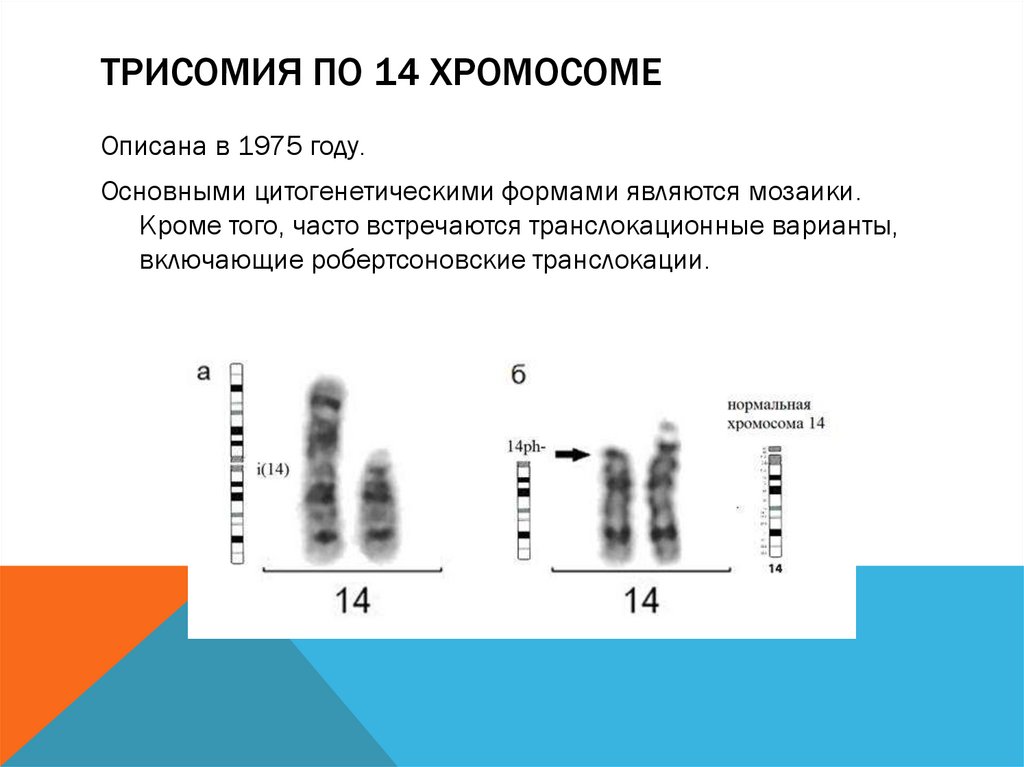
ТРИСОМИЯ ПО 14 ХРОМОСОМЕОписана в 1975 году.
Основными цитогенетическими формами являются мозаики.
Кроме того, часто встречаются транслокационные варианты,
включающие робертсоновские транслокации.
68.
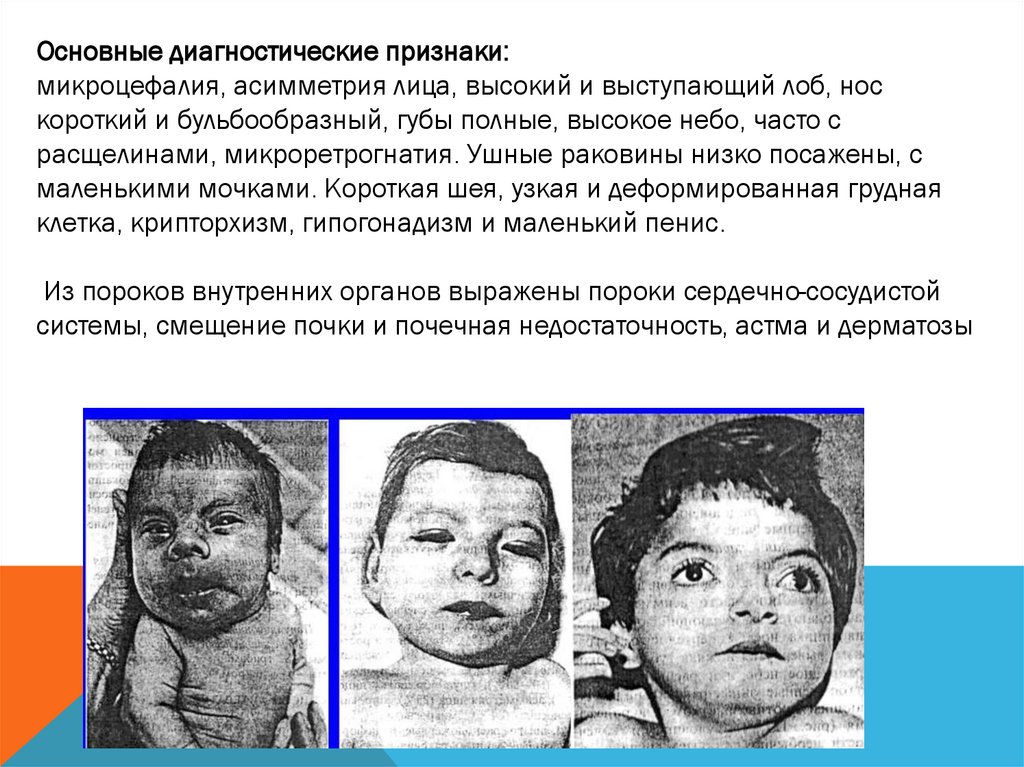
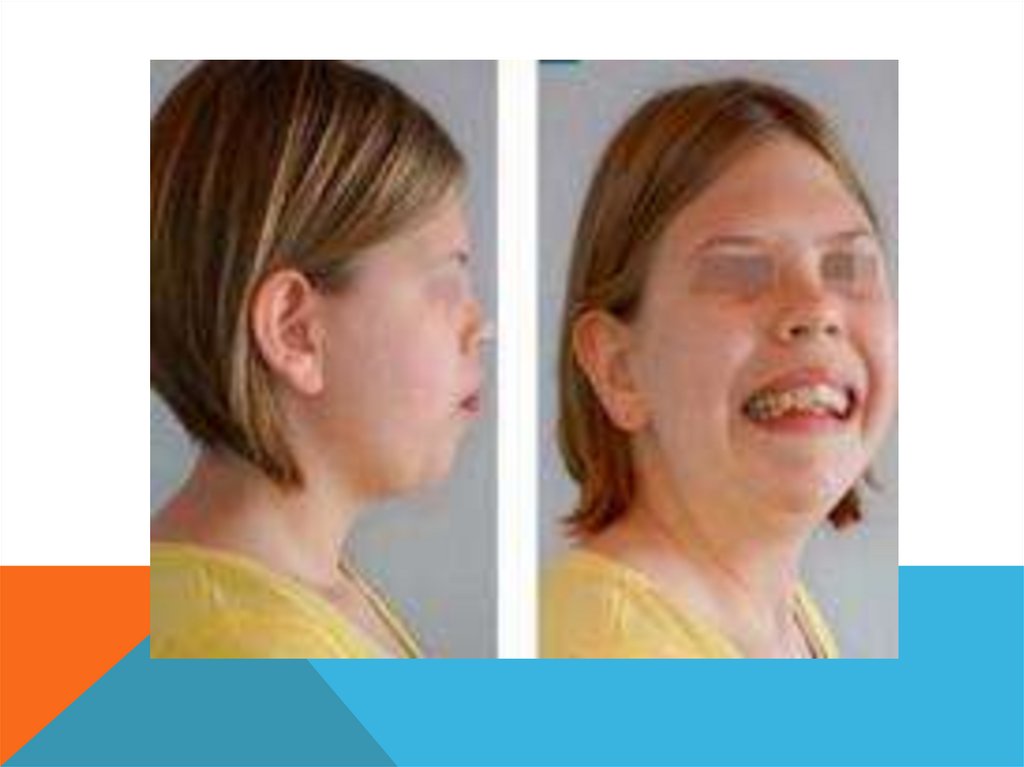
Основные диагностические признаки:микроцефалия, асимметрия лица, высокий и выступающий лоб, нос
короткий и бульбообразный, губы полные, высокое небо, часто с
расщелинами, микроретрогнатия. Ушные раковины низко посажены, с
маленькими мочками. Короткая шея, узкая и деформированная грудная
клетка, крипторхизм, гипогонадизм и маленький пенис.
Из пороков внутренних органов выражены пороки сердечно-сосудистой
системы, смещение почки и почечная недостаточность, астма и дерматозы
69.
Прогноз жизни неблагоприятный, однако отмечены больные ввозрасте 13 лет.
70.
71.
ПОЛИСОМИЯ ПО Х ХРОМОСОМЕПопуляционная частота 1 на 1000 новорожденных девочек.
Цитогенетическивыявляются формы 47,ХХХ, 48.ХХХХ и 49.ХХХХХ.
С увеличением числа X хромосомы нарастает степень отклонений от
нормы.
У женщин с тетра- и пентасомией X описаны отклонения в умственном
развитии, аномалии скелета и половых органов.
Женщины с кариотипом 47.ХХХ в полной или мозаичной форме в
основном имеют нормальное физическое и психическое развитие, а
интеллект - в пределах нижней границы нормы. У этих женщин имеется
нерегулярный менструальный цикл и вторичная аменорея, однако они
могут иметь потомство.
72.
73.
ПОЛИСОМИЯ ПО У ХРОМОСОМЕПопуляционная частота 1 на 1000
мальчиков.
Цитогенетически отмечены полные и
мозаичные формы.
Большинство по физическому и
умственному развитию не
отличается от здоровых
. Обычно они высокого роста (+15см)
и в 50% случаев могут иметь
нормальное потомство.
При данном синдроме имеются
особенности поведения: склонность
к агрессии, асоциальному
поведению, гомосексуализму.
74.
75.
СИНДРОМ ВОЛЬФА-ХИРШХОРНАОписан в 1965 году.
Популяционная частота 1 на 100 000.
Цитогенетически обусловлен частичной делецией короткого плеча 4
хромосомы. Наследственная форма составляет 10%, а 90% случаев
представлены мутациями de novo. Отмечены кольцевые и изохромосомы.
76.
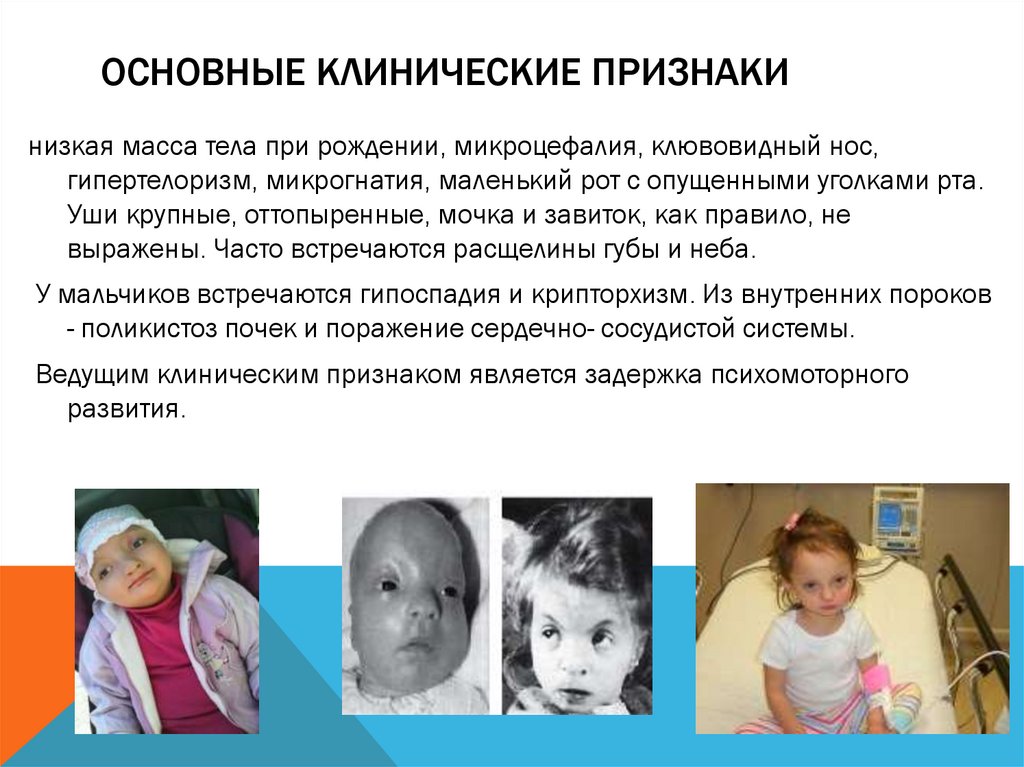
ОСНОВНЫЕ КЛИНИЧЕСКИЕ ПРИЗНАКИнизкая масса тела при рождении, микроцефалия, клювовидный нос,
гипертелоризм, микрогнатия, маленький рот с опущенными уголками рта.
Уши крупные, оттопыренные, мочка и завиток, как правило, не
выражены. Часто встречаются расщелины губы и неба.
У мальчиков встречаются гипоспадия и крипторхизм. Из внутренних пороков
- поликистоз почек и поражение сердечно- сосудистой системы.
Ведущим клиническим признаком является задержка психомоторного
развития.
77.
78.
СИНДРОМ "КОШАЧЬЕГО КРИКА"Описан в 1963 году.
Популяционная частота 1 на 50 000.
Цитогенетические варианты варьируют от частичной до полной делеции
короткого плеча хромосомы 5 (так же отмечены кольцевые хромосомы
5, мозаичные формы, а также транслокации между коротким плечом
хромосомы 5 (с потерей критического сегмента) и другой аутосомой.))
79.
ДИАГНОСТИЧЕСКИЕ ПРИЗНАКИЗАБОЛЕВАНИЯ
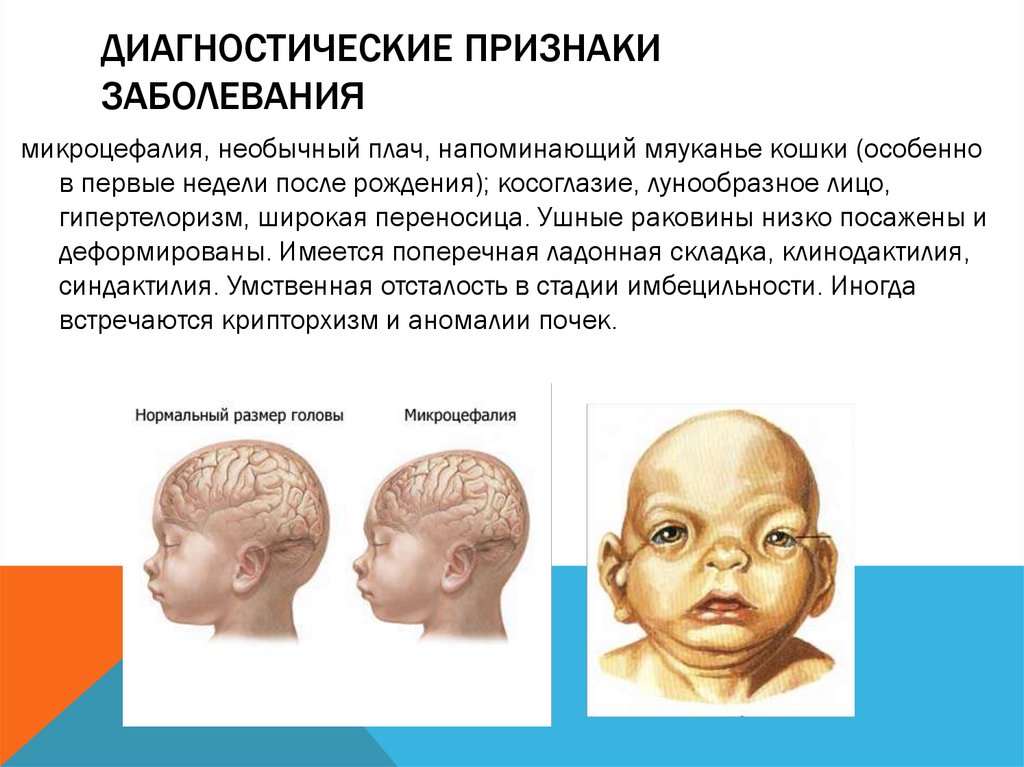
микроцефалия, необычный плач, напоминающий мяуканье кошки (особенно
в первые недели после рождения); косоглазие, лунообразное лицо,
гипертелоризм, широкая переносица. Ушные раковины низко посажены и
деформированы. Имеется поперечная ладонная складка, клинодактилия,
синдактилия. Умственная отсталость в стадии имбецильности. Иногда
встречаются крипторхизм и аномалии почек.
80.
Лунообразное лицо и кошачий крик с возрастом сглаживаются, амикроцефалия и косоглазие выявляются более отчетливо.
Продолжительность жизни зависит от тяжести врожденных
пороков развития внутренних органов. Большинство больных
погибают в первые годы жизни.
81.
82.
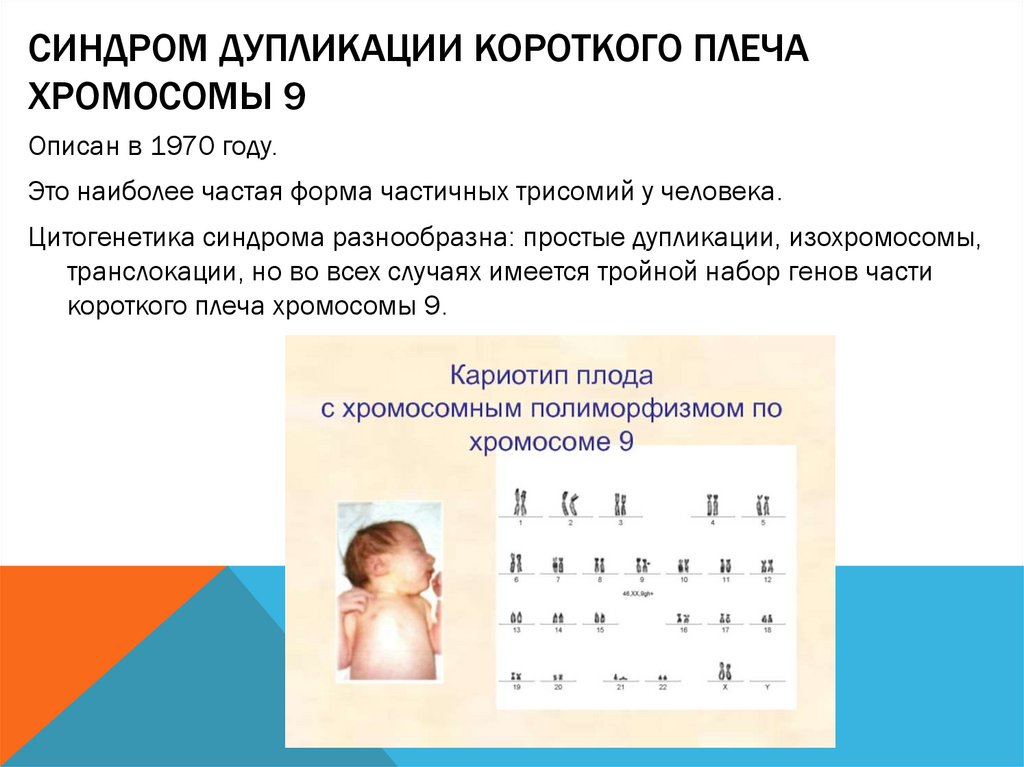
СИНДРОМ ДУПЛИКАЦИИ КОРОТКОГО ПЛЕЧАХРОМОСОМЫ 9
Описан в 1970 году.
Это наиболее частая форма частичных трисомий у человека.
Цитогенетика синдрома разнообразна: простые дупликации, изохромосомы,
транслокации, но во всех случаях имеется тройной набор генов части
короткого плеча хромосомы 9.
83.
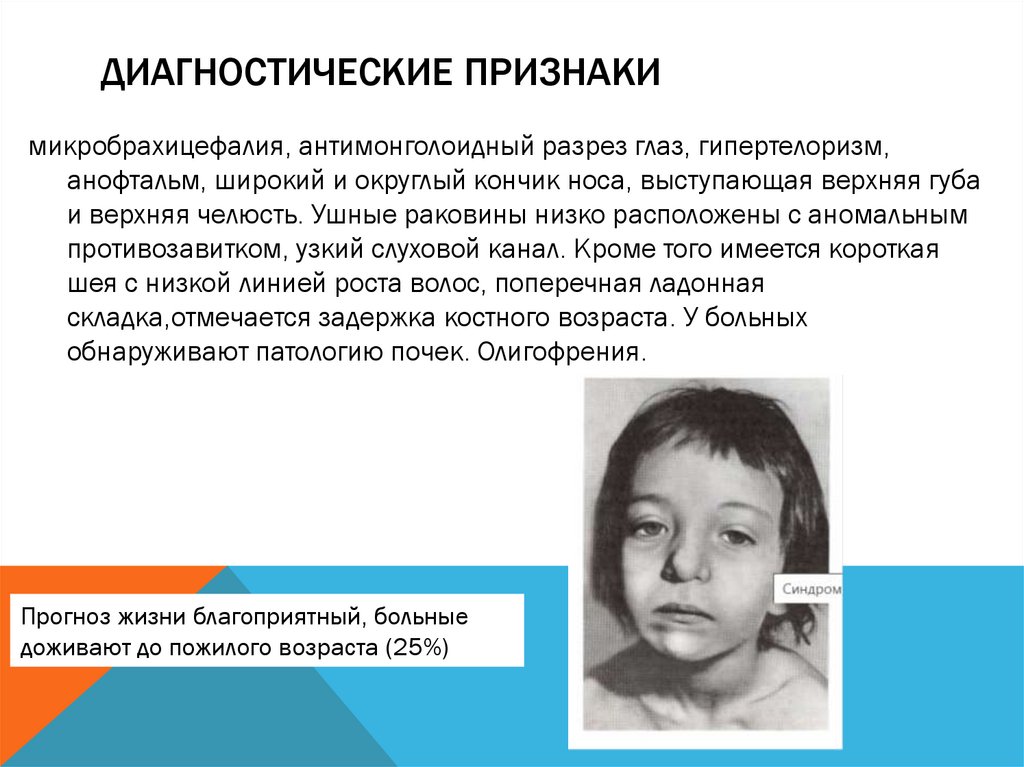
ДИАГНОСТИЧЕСКИЕ ПРИЗНАКИмикробрахицефалия, антимонголоидный разрез глаз, гипертелоризм,
анофтальм, широкий и округлый кончик носа, выступающая верхняя губа
и верхняя челюсть. Ушные раковины низко расположены с аномальным
противозавитком, узкий слуховой канал. Кроме того имеется короткая
шея с низкой линией роста волос, поперечная ладонная
складка,отмечается задержка костного возраста. У больных
обнаруживают патологию почек. Олигофрения.
Прогноз жизни благоприятный, больные
доживают до пожилого возраста (25%)
84.
85.
СИНДРОМ АЛЬФИВпервые описан в 1973 году.
Цитогенетические варианты могут быть различны: частичная
делеция короткого плеча 9 хромосомы, изохромосома 9q,
несбалансированные транслокации, однако во всех случаях
наблюдается потеря сегмента.
86.
ДИАГНОСТИЧЕСКИЕ ПРИЗНАКИрезко выступающий лоб, эпикант, экзофтальм, гипертелоризм, уплощенная и
широкая переносица, маленький рот с большой верхней губой, высокое
небо.
Ушные раковины без мочки или она недоразвита, со сглаженным завитком.
Шея короткая, отмечен гипертелоризм сосков. Пальцы рук и ног длинные,
с дополнительными сгибательными складками; ногти широкие, выпуклые,
квадратной формы.
У девочек выражена гипоплазия малых и больших половых губ, у мальчиков гипоплазия мошонки и полового члена.
Из пороков внутренних органов отмечено поражение сердечно-сосудистой
системы и гидронефроз почек.
87.
Умственная отсталость встадии имбецильности,
реже - дебильности.
По характеру ласковые,
спокойные, послушные.
Прогноз жизни
благоприятный.
88.
В результате интенсивного изучения хромосом человека и хромосомныхболезней на протяжении 35–40 лет сложилось учение о хромосомной
патологии, которая имеет большое значение в современной медицине.
89.
Хромосомные болезни - это большая группа врожденных наследственныхболезней. Хромосомные болезни занимают одно из ведущих мест в структуре
наследственной патологии человека. По данным цитогенетических исследований
среди новорожденных детей частота хромосомной патологии составляет 0,6-1,0%.
Самая высокая частота хромосомной патологии (до 70%) зафиксирована в
материале ранних спонтанных абортусов. Следовательно, большинство
хромосомных аномалий у человека несовместимо даже с ранними этапами
эмбриогенеза. Такие зародыши элиминируются во время имплантации (7-14-е дни
развития), что клинически проявляется как задержка или выпадение
менструального цикла. Некоторая часть эмбрионов гибнет вскоре после
имплантации (ранние выкидыши). Сравнительно немногие варианты числовых
аномалий хромосом совместимы с постнатальным развитием и ведут к
хромосомным заболеваниям (Кулешов Н.П., 1979).