


")


")
")













")


























 Биология
Биология Химия
ХимияПохожие презентации:










Клінічна хімія
1. КЛІНІЧНА ХІМІЯ
вступЛекцій – 28 год.
Лабораторні заняття - 30 год.
Cамостійна робота – 60 год.
2. ПЕРЕЛІК ЛЕКЦІЙ
Лекція 1. Мета і завдання курсу «Клінічна хімія». Обмін вуглеводів та його порушенняЛекція 2. Обмін ліпідів та його порушення
Лекція 3. Обмін азот-умісних сполук та його порушення
Лекція 4. Генетичні ушкодження функцій ферментів як причини спадкових розладів
Лекція 5. Основи ензимодіагностики та принципи застосування ферментних
препаратів у терапії
Лекція 6. Клініко-біохімічні характеристики дисвітамінозів та їх корекція
Лекція 7. Водно-сольовий та мінеральний обмін та його порушення
Лекція 8. Біологічна роль гормонів. Порушення гормонального статусу організму.
Лекція 9. Клінічна хімія печінки.
Лекція 10. Клінічна хімія крові
Лекція 11. Клінічна хімія нирок і сечі
Лекція 12. Хіміко-функціональні особливості нервової системи. Порушення
функціонального стану нервової системи, принципи їх діагностики
Лекція 13. Хіміко-функціональні особливості м’язової та сполучної тканин. Порушення
функціонального стану цих тканин, принципи їх діагностики
3. РОЗПОДІЛ БАЛІВ
Іспит – 40 балів
1. Модульна конт рольна робот а 1 – 15 балів/ 7,5 бала (теми 1 – 8)
2. Модульна конт рольна робот а 2 – 15 балів/ 7,5 бала (теми 9 –
13)
3. Лаборат орні заняття – 30 балів/ 15 балів
4. Рекомендована література (в Інтернеті у вільному доступі є раніші видання)
1. Хоперія В. Г., Харченко О. І., Синельник Т.Б., Костюк О.С., Остапченко Л.І. Клінічналабораторна діагностика. Клінічна біохімія: підручник. – К.: ВПЦ "Київський
університет", 2022. – 600 с.
https://biomed.knu.ua/images/stories/Kafedry/Biochimiya/Biblioteka/Klinichna_laboratorna_di
agnostyka.pdf
2. Синельник Т.Б., Конопельнюк В.В., Остапченко Л.І. Збірка тестів для підготовки до
іспиту з дисциплін «Клінічна хімія» та «Клінічна лабораторна діагностика» (розділ
«Клінічна біохімія») для студентів 3го курсу (спеціальність 224 «Технології медичної
діагностики та лікування», ОПП «Лабораторна діагностика» та «Дієтологія). – 2022 - 113
стор. (електронне видання)
3. Біологічна і біоорганічна хімія : у 2 кн.: підручник. Кн. 2. Біологічна хімія / Ю.І. Губський,
І.В. Ніженковська, М.М. Корда та ін.; за ред. Ю.І. Губського, І.В. Ніженковської. – 3-є вид. – К.:
ВСВ “Медицина”, 2021. - 544 c.
4. Біохімія людини : підручник / Я. І. Гонський, Т. П. Максимчук ; за ред. Я. І. Гонського. — 3тє вид., випр. і допов. — Тернопiль : Укрмедкнига, 2019. — 732 с.
5. Основи біохімії за Ленінджером / Дейвід Л. Нельсон, Майкл М. Кокс ; [пер. з англ. О.
Матишевська та ін. ; наук. ред. пер. С. Комісаренко та ін. ; ред. М. Мартиняк]. Львів: БаК, 2015.
– 1256 с.
6. Остапченко Л. І., Андрійчук Т.Р., Бабенюк Ю.Д. та ін. Біохімія: підручник. – К.: ВПЦ
“Київський університет”, 2012. — 796 с.
7. Клінічна біохімія: підручник / Д.П. Бойків, Т.І. Бондарчук, О.Л. Іванків та ін.; за ред. О.Я.
Склярова. - К.: Медицина, 2006. - 432 с.
5. ПОРУШЕННЯ ОБМІНУ ВУГЛЕВОДІВ
6. Основні харчові вуглеводи
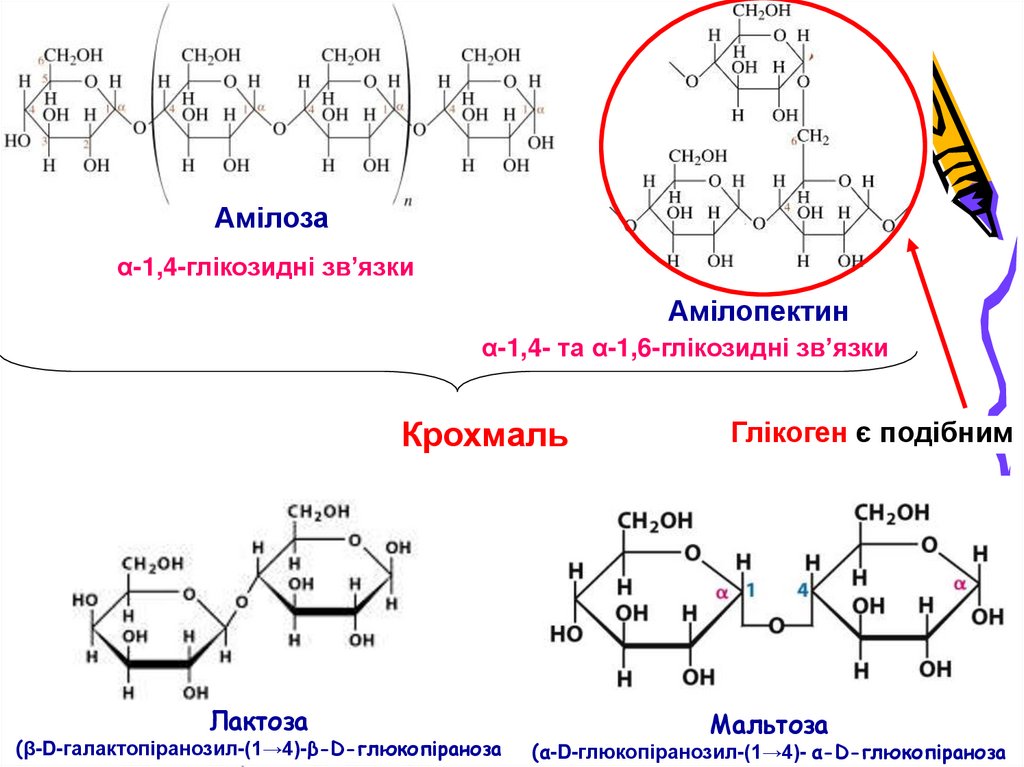
Крохмаль
Глюкоза
Фруктоза
Сахароза (глюкоза+фруктоза)
Лактоза (галактоза + глюкоза)
Мальтоза (глюкоза + глюкоза; складова солоду й пива)
Більшість рослинних вуглеводів - целюлоза, лігнін, пектини, ксилани - через
відсутність необхідних ферментів у організмі людини не засвоюються.
• Роль рослинних волокон:
- адсорбують воду і збільшують об’єм харчової грудочки, що стимулює
перистальтику кишечнику;
- абсорбують ряд токсичних сполук, у тому числі канцерогенів, що полегшує їх
виведення з організму
- неперетравлена целюлоза сприяє формуванню калових мас.
У людини рослинні волокна сприяють профілактиці порушень функцій товстого
кишечнику, попереджують виникнення ряду його захворювань, у тому числі і
раку.
7.
Амілозаα-1,4-глікозидні зв’язки
Амілопектин
α-1,4- та α-1,6-глікозидні зв’язки
Крохмаль
Лактоза
(β-D-галактопіранозил-(1→4)-β-D-глюкопіраноза
Глікоген є подібним
Maltose
Мальтоза
(α-D-глюкопіранозил-(1→4)- α-D-глюкопіраноза
8. Ди- і моносахариди: шлунково-кишковий тракт (ШКТ)
Ротова порожнина:
- фермент слинних залоз амілаза, що каталізує гідроліз α-1,4глікозидних зв’язків полісахаридів.
- бактеріальна глікозилтрансфераза, яка гідролізує вуглеводи
їжі у декстрини.
В результаті утворюється суміш декстринів (олігосахаридів), і
дисахаридів мальтози та ізомальтози, а також незначна кількість
вільної глюкози.
Шлунковий сік не містить глікозидаз, а амілаза слини інактивується
кислим рН, тому перетравлення вуглеводів тут зупиняється.
9. Ди- і моносахариди: шлунково-кишковий тракт (ШКТ)
12-пала кишка (порожнинне травлення!!!):
- α-амілаза підшлункової залози (також гідролізує α-1,4-глікозидні
зв’язки);
– оліго-1,6-глікозидаза (термінальна декстриназа) гідролізує α1,6-глікозидні зв’язки, які містяться у точках розгалуження глікогену й
амілопектинової фракції крохмалю (точніше – вже в декстринах).
Мукозна поверхня клітин кишечнику (пристінкове травлення!!!):
- дисахаридази (синтезуються епітеліальними клітинами слизової
оболонки і функціонують, зв’язані із зовнішньою стороною мембран цих
клітин): мальтаза (α-глюкозидаза) каталізує утворення 2-х молекул
глюкози; лактаза (β-галактозидаза) – молекул глюкози і галактози,
сахараза – глюкози і фруктози.
Моносахариди всмоктуються у кишечнику без попередньої
ферментативної обробки.
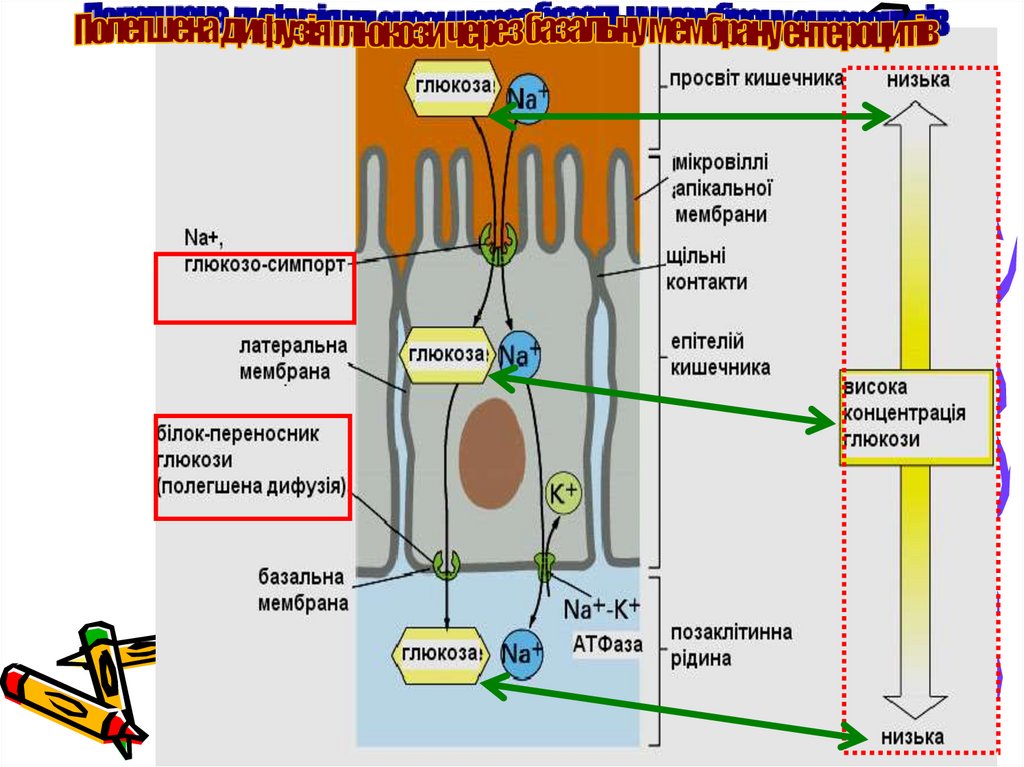
10. Всмоктування моносахаридів і потрапляння їх до печінки
Суміш моносахаридів (глюкози та меншої кількості фруктози та галактози)
поглинається епітеліальними клітинами, що вистилають тонкий кишечник:
вторинно-активним транспортом (симпорт з натрієм) - при низьких
концентраціях глюкози чи галактози у просвіті кишечнику;
полегшеною дифузією через транспортні білки GLUTs
(стимулюється інсуліном) або простою дифузією (в
інсулінонезалежних тканинах) - коли вміст моносахаридів у просвіті
кишечнику більший за їх вміст у клітині
З током крові глюкоза, галактоза та фруктоза переносяться у печінку.
55% глюкози захоплюється її клітинами, а решта з током крові переноситься
до:
інсулінозалежних клітин жирової тканини і скелетних м’язів (15%);
інсулінонезалежними тканинами (мозком, нервами, еритроцитами,
мозковою частиною нирок) – 25%;
5% залишається у рідинах організму;
11.
12. PI3K/Akt сигнальний каскад у інсуліновій регуляції поглинання глюкози клітинами
В інсулінозалежних клітинах - адипоцитах і м’язових клітинах – інсулін стимулює
поглинання глюкози з крові через активацію PI3K/Akt-залежного шляху.
Активація PKB/Akt стимулює транслокацію транспортера глюкози GLUT4 із
внутрішньоклітинних компартментів до плазматичної мембрани і посилення поглинання
глюкози
РІ3К-кінази активуються
інсуліновим рецептором і
фосфорилюють мембранний
фосфоліпід фосфатидилінозитол4,5-дифосфат (РІ-4,5-Р2, де РІ фосфатидилінозитол) в 3
положенні, продукуючи РІ-3,4,5Р3 (фосфатидилінозитол-3,4,5трифосфат).
Фосфорильовані інозитоли є
вторинними посередниками, що
залучаються у активацію великої
кількості різних білків, зокрема,
протеїнкінази PKB/Akt
13. Поглинання глюкози клітинами
Біохімічні основи порушення перетвореннявуглеводів у ШКТ та їх всмоктування
Порушення роботи К, Na-АТФази (природжений дефект, наявність інгібіторів –
напр., уабаїн, енергетичний голод) – через унеможливлення вторинно-активного
транспорту
Вроджене або набуте порушення роботи самого Na+-залежного переносника.
флоридзиновий діабет – ця сполука є отруйним глікозидом, інгібітором
переносника моносахаридів; при цьому порушується надходження
глюкози в ентероцити.
глюкозо-галактозна мальабсорбція – дефіцит натрій-залежного
транспортера глюкози і галактози (супроводжується проносом, нирковою
глюкозурією)
Синдром, пов’язаний з недостатністю дисахаридаз – лактази, інвертази,
мальтази, трегалази (вроджена алактазія – симптоми при першому вживанні
молока; недостатність інвертази і мальтази виявляється при переведенні дитини на
штучне вигодовування). З віком в людини експресія гена лактази може знижуватися
під впливом компонентів мішаного харчування. Мальтазна недостатність у
дорослої людини – неперенесення крохмалю, круп, пива та інших напоїв на основі
солоду. Недостатність трегалази – неперенесення грибів, що містять трегалозу.
14. Біохімічні основи порушення перетворення вуглеводів у ШКТ та їх всмоктування
15.
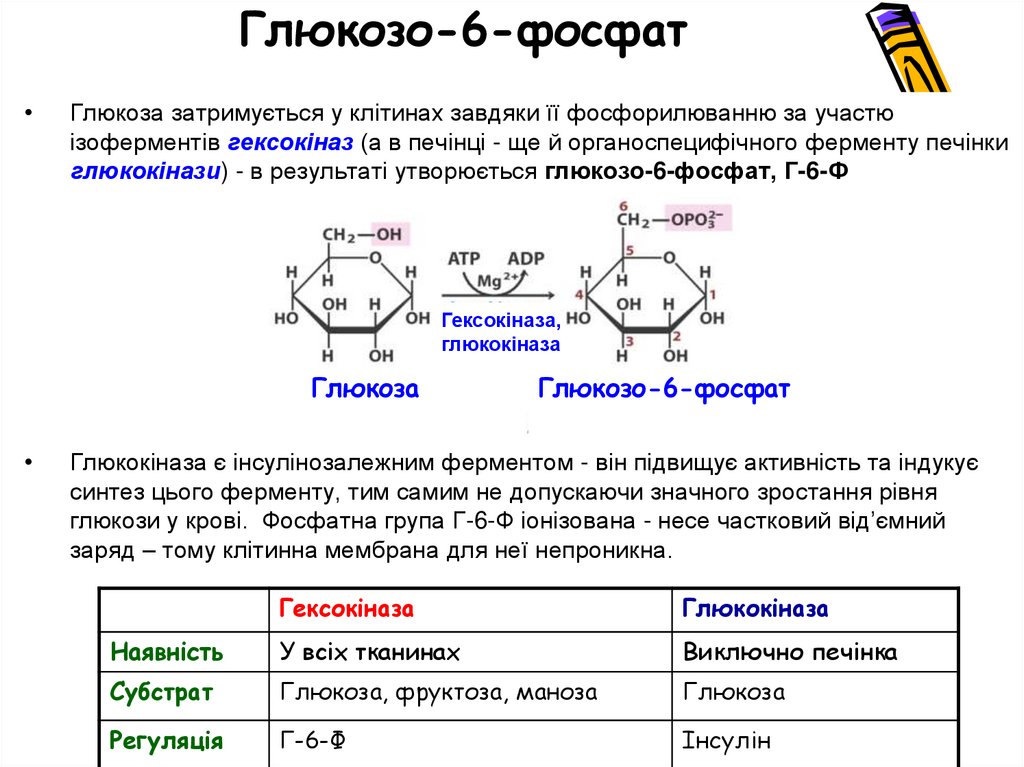
Глюкозо-6-фосфатГлюкоза затримується у клітинах завдяки її фосфорилюванню за участю
ізоферментів гексокіназ (а в печінці - ще й органоспецифічного ферменту печінки
глюкокінази) - в результаті утворюється глюкозо-6-фосфат, Г-6-Ф
Гексокіназа,
глюкокіназа
Глюкоза
Глюкозо-6-фосфат
Глюкокіназа є інсулінозалежним ферментом - він підвищує активність та індукує
синтез цього ферменту, тим самим не допускаючи значного зростання рівня
глюкози у крові. Фосфатна група Г-6-Ф іонізована - несе частковий від’ємний
заряд – тому клітинна мембрана для неї непроникна.
Гексокіназа
Глюкокіназа
Наявність
У всіх тканинах
Виключно печінка
Субстрат
Глюкоза, фруктоза, маноза
Глюкоза
Регуляція
Г-6-Ф
Інсулін
16.
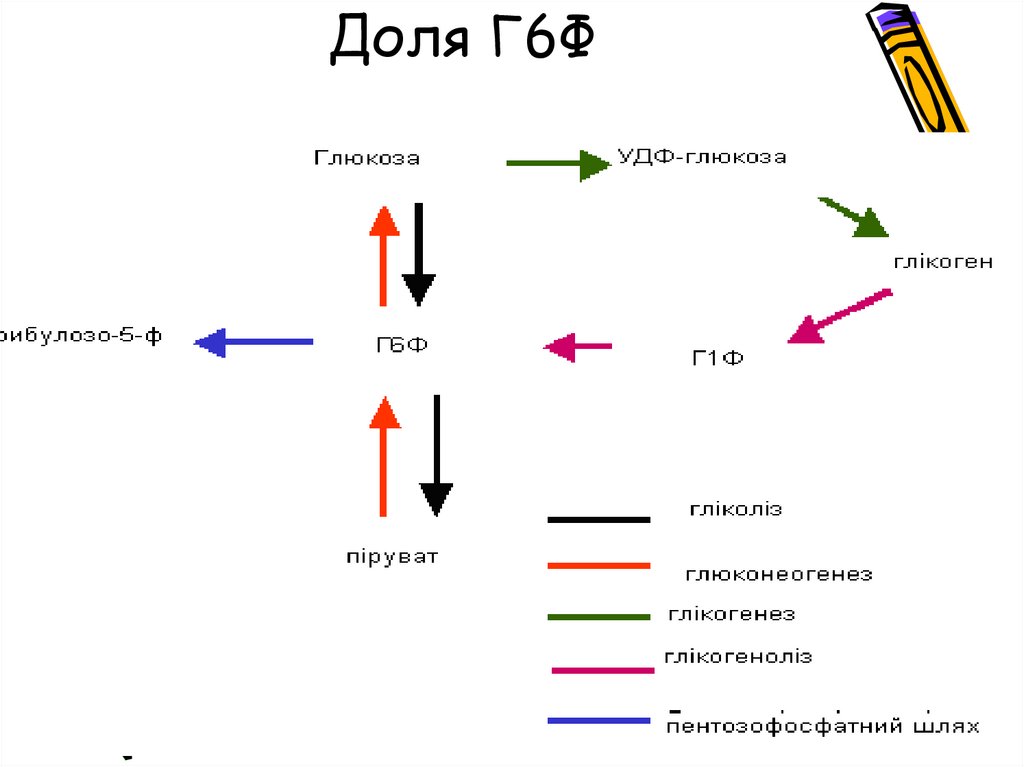
Доля Г6Ф17. Доля Г6Ф
Основні ферменти синтезу глікогенуТак утворюються α-(1→4)-глікозидні зв’язки; надалі фермент розгалуження формує α(1→6)-глікозидні зв’язки у точках розгалуження
18. Основні ферменти синтезу глікогену
Основні депо глікогену:скелетні м’язи (400 г у стані спокою) - джерело енергії при
м’язовому скороченні;
печінка (100г, 2-8 % маси органа) - регулятор постійності
концентрації глюкози у крові.
19.
Регуляторний фермент синтезу глікогену – глікогенсинтаза:неактивна форма – фосфорильована;
активна – нефосфорильована
Реакцію фосфорилювання здійснює цАМФ-залежна протеїнкіназа А,
протилежну реакцію – дефосфорилювання ферменту – здійснюють
протеїнфосфатази.
Активності ферментів фосфорилювання і дефосфорилювання (цАМФзалежної протеїнкінази та протеїнфосфатази) перебувають під гормональним
контролем:
Інсулін стимулює розщеплення цАМФ фосфодіестеразою,
знижуючи цАМФ-залежне фосфорилювання глікогенсинтази – і
тому стимулює синтез глікогену; інший шлях – РІ3К/Aktзалежне інгібування кінази глікогенсинтази 3 (GSK-3), що
також веде до збереження активності глікогенсинтази)
адреналін і глюкагон – стимулюють синтез цАМФ,
протеїнкіназу А, фосфорилювання глікогенсинтази, гальмуючи
синтез глікогену. Одночасно стимулюють розпад глікогену
(цАМФ-опосередковано).
20. Регуляція синтезу глікогену
Основні ферменти розпаду глікогенуТак гідролізуютьсяα-(1→4)-глікозидні зв’язки; надалі біфункціональний
дерозгалудужуючий фермент, також відомий як α (1→6) глікозидаза, гідролізує α(1→6)-глікозидні зв’язки у точках розгалуження
21. Основні ферменти розпаду глікогену
Регуляторним ферментом розпаду глікогену є глікогенфосфорилаза:Неактивна форма ферменту (фосфорилаза b) - нефосфорильована;
активна (фосфорилаза а) – фосфорильована.
Реакцію фосфорилювання здійснює цАМФ-залежна кіназа фосфорилази b,
яка активується цАМФ-залежною протеїнкіназою А.
Адреналін і глюкагон стимулюють утворення цАМФ, а, отже,
фосфорилювання глікогенфосфорилази і розпад глікогену
Відщеплення фосфатних груп здійснюють, як і у випадку глікогенсинтази,
протеїнфосфатази
22. Регуляція розпаду глікогену
Глікогенози тааглікогенози
Обмін глікогену в печінці
Основні ферменти синтезу глікогену:
фосфоглюкомутаза
глюкозо-1-фосфатуридилтрансфераза
глікогенсинтаза
Основні ферменти розпаду глікогену:
глікогенфосфорилаза
глюкозо-6-фосфатаза
Аглікогенози
Дефектні ферменти синтезу глікогену
Глікогенози
Дефектні ферменти розпаду глікогену
ОЗНАКА ОБОХ ПАТОЛОГІЙ – ГІПОГЛІКЕМІЯ!!!
23. Глікогенози та аглікогенози
• Глікогенози - спадкові ензимопатії, викликані порушенням обмінуглікогену, що характеризуються надлишковим накопиченням
глікогену в клітинах. Їх відносять до хвороб накопичення. Також
супроводжуються гіпоглікемією через нездатність глікогену
вивільнювати глюкозу, гепатомегалією, жировою дистрофією печінки,
цирозом. У м’язах – судоми за фізичних навантажень
• Протилежні стани, які характеризуються майже повною відсутністю в
клітинах глікогену, мають назву аглікогенозів. Причиною
аглікогенозів є знижена активність глікогенсинтази печінки або повна
відсутність активності цього ферменту.
24. Глікогенози та аглікогенози
Глікогенози: типиВ залежності від того, дефект якого ферменту є причиною ушкодження обміну
глікогену, розрізняють глікогенози різних типів. Їх поділяють на групи за локалізацією
ферментного дефекта (печінкові, м’язові, мішані).
Хвороба Гірке (І тип) - генетичний дефект глюкозо-6-фосфатази (Іа) або
транслокази Г-6-Ф (Іb).
Хвороба Помпе (ІІ тип) є наслідком недостатності або відсутності лізосомної
кислої α-глюкозидази (γ-амілази), аномальне накопичення глікогену спостерігають
майже в усіх органах хворого (генералізована форма глікогенозу).
Хвороба Корі ( ІІІ тип) - дефект аміло-1,6-глікозидази – фермента, який
розщеплює зв’язки у точках розгалужень в молекулах глікогену, характеризується
накопиченням структурно-аномальних глікогенів – ліміт-декстринів, які можуть
накопичуватися в різних органах, що і визначає існування різних підтипів
захворювання.
Хвороба Андерсена (4-й тип) - недостатність ферменту розгалуження глікогену;
Захворювання Мак-Ардла (5-й тип) - аномальне накопичення глікогену
відбувається лише у скелетних м’язах через дефект фосфорилази м’язів. Підтипи недостатність фосфоглюкомутази, дефект М-субодиниці ЛДГ (піруватемія).
хвороба Герса (6-й тип) – недостатність фосфорилази печінки (6а) або кінази
фосфорилази b (6b);
Хвороба Таруї (7-й тип) – дефект фосфофруктокінази м’язів;
25. Глікогенози: типи
Глікогенози (продовження)Печінкові – уражують метаболічний шлях, яким іде утилізація глікогену для підтримки
рівня глюкози в крові: Іа, Іb, III, IV, VІa, VІb;
М’язові: домінують порушення в енергозабезпеченні скелетних м’язів, що найчастіше
виявляються при фізичному навантаженні і супроводжуються слабкістю,
міоглобінурією, міалгією – V, VII, ІІ
Залежно від появи перших симптомів деякі типи глікогенозів мають форми:
- рання,
- юнацька
- доросла.
Клінічна картина ряду глікогенозів часто схожа із клінічною картиною інших
захворювань:
- гепатитів, пухлин печінки (глікогенози печінкової форми);
- прогресуючою м’язовою дистрофією, міопатіями (м’язові форми
захворювання);
- фіброеластозом ендокарду, міокардитом (генералізовані форми).
Тому кінцевий діагноз може бути поставлений лише після виявлення ензиматичного
дефекту у тканині, яку отримали при біопсії ушкодженого органу.
При генералізованій формі захворювання лікування поки що не є ефективним.
26. Глікогенози (продовження)
Гліколіз – анаеробний процес. Кінцевий його продукт – молочна
кислота. В цьому процесі утворюється АТФ.
• Сумарне рівняння:
С6Н12О6 + 2АДФ + 2ФН –> 2СН3СН(ОН)СООН + 2АТФ + 2Н2О
Глюкоза
→ Молочна кислота
Він забезпечує виконання фізіологічних функцій при нестачі кисню.
Аеробний гліколіз – це гліколітичне розщеплення глюкози до
піровиноградної кислоти – є І стадією окиснення глюкози до
кінцевих продуктів – СО2 і води.
27. Гліколіз
І – фосфорилювання глюкози за рахунок АТФ з утворенням Г6Ф (ферментгексокіназа). Реакція необернена:
α-D-глюкоза + ATФ →Глюкозо-6-фосфат + AДФ
ІІ – перетворення Г6Ф на Ф6Ф ( фермент глюкозо-6-фосфат ізомераза).
D-глюкозо-6-фосфат ↔D-фруктозо-6-фосфат
ІІІ – Ф6Ф фосфорилюється за рахунок другої молекули АТФ з утворенням Ф1,6-дифосфату (фермент фосфофруктокіназа). Реакція необернена. Ця
реакція визначає швидкість всього процесу гліколізу.
D-фруктозо-6-фосфат + ATФ → D-фруктозо-1,6-дифосфат + АДФ
ІV – розщеплення Ф-1,6-дифосфату на 2 фосфотріози (діоксиацетонфосфат
та гліцеральдегід-3-фосфат), фермент альдолаза.
D-фруктозо-1,6-дифосфат ↔ діоксіацетонфосфат + гліцеральдегід-3-фосфат
28.
Стадії гліколізуV – реакція ізомеризації тріозофосфатів (фермент тріозофосфатізомераза).
Рівновага реакції зсунута до утворення диоксиацетонфосфату, але в наступні
реакції гліколізу включається лише гліцеральдегід-3-фосфат, і по мірі його
зменшення диоксиацетонфосфат перетворюється на гліцеральдегід-3фосфат.
діоксіацетонфосфат ↔ гліцеральдегід-3-фосфат
VІ – гліцеральдегід-3-фосфат окиснюється з утворенням 1,3дифосфогліцерату, фермент гліцеральдегідфосфатдегідрогеназа.
Утворена сполука є макроергічною.
Гліцеральдегід-3-фосфат + НАД+ + Н3РО4 ↔ 1,3-дифосфогліцерат + НАДН + Н+
VІІ – передача макроергічного фосфатного залишку від 1,3дифосфогліцерату на АДФ з утворенням АТФ і 3-фосфогліцерату.
Фермент – фосфогліцераткіназа. Це реакція субстратного
фосфорилювання
1,3-дифосфогліцерат + АДФ → 3-фосфогліцерат + АТФ
VІІІ – внутрішньомолекулярний перенос фосфатної групи, яка залишилася, з
утворенням 2-фосфогліцерату. Фермент – фосфогліцеромутаза.
3-фосфогліцерат ↔ 2-фосфогліцерат
29. Стадії гліколізу
ІХ – 2-фосфогліцерат в результаті відщеплення молекуливоди перетворюється на фосфоенолпіровиноградну
кислоту (фосфоенолпіруват), а фосфатний зв’язок у ІІ
положенні стає макроергічним. Фермент – енолаза.
2-фосфогліцерат ↔ фосфоенолпіруват + Н2О
Х – розрив макроергічного зв’язка і перенос фосфатного залишку від
фосфоенолпірувата на АДФ (субстратне фосфорилювання).
Фермент – піруваткіназа. Реакція необернена.
фосфоенолпіруват + АДФ → піруват + АТФ
ХІ – відновлення пірувату до лактату за участю НАДН2,
утвореного в реакції 6. Фермент – лактатдегідрогеназа.
піруват + НАДН + Н+ ↔ L-лактат + НАД+
30. Стадії гліколізу
Біологічне значення гліколізу – утворення
високоергічних сполук (АТФ).
Енергетична цінність гліколізу – 2 молекули АТФ з 1
молекули глюкози.
Необернених реакцій три: №№ 1, 3, 10.
Швидкість гліколізу лімітується реакціями № 3
(основна), 1, 11.
31. Біологічне значення гліколізу
Включення інших вуглеводів у гліколіз: фруктозаА) У м’язах, нирках, жировій тканині фруктоза перетворюється на фруктозо-6фосфат (Ф-6-Ф) (фермент гексокіназа).
•D-фруктоза + АТФ → D-фруктозо-6-фосфат + АДФ
Ф6Ф є проміжним метаболітом гліколізу (утворюється на його ІІ стадії), який
надалі у ІІІ стадії гліколізу фосфорилюється з утворенням фруктозо-1,6дифосфату (фермент 6-фосфофруктокіназа):
АТФ
АДФ
Фруктозо-1,6-дифосфат на IV стадії гліколізу розщеплюється на
діоксиацетонфосфат і гліцеральдегід-3-фосфат (фермент фруктозо-1,6дифосфатальдолаза), вроджене зниження активності може стати причиною
спадкової непереносимості фруктози.
32.
Включення інших вуглеводів у гліколіз:фруктоза
Б) В печінці фермент фруктокіназа фосфорилює фруктозу не по 6-му, а по 1-му
атому вуглецю з утворенням фруктозо-1-фосфату (Ф1Ф).
D-фруктоза + АТФ → D-фруктозо-1-фосфат + АДФ
При вродженій недостачі фруктокінази (хвороба «ессенціальна фруктозурія») в
організмі не утворюється Ф1Ф.
Надалі Ф1Ф розщеплюється під дією фруктозо-1-фосфатальдолази (у ряді
випадків спадкової непереносимості фруктози цей фермент теж може бути
дефектним) на дві тріози - диоксиацетонфосфат і D-гліцеральдегід:
D-фруктозо-1-фосфат → гліцеральдегід + діоксіацетонфосфат
Диоксиацетонфосфат є проміжним продуктом гліколізу, а гліцеральдегід
фосфорилюється з утворенням іншого метаболіту гліколізу - гліцеральдегід-3фосфату (фермент тріозокіназа).
Гліцеральдегід + АТФ → гліцеральдегід-3-фосфат + АТФ
33.
Генет ичні дефект и обміну моноцукрів. Спадкованепереносиміст ь фрукт ози т а есенціальна фрукт озурія
Спадкова непереносиміст ь фрукт ози
• Характеризується ушкодженням печінки і нирок
• Клінічно проявляється судомами, блювотою, іноді коматозними станами.
• Навантаження фруктозою дає різку гіпоглікемію.
• Видалення з раціону фруктозовмісних продуктів дає терапевтичний ефект.
Ессенціальна фруктозурія
• супроводжується фруктоземією, фруктозурією, але не має клінічних проявів. В
зв’язку з тим, що в цьому випадку блокується синтез фруктокінази, порушення
обміну фруктози не супроводжується накопиченням у крові неперетвореного
субстрату ферментативної реакції – фруктозо-6-фосфату, а при навантаженні
фруктозою не спостерігається гіпоглікемія.
34. Генетичні дефекти обміну моноцукрів. Спадкова непереносимість фруктози та есенціальна фруктозурія
Включення інших вуглеводів у гліколіз:галактоза
Спочатку галактоза перетворюється на галактозо-1-фосфат (фермент
галактокіназа):
D-галактоза + АТФ → D-галактозо-1-фосфат + АДФ
Далі у присутності УДФ-глюкози фермент гексозо-1фосфатуридилілтрансфераза каталізує перетворення галактозо-1фосфату на глюкозо-1-фосфат, одночасно утворюється із УДФглюкози синтезується УДФ-галактоза.
D-галактозо-1-фосфат + УДФ-глюкоза → D-глюкозо-1-фосфат + УДФ-галактоза
Спадковий дефект синтезу гексозо-1-фосфат уридилт рансферази
проявляєт ься уродженою ензимопатією — галакт оземією
35.
Включення інших вуглеводів у гліколіз:галактоза
Утворений глюкозо-1-фосфат:
- або перетворюється на глюкозо-6-фосфат (фермент –
фосфоглюкомутаза), що є ключовим метаболітом гліколізу,
- або під впливом фосфатази утворює вільну глюкозу, яка також
надходить у гліколіз
Утворена УДФ-галактоза перетворюється на УДФ-глюкозу (фермент
УДФ-глюкозо-4-епімераза):
УДФ-галактоза → УДФ-глюкоза
Ознаки галактоземії відмічаються також при генетичному дефекті УДФглюкозо-4-епімерази.
УДФ-глюкоза із залученням пірофосфату розщеплюється з утворенням
глюкозо-1-фосфату і УТФ (фермент УДФ-глюкозо-пірофосфорилаза):
УДФ-глюкоза + пірофосфат→ Глюкозо-1-фосфат + УТФ
Глюкозо-1-фосфат надалі йде у загальну схему.
36.
Генетичні дефекти обмінумоноцукрів. Галактоземія
Характерні ознаки:
- гіпоглікемія,
- галактозурія,
- катаракта (через перетворення галактози на галактіол)
- поява і накопичення в крові поряд з галактозою галактозо-1фосфату;
- знижується маса тіла,
- розвиваються жирова дистрофія та цироз печінки,
- затримується психомоторний розвиток.
Виражений терапевтичний ефект дають виключення з
раціону галактози і відповідна дієтотерапія на ранніх етапах
захворювання.
• Генетичні дефекти обміну олігосахаридів (у т.ч. дисахаридів)
асоційовані з порушенням травлення і абсорбції вуглеводів їжі у тонкому
кишечнику (були розглянуті раніше)
37. Генетичні дефекти обміну моноцукрів. Галактоземія
Генетичні дефекти олігоцукрів.Полягають у порушенні розщеплення і всмоктування вуглеводів їжі, що
відбувається головним чином у тонкій кишці.
Відсутність або зниження активності дисахаридаз в слизовій оболонці
тонкої кишки є головною причиною непереносимост і відповідних
дисахаридів, що часто веде до ушкодження печінки і нирок, є причиною
діареї та метеоризму.
Спадкова непереносимість лактози:
- вроджена відсутність лакт ази – ферменту, що міститься в кишечнику
немовлят і забезпечує засвоєння лактози грудного молока, розщеплюючи її
на глюкозу і галактозу;
- симптоми - діарея, метеоризм, кишкові коліки;
Із віком активність лактази в кишечнику зменшується, а у деяких людей
щезає повністю. Лактаза відсутня приблизно у 15% дорослих людей
європейських країн і у 80% східних країн, афроамериканців, індійців тощо.
Дуже часто непереносимість лактози є набутою, тимчасовою: наприклад,
при багатьох кишково-шлункових захворюваннях, інфекційних хворобах,
після резекції шлунку. Після виліковування основного захворювання
недостатність лактази щезає.
38. Генетичні дефекти олігоцукрів.
Діагностика неперенесеннямоно- і олігосахаридів
Метод глюкозного (галактозного, фруктозного тощо) навантаження із побудовою
відповідних кривих (глікемічні криві). Натще перорально вводять вуглевод,
непереносимість якого підозрюють.
Суть навантажувальної проби (для глюкози): у пацієнта натще визначають рівень
глюкози у крові, потім дають йому випити розчин глюкози (1 г/кг маси тіла у 200 мл теплої
води) і через кожні 0,5 годин визначають рівень глюкози у крові. При оцінці побудованих
глікемічних кривих визначають: час максимального підйому, висоту цього підйому,
час повернення концентрації глюкози до вихідного рівня.
У здорової людини через 15 хвилин після вживання глюкози спостерігається збільшення
концентрації глюкози у крові із максимумом у межах першої години (між 30-ю та 60-ю
хвилинами). Після цього починається зниження цукру у крові, яке до 120-ї хвилини може
повернутися до вихідного рівня чи трохи знизитись. Через 3 години після навантаження
глюкозою вміст глюкози у крові завжди повертається до вихідного рівня.
39. Діагностика неперенесення моно- і олігосахаридів
Типи глікемічних кривих за порушеннятолерантності до глюкози і цукрового діабету
Більш точна
діагностика: біопсія
слизової оболонки
кишечнику із
визначенням в
біопсійному матеріалі
активності
дисахаридаз.
Лікування –
виключення з раціону
продуктів, які містять
відповідні дицукри;
застосування
ферментних
препаратів.
18
В
16
14
Вміст глюкози у плазмі крові, ммоль/л
Глікемічні криві у
хворих із
непереносимістю
сахаридів сплощені
порівняно з
нормальними.
А – норма
12
Б – порушення
толерантнос ті до
глюкози
Б
10
8
В – цукровий діабе т
6
А
4
2
0
0
0,5
1
1,5
2
2,5
3
Час, години
Порушення толерантності до глюкози не є
захворюванням – це граничний стан між нормою і ЦД,
фактор ризику.
Таким пацієнтам треба щороку обстежуватися і
дотримуватися лікувального харчування
40. Типи глікемічних кривих за порушення толерантності до глюкози і цукрового діабету
При оцінці побудованих глікемічних кривих звертають увагу
на:
- час максимального підйому,
- висоту цього підйому
- час повернення концентрації глюкози до вихідного
рівня.
Для оцінки глікемічних кривих використовують коефіцієнт
Бодуена:
де А – рівень глюкози в крові натще; В - максимальний вміст
глюкози в крові після навантаження глюкозою.
В нормі цей показник складає біля 50 %.
Значення, що перевищують 80%, свідчать про серйозне
порушення обміну вуглеводів
41.
Це синтез глюкози із невуглеводних продуктів (молочна,
піровиноградна кислоти, глікогенні амінокислоти, гліцерол).
Відбувається у клітинах нирок і печінки.
Більшість стадій – це обернені реакції гліколізу; необернені
реакції йдуть «обхідним» шляхом із залученням інших
ферментів.
42. Глюконеогенез
Обхід реакції №10 (фосфоенолпіруват + АДФ → піруват + АТФ)– синтез фосфоенолпірувату із пірувату - в кілька етапів:
А) у мітохондріях піруват карбоксилюється з утворенням
оксалоацетату (фермент піруваткарбоксилаза):
піруват + СО2 + АТФ → оксалоацетат + АДФ + Фн (регуляторна
реакція)
Б) Мітохондріальна мембрана є непроникною для
оксалоацетату. Оксалоацетат у мітохондріях із
залученням НАДН2 відновлюється до малату (фермент –
НАД-залежна мітохондріальна малатдегідрогеназа).
Малат виходить із мітохондрій. В цитоплазмі малат
окиснюється до оксалоацетату (фермент – НАД-залежна
цитоплазматична малатдегідрогеназа):
оксалоацетат + НАДН + Н+ → малат + НАД+,
малат + НАД+ → оксалоацетат + НАДН + Н+
В) Оксалоацетат у цитоплазмі декарбоксилюється і
фосфорилюється з утворенням фосфоенолпірувату
(фермент фосфоенолпіруват-карбоксикіназа)
оксалоацетат + ГТФ → ФЕП + СО2 + ГДФ
43.
Обхід реакції №3 (фруктозо-6-фосфат + АТФ → фруктозо-1,6-дифосфат+ АДФ): перетворення фруктозо-1,6-дифосфату на фруктозо-6фосфат за участю фруктозодифосфатази
фруктозо-1,6-дифосфат + Н2О → фруктозо-6-фосфат + Фн
(регуляторна реакція)
Обхід реакції №1 (глюкоза + АТФ → глюкозо-6-фосфат + АДФ):
дефосфорилювання Г6Ф ферментом глюкозо-6-фосфатазою:
глюкозо-6-фосфат + Н2О → глюкоза + Фн
Інсулін-індуковане інгібування печінкового глюконеогенезу також
зумовлюється впливом PKB/Akt, а саме, PKB/Akt-опосередкованим
фосфорилюванням транскрипційного фактору FoxO1 (forkhead
box O). Фосфорильований FoxO1 не здатний транслокуватися в ядро
і активувати транскрипцію генів, необхідних для глюконеогенезу,
зокрема, генів фосфоенолпіруваткарбоксикінази та глюкозо-6фосфатази.
44.
Розходження шляхів окиснення вуглеводів – циклу Кребса та ПШ –починається зі стадії утворення глюкозо-6-фосфату (Г6Ф).
Роль ПШ:
• постачання відновленого НАДФН для біосинтезу жирних
кислот, холестеролу і пр., а також для відновлювальніх реакцій
антиоксидантної системи. Утворений НАДФН використовується
у цитозолі на відновні синтези і у нормі не бере участі в окисному
фосфорилюванні у мітохондріях.
• постачання пентозофосфатів для синтезу нуклеїнових кислот і
ряду коферментів.
В печінці, надниркових залозах, ембріональній тканині, молочній
залозі при лактації.
45. Пентозофосфатний шлях окиснення вуглеводів.
Захворювання, викликані порушеннямобміну різних глікокон’югат ів.
• Є наслідком вроджених порушень розпаду гліколіпідів,
глікопротеїнів або глікозаміногліканів (мукополісахаридів) у різних
органах і також належать до хвороб накопичення.
• В залежності від того, яка сполука аномально накопичується в
організмі, розрізняють:
- гліколіпідози,
- глікопротеїнози,
- мукополісахаридози.
46. Захворювання, викликані порушенням обміну різних глікокон’югатів.
Захворювання, викликані порушеннямобміну різних глікокон’югат ів.
Багато з лізосомальних глікозидаз, дефект яких лежить в основі спадкового
порушення вуглеводного обміну, існують в різних молекулярних формах (мають
ізоферменти) і захворювання може бути викликане дефектом якогось одного і
ізоферментів.
Наприклад, хвороба Тея-Сакса (одна із форм гангліозидозів - спадкових
порушень обміну гангліозидів) є наслідком дефекту N-ацетилгексозамінідази
форми А ( гексоамінідази А), у той час коли дефект форм А і В цього
ферменту веде до хвороби Сандгоффа (іншої форми гангліозидозів).
Перебіг більшості хвороб накопичення вкрай тяжкий, багато з них є
невиліковними.
Для діагностики хвороб накопичення в сечі виділяють та ідентифікують різні за
структурою олігосахариди. Для постановки пренатального діагнозу визначають
активність ферментів в культурі клітин із амніотичної рідини, яку отримують при
амніоцентнезі при підозрі на хвороби накопичення.
47. Захворювання, викликані порушенням обміну різних глікокон’югатів.
Мукополісахаридози: структураглікозаміногліканів
Глікозаміноглікани, або мукополісахариди – нерозгалужені полімери, побудовані
із дисахаридних одиниць, які повторюються.
До складу дисахаридної одиниці входять 2 компоненти:
- представник уронових кислот (глюкуронова або ідуронова),
- N-ацетилпохідне гексозамінів (глюкозаміну, галактозаміну)
• Глікозаміноглікани є поліаніонами: наявність карбоксильних і (або) сульфатних
груп забезпечує їх здатність утримувати в біологічних тканинах значну кількість
води (до аніонної групи притягуються іони натрію, які є осмотичноактивними і
“притягують” на себе воду, що веде до збільшенню об”єму води і протидіє
стисканню).
48. Мукополісахаридози: структура глікозаміногліканів
Зв’язки у глікозаміногліканах49. Зв’язки у глікозаміногліканах
Мукополісахаридози: протеогліканиМайже всі глікозамінглікани виконують свої біохімічні та
фізіологічні функції,будучи зв’язаними з білками.
Ковалентні комплекси глікозаміногліканів сполучної тканини
(хондроїтинсульфатів, кератансульфатів тощо) із білками отримали
назву прот еогліканів – на відміну від справжніх глікопротеїнів, у
них вуглеводна частина переважає над білковою
Білкові компонент и різних протеогліканів (інша назва яких – корові
білки) відрізняються між собою; вони поєднуються із
глікозаміногліканами через залишки серину; молекули
хондроїтинсульфату приєднуються до „корового” білку зв’язком між
серином та посередником – ксилозою, яка, не входячи до складу
глікозаміноглікану, служить для його зв’язування із білком. При
гідролізі протеогліканів їх білкова частина розщеплюється
кат епсинами.
50. Мукополісахаридози: протеоглікани
Катаболізм глікозаміногліканівЕкзо- і ендоглікозидази (β-гіалуронідаза, глюкуронідаза,
галактозидаза, ідуронідаза);
Наприклад, β-гіалуронідаза гідролізує β-1,4-глікозидний зв язок
між дисахаридними одиницями гіалуронової кислоти з
утворенням дисахариду – глюкуронова кислота (β1–>3) Nацетилглюкозамін. Цей дисахарид надалі гідролізується під
впливом лізосомальної β-глікозидази.
Хондроїтин-сульфати теж здатні розщеплюватися під впливом βгіалуронідази.
Сульфатази
Із позаклітинного простору глікозаміноглікани надходять у клітину
(ендоцитоз). Надалі лізосомальні гідролази поступово розщеплюють
їх до мономерів.
51. Катаболізм глікозаміногліканів
МукополісахаридозиМПС пов’язані зі спадковою недостатністю ферментів розщеплення
вуглеводних компонентів протеогліканів
МПС І – дефект α-L-ідуронідази (фермента, що каталізує гідроліз зв язка між
несульфатованою L-ідуроновою кислотою в дермантан- і гепарансульфатах (в сечі
виявляють дермантан- і гепарансульфати);
МПС ІІ – дефект ідуроносульфатсульфатази (в сечі виявляють дермантан- і
гепарансульфати);
МПС ІІІ – гетерогенна група захворювань (дефекти гепарансульфатсульфатази
(сульфамідази) (типА), α-N-ацетил-глюкозамінідаза (тип В), глюкозамін-Nацетилтрансфераза (тип С), глюкозамін-6-сульфатаза (тип D), Nсульфоглюкозамін-3-О-сульфатаза (тип Е) - в сечі виявляють гепарансульфати;
МПС ІV – дефект N-ацетилгексозамін-6-сульфатсульфатази або βгалактозидази (в сечі виявляють кератансульфати);
МПС VІ – дефект арилсульфатази В (в сечі виявляють дермантансульфати);
МПС VІІ – дефект β-глюкуронідази (в сечі виявляють дермантансульфати);
Діагностика базується на визначенні активності лізосомальних гідролаз.
Не підлягають лікуванню