












 Медицина
МедицинаПохожие презентации:


")







Миотоническая дистрофия Куршмана-Баттена-Штейнерта
1. Миотоническая дистрофия Куршмана-Баттена-Штейнерта
МИОТОНИЧЕСКАЯДИСТРОФИЯ
КУРШМАНА-БАТТЕНАШТЕЙНЕРТА
Работу выполнил Макаров Виталий
2. ВВедение
ВВЕДЕНИЕ
Мышечная дистрофия Штейнерта
(миотоническая дистрофия;
дистрофическая миотония
Россолимо – Штейнерта – Куршмана
– Баттена; атрофическая атония;
DM1; МКБ 10 – G71.1) — редкое
наследственное заболевание,
проявляется сочетанием
отсроченного расслабления мышц
после сокращения (миотонии) и
прогрессирующей мышечной
слабости (дистрофии). Заболевание
вызвано удлинением некодирующей
части гена DMPK.
3. Историческая справка
ИСТОРИЧЕСКАЯ СПРАВКА
Древнеегипетский фараон
Эхнатон (Аменхотеп IV), умерший
в возрасте 36 лет. Возможно, изза религиозной реформы
изображения фараона сильно
отличаются от традиционных
идеализированных изображений
правителей. На статуях можно
видеть, что у Эхнатона
вытянутое лицо со впалыми
щеками, полуоткрыт рот и
опущены веки.
4.
5. Миотическая дистрофия это гетерогенное заболевание, представленное тремя подтипами:
DM1 (мутация 19q13.3),DM2 (мутация 3q21),
DM3 (мутация 15q21-q24).
Существуют отдельные исследования, подтверждающие наличие
четвёртого подтипа DM и др. (DM4, DMX).
6. Распространённость и тип наследования.
• В среднем около 12,5 случаев на 100 тыс. населения(около1 на 8000 человек).
• В России заболевание наиболее широко распространено в
Красноярском крае: 14,17 на 100 тыс.(около 1 на 7000
человек).
• Болезнь наследуется по аутосомно-доминантному типу, т. е.
одной копии «дефектного» гена достаточно, чтобы
развилось заболевание. Вероятность развития
миотонической дистрофии у детей больных составляет 50
%.
7. Выделяют четыре формы течения болезни Штейнерта:
Врождённая (конгенитальная ДМ, клиническая симптоматикаразвивается сразу после рождения)
Юношеская (ювенильная ДМ, с дебютом от 1 года до подросткового
возраста)
Классическая (ДМ взрослых, с дебютом у индивидуумов старше 20,
но моложе 40 лет)
Минимальная (ДМ с поздним дебютом, у индивидуумов старше 40
ле (как правило, до 60 лет) и с более лёгким течением)
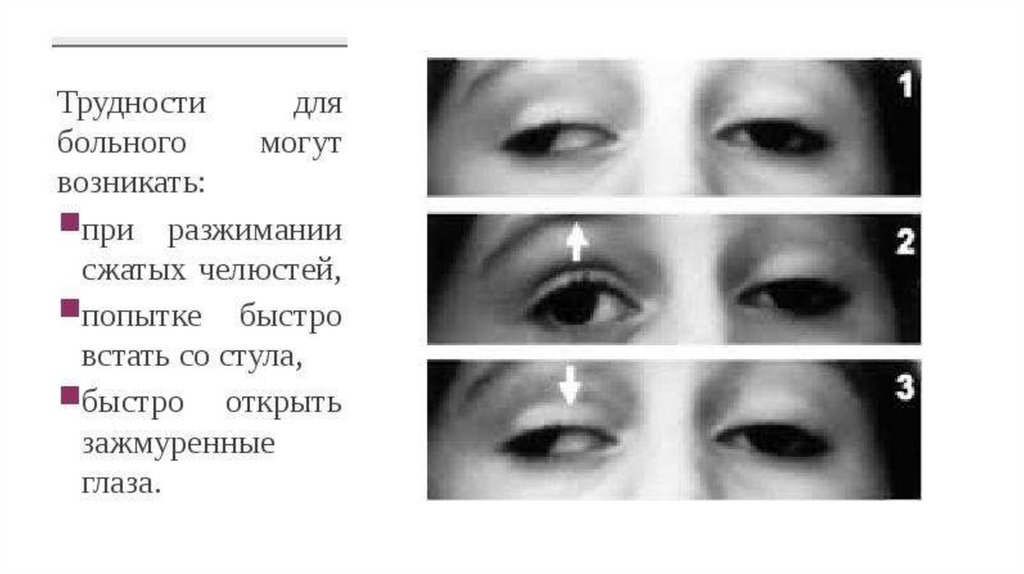
8. Симптоматика
Миотония: Спазмыжевательных мышц, кистей.
Мышечная атрофия: Лицо
(«маскообразное»), шея,
конечности.
Осложнения:
Сердечные аритмии (блокада
ножек пучка Гиса).
Эндокринные нарушения
(гипогонадизм, ранний
климакс).
Умственная отсталость,
гиперсомния.
9.
10.
11.
12. Лечение
• Радикального лечения миотопии не существует. Дляуменьшения выраженности миотонических проявлений
используют фенитоин в дозе 200-400 мг/сут перорально.
Курсы диуретиков позволяют снизить уровень калия в крови
и уменьшить миотонические проявления.
• Нейромиотонические проявления уменьшаются при
назначении карбамазепина.
• Ремиссии иногда удаётся достичь с помощью
иммуносупрессивной терпии.
13. Молекулярные механизмы
• Ген DMPK: Экспансия CTG-повторов в 3’-нетранслируемойобласти.
• Последствия:
o Накопление токсичной мРНК → гибель клеток.
o Нарушение функции миотонинпротеинкиназы
• Клиническая корреляция:
o Норма: 5–37 повторов.
o DM1: 50–1000+ повторов.
14. Список наиболее частых мутаций гена DMPK:
• NM_001081563.2(DMPK): c.224-226CTG[28]CCG[2]CTG[2]CCG[1]CTG[88];• NM_001081563.2(DMPK): c.224-226CTG[30]CCG[2]CTG[2]CCG[1]CTG[105];
• NM_001081563.2(DMPK): c.961C>T (p.Arg321Ter);
• NM_004409.4: c.224-283CTG[(260-320)]CCGCTGCTG[(10-14)]CTG[(15-23)];
• NM_004409.4: c.224-283CTG[(200-300)]CCG[1]CTG [(41-59)].
15. Спасибо за внимание!
СПАСИБО ЗАВНИМАНИЕ!