





")
")




")













 Медицина
МедицинаПохожие презентации:



")

")




Липодистрофии. Патогенез и клинические проявления липодистрофий
1. Липодистрофии
ФГАОУ ВО Первый Московский Государственный Университетим. И.М. Сеченова Министерства Здравоохранения РФ
(Сеченовский Университет)
СНК Эндокринологии
Липодистрофии
Докладчик:
Шумская Юлия
лечебный факультет 6 курс 29 группа
Научные руководители СНК:
доцент кафедры, к.м.н. Моргунова Т.Б.
ассистент кафедры, к.м.н. Рунова Г.Е.
2. Липодистрофии
Гетерогенная группа редкихзаболеваний, характеризующихся
полной или частичной потерей
подкожной жировой клетчатки, а также
неправильным ее распределением (при
условии отсутствия предшествующего
голодания или катаболического
состояния) и предрасположенностью к
развитию связанных с
инсулинорезистентностью
метаболических осложнений.
3. Эпидемиология
США 1:1 000 000 [Lightbourne M., Brown R.J.]1 - 60:1 000 000 [Brown R.J., Rother K.I.]
был описан 1141 пациент в 351
исследовании с дебютом (на май 2016г)
[Gupta N. et al.]
4. Классификация
По степени потери жировой ткани• Генерализованные
• Парциальные
По этиологии
• Наследственные
• Приобретенные
[Handelsman Y. et al., 2013]
5.
Врожденные генерализованные липодистрофииПриобретенные генерализованные липодистрофии
Семейные парциальные липодистрофии
Приобретенные парциальные липодистрофии
Липодистрофии в рамках других генетических
синдромов
Локальные липодистрофии
ВААРТ-индуцированная липодистрофия у ВИЧинфицированных пациентов
Handelsman Y. et al.; Hussain I., Garg A.; Thauvin-Robinet C. et al.
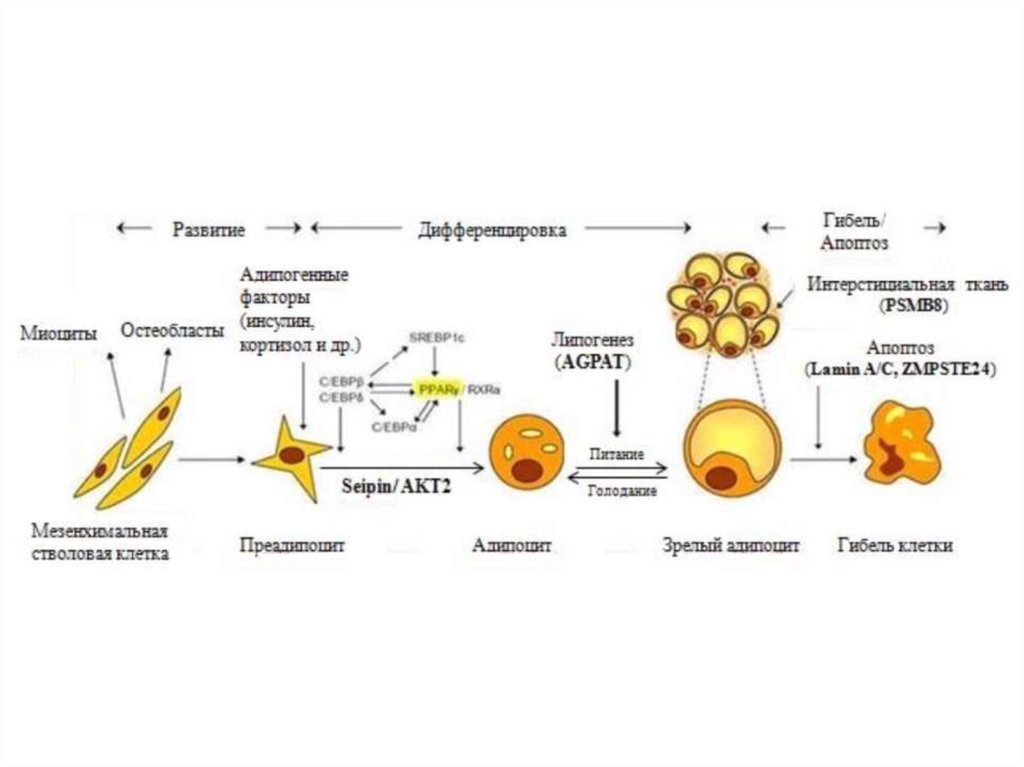
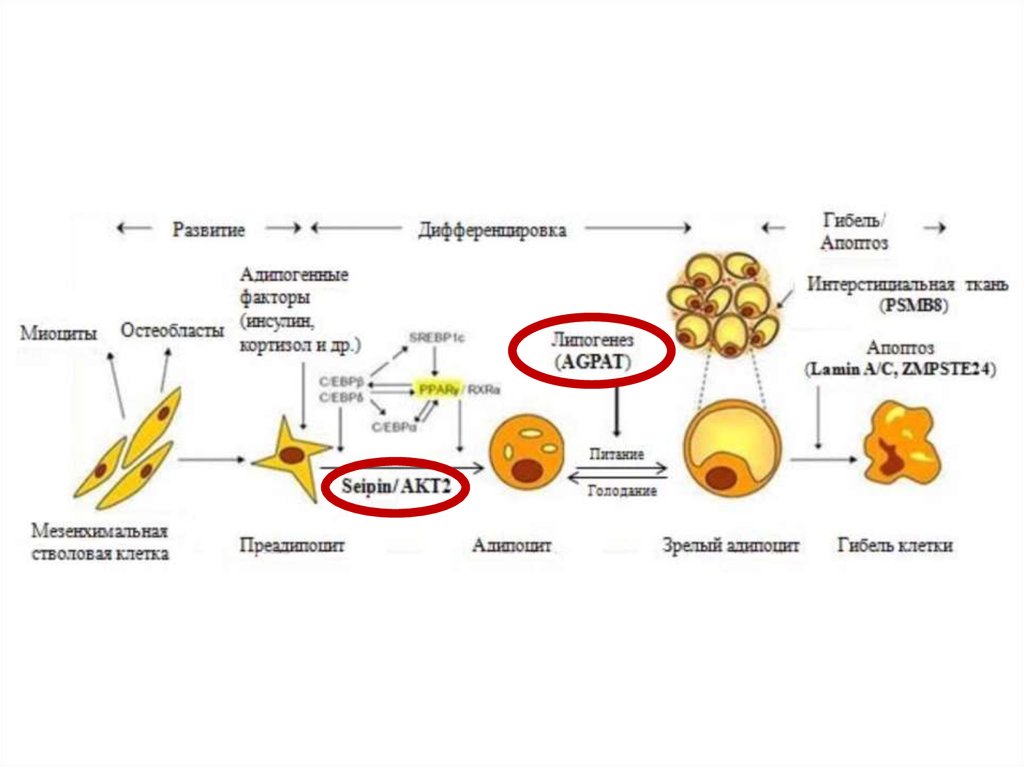
6. Патогенез и клинические проявления липодистрофий
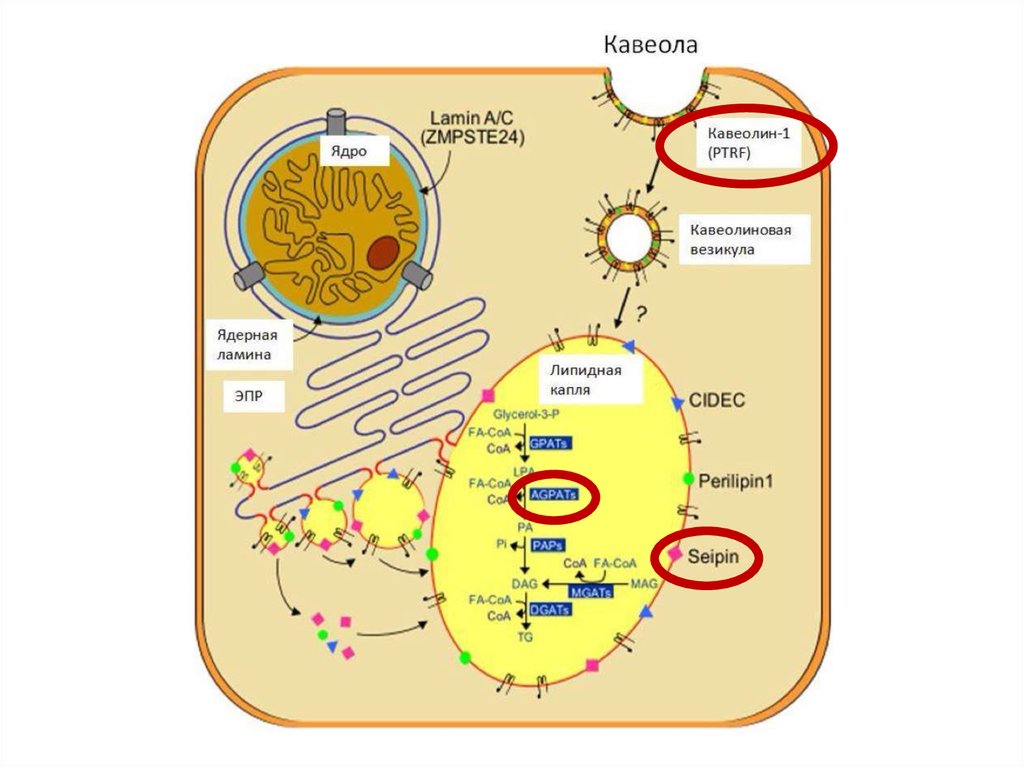
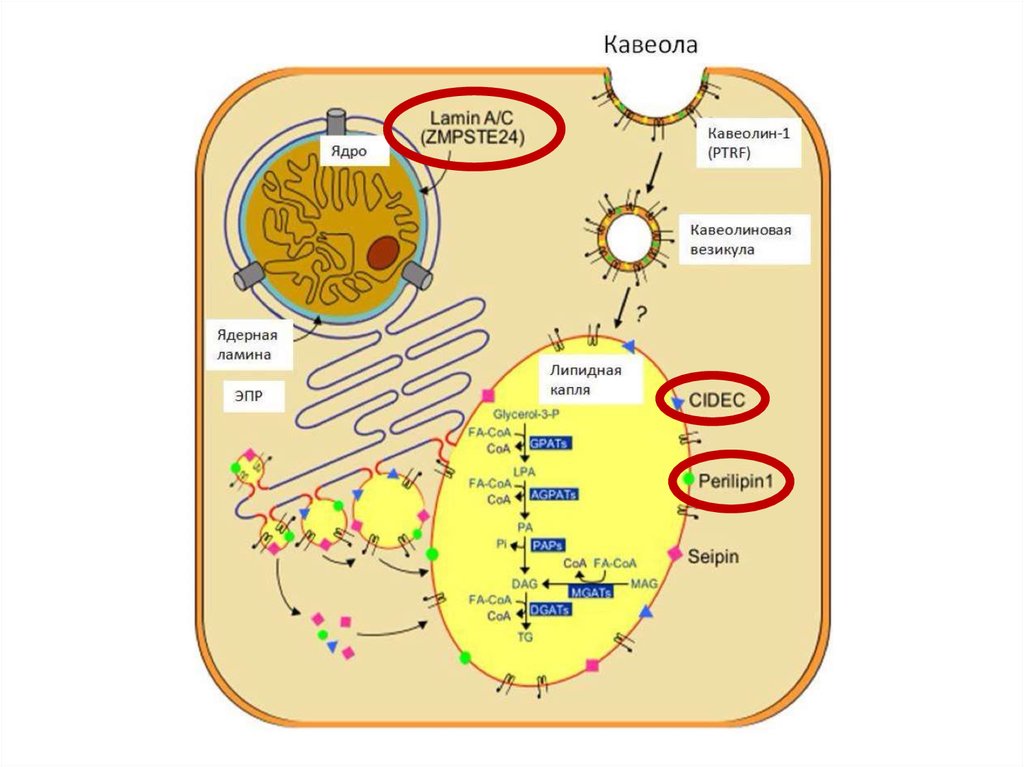
нарушение адипогенеза на различных уровнях7.
8.
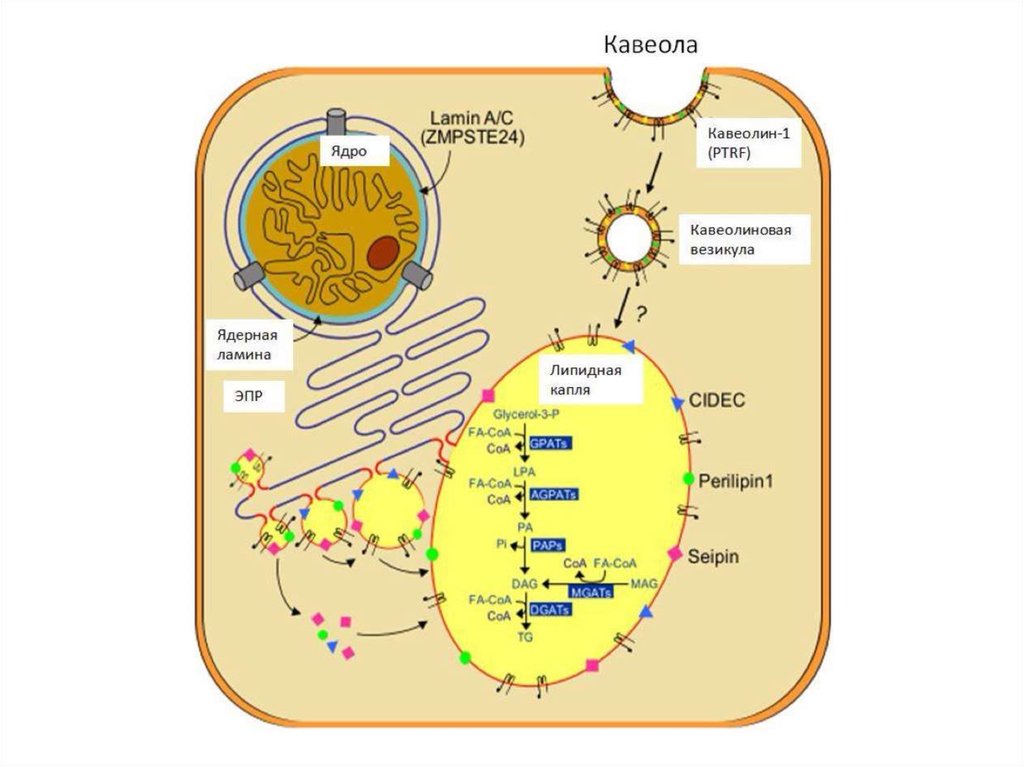
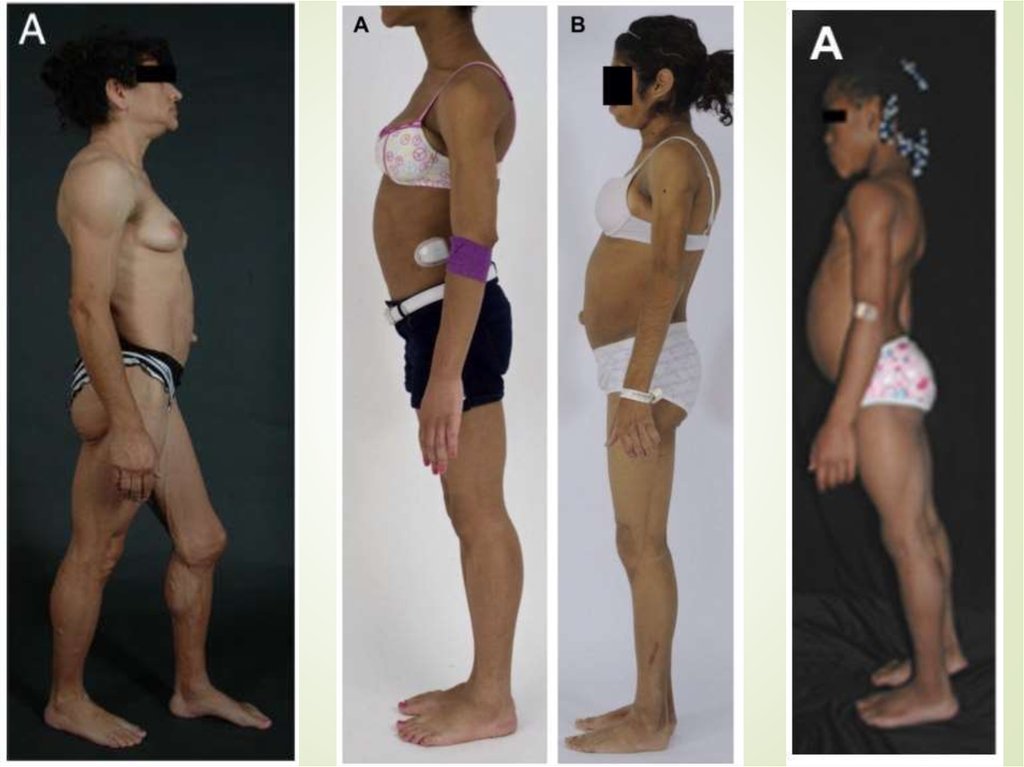
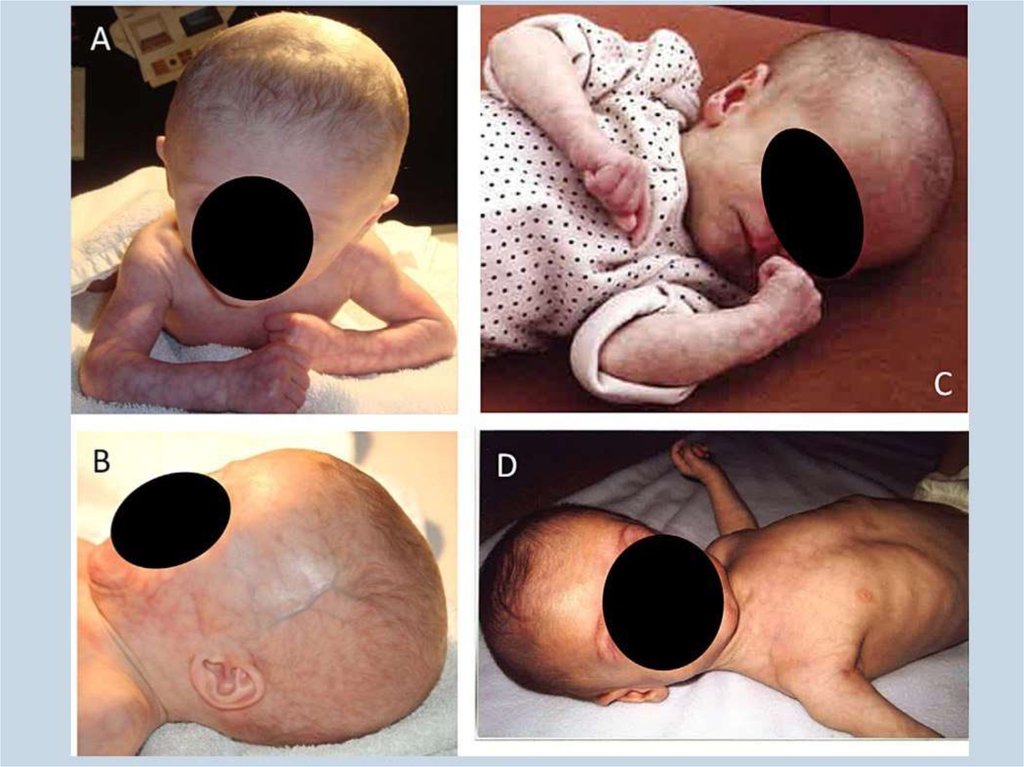
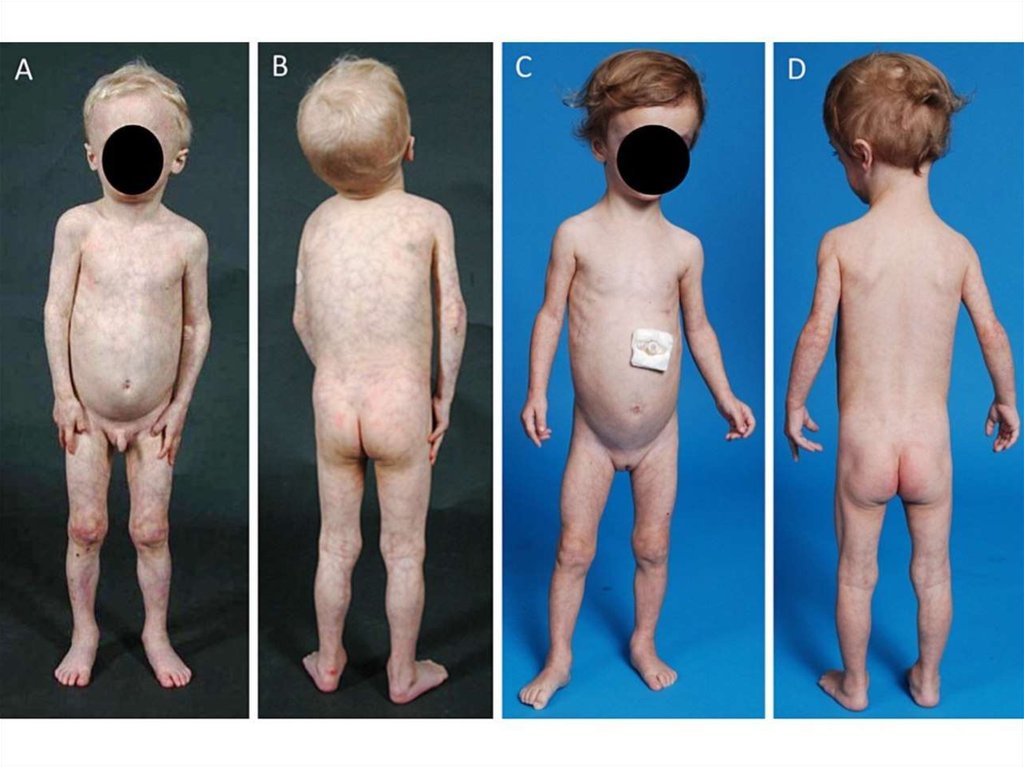
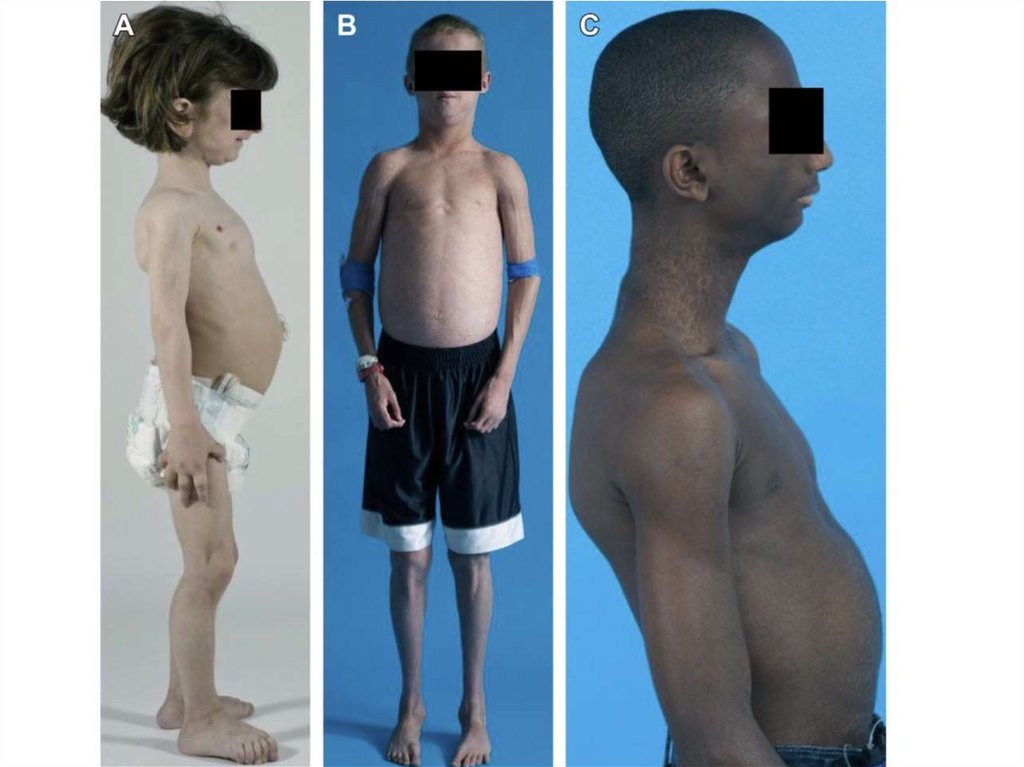
9. Врожденная генерализованная липодистрофия (синдром Берардинелли-Сейпа)
Полное отсутствие ПЖКВыраженная рельефность скелетных мышц
Усиленный венозный рисунок на конечностях
Пупочная грыжа
Гепато- и спленомегалия
Гиперфагия
Акромегалоидные черты
Инсулинорезистентность
НАЖБП
Гиперандрогения
10.
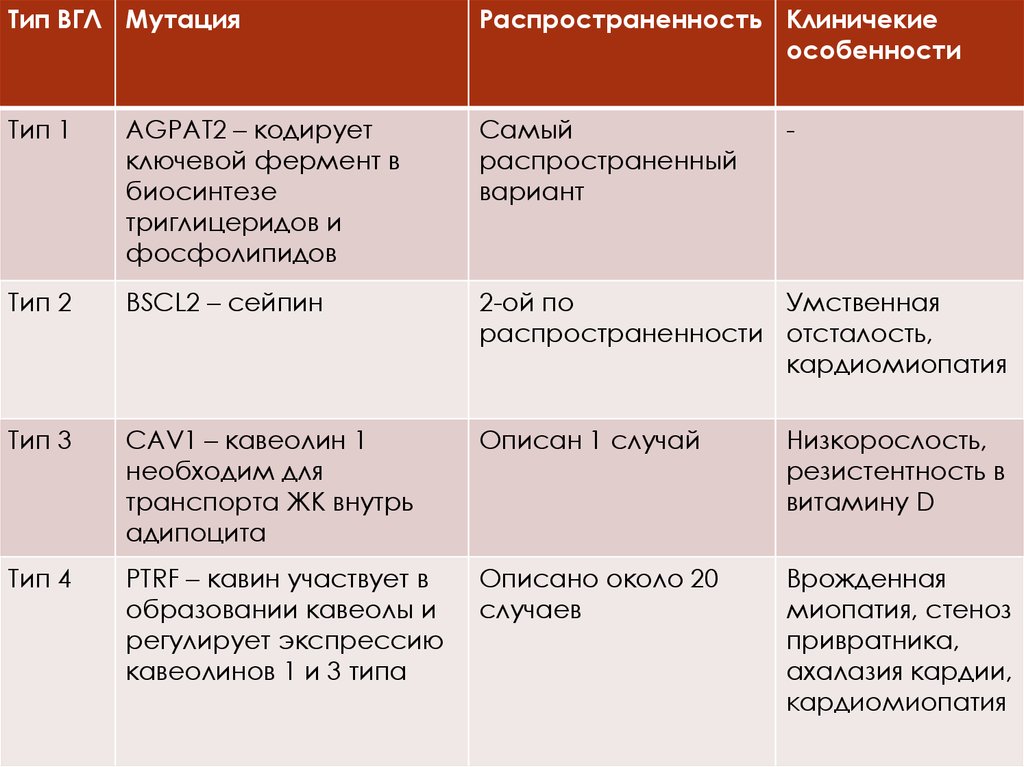
Тип ВГЛ МутацияРаспространенность Клиничекие
особенности
Тип 1
AGPAT2 – кодирует
ключевой фермент в
биосинтезе
триглицеридов и
фосфолипидов
Самый
распространенный
вариант
Тип 2
BSCL2 – сейпин
2-ой по
Умственная
распространенности отсталость,
кардиомиопатия
Тип 3
CAV1 – кавеолин 1
необходим для
транспорта ЖК внутрь
адипоцита
Описан 1 случай
Низкорослость,
резистентность в
витамину D
Тип 4
PTRF – кавин участвует в
образовании кавеолы и
регулирует экспрессию
кавеолинов 1 и 3 типа
Описано около 20
случаев
Врожденная
миопатия, стеноз
привратника,
ахалазия кардии,
кардиомиопатия
-
11.
12.
13.
14.
15.
16.
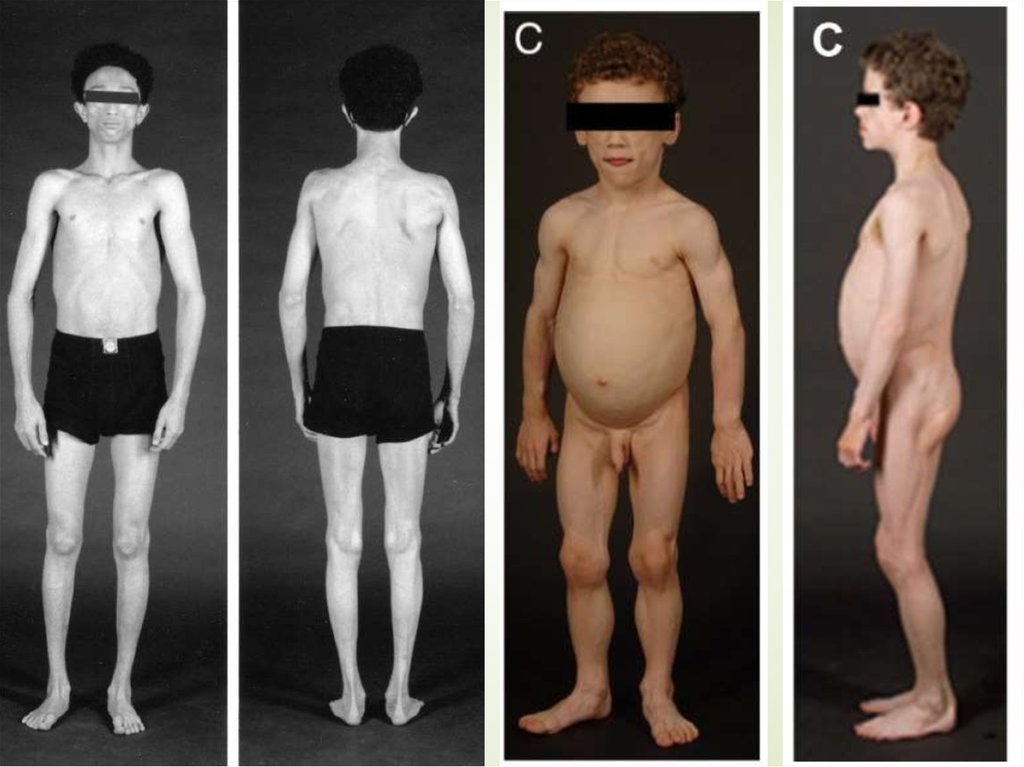
17. Приобретенная генерализованная липодистрофия (синдром Лоуренса)
Дебют в детском или подростковом возрастеПрогрессирующее постепенное исчезновение
ПЖК
Ж:М 3:1
Жировая ткань костного мозга и
ретроорбитальная жировая клетчатка не
страдают
Степень потери внутрибрюшного жира
варьирует
Инсулинорезистентность
18.
ПодтипРаспространенность
Клинические особенности
Ассоциированная
с панникулитом
~ 25%
Начальная стадия панникулита
сопровождается потерей
жировой клетчатки в локально в
местах воспаления.
Продолжающееся воспаление
приводит к генерализованной
потере ПЖК
Аутоиммунная
~ 25%
Постепенная повсеместная
потеря подкожной жировой
клетчатки, ассоциированная с
аутоиммунными
заболеваниями, в особенности
с ювенильным
дерматомиозитом. Некоторые
пациенты имеют сниженную
концентрацию С4
Идиопатическая
~ 50%
Постепенная повсеместная
потеря подкожной жировой
клетчатки неясной этиологии
19.
20.
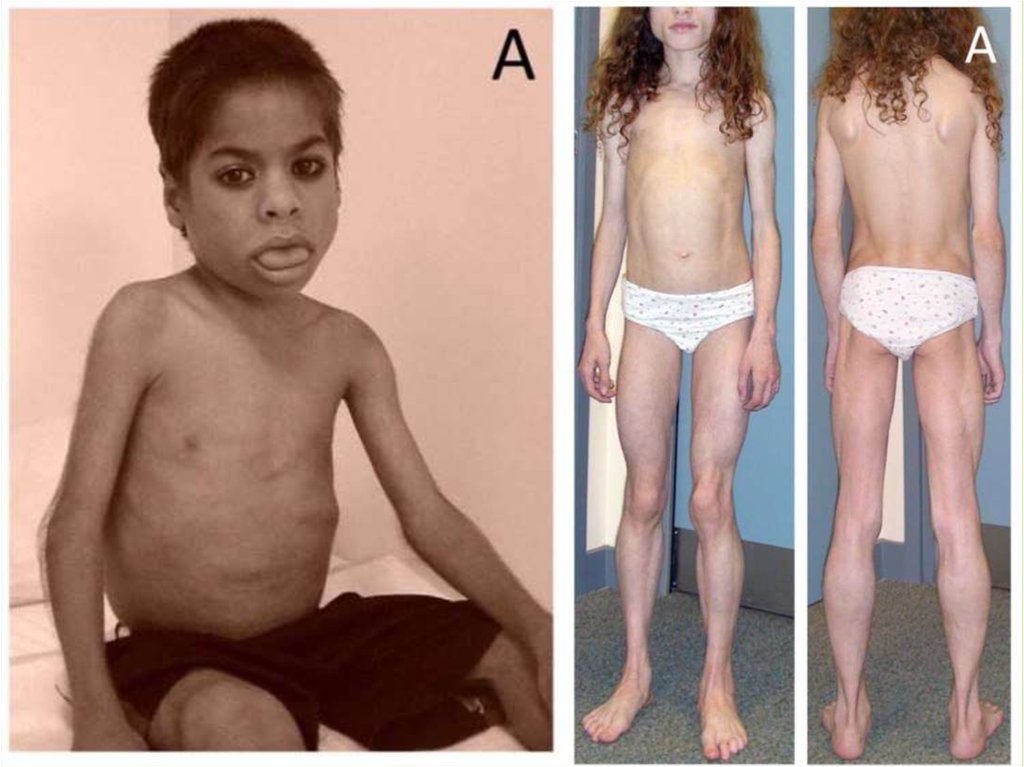
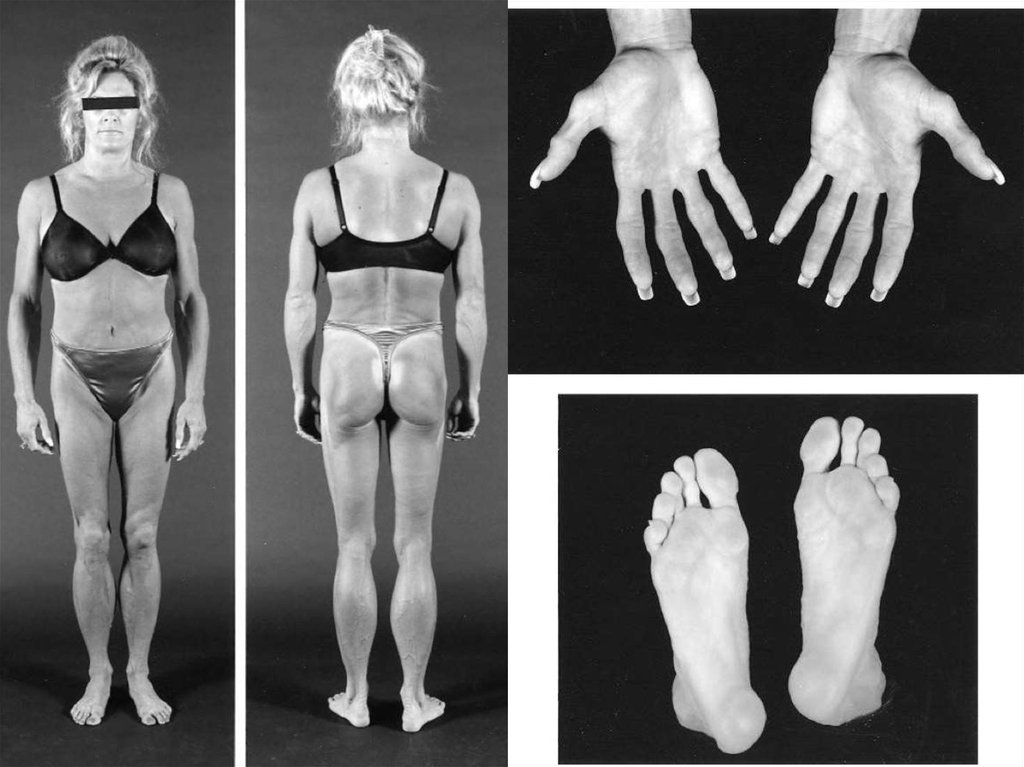
21. Семейные парциальные липодистрофии
Дебют в детском или подростковом возрастеПотеря ПЖК в специфических анатомических
областях с перераспределением ее в другие
области
Инсулинорезистентность
Гиполептинемия
НАЖБП
Дислипидемия
22.
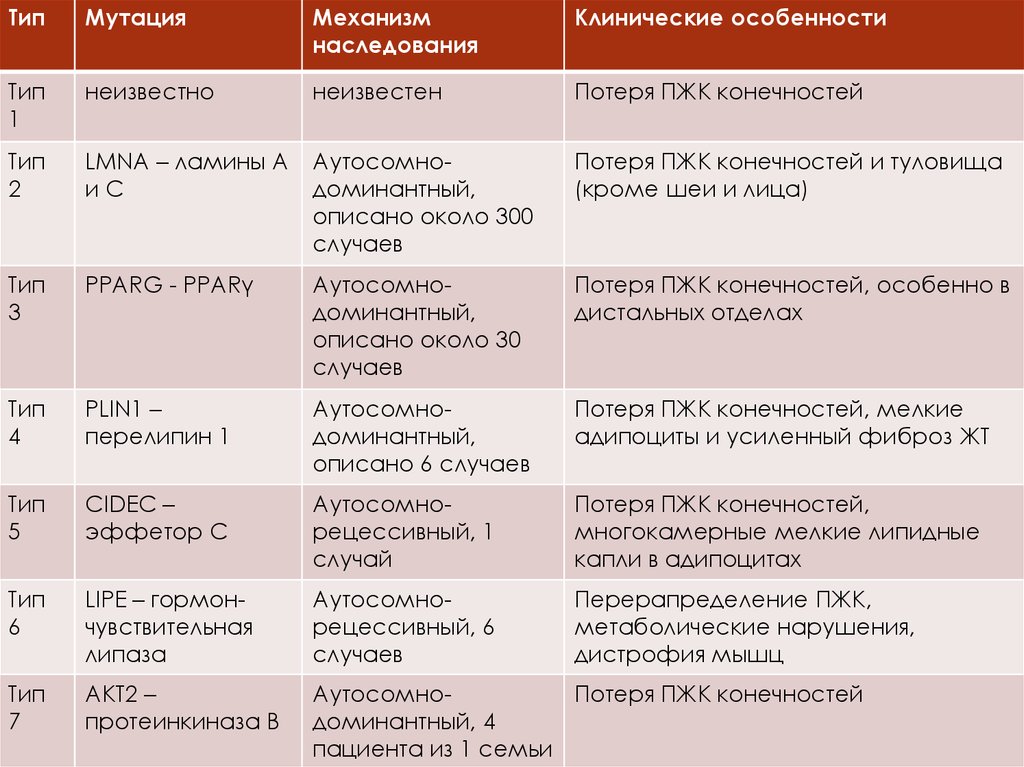
ТипМутация
Механизм
наследования
Клинические особенности
Тип
1
неизвестно
неизвестен
Потеря ПЖК конечностей
Тип
2
LMNA – ламины А
иС
Аутосомнодоминантный,
описано около 300
случаев
Потеря ПЖК конечностей и туловища
(кроме шеи и лица)
Тип
3
PPARG - PPARγ
Аутосомнодоминантный,
описано около 30
случаев
Потеря ПЖК конечностей, особенно в
дистальных отделах
Тип
4
PLIN1 –
перелипин 1
Аутосомнодоминантный,
описано 6 случаев
Потеря ПЖК конечностей, мелкие
адипоциты и усиленный фиброз ЖТ
Тип
5
CIDEC –
эффетор С
Аутосомнорецессивный, 1
случай
Потеря ПЖК конечностей,
многокамерные мелкие липидные
капли в адипоцитах
Тип
6
LIPE – гормончувствительная
липаза
Аутосомнорецессивный, 6
случаев
Перерапределение ПЖК,
метаболические нарушения,
дистрофия мышц
Тип
7
AKT2 –
протеинкиназа В
АутосомноПотеря ПЖК конечностей
доминантный, 4
пациента из 1 семьи
23.
24.
25.
26. Приобретенная парциальная липодистрофия (синдром Барракера-Симонса)
Прогрессирующая потеря ПЖК в верхнейчасти туловища
Метаболические нарушения менее
характерны
Часто ассоциирована с заболеваниями почек
Низкий уровень С3
Ж:М 4:1
27.
28. ВААРТ-ассоциированная липодистрофия у ВИЧ-инфицированных пациентов
29. Генетические синдромы, сопровождающиеся липодистрофией
Истинная прогерия Хатчинсона-ГилфордаСиндром Вернера
Атипичный синдром Вернера
Мандибуло-акральная дисплазия
Прогероидный синдром
Синдром Нестера-Гильермо
Синдром Марфана с липодистрофией
Синдром Keppen-Lubinsky
SHORT-синдром
CANDLE-синдром
30.
31. Локальные липодистрофии
32. Диагностика
АнамнезФизикальный осмотр
Оценка состава тела
Метаболический статус
Генетическое исследование в половине случаев не
информативно
Методика оценки сывороточного лептина не
стандартизирована
При подозрении на приобретенную ЛД исследуется
С3, С4 и АТ в зависимости от фонового
аутоиммунного заболевания
[Rother K.I., Brown R.J. 2013], [Dagogo-jack S. 2015], [Valerio C.M. et al., 2012]
33. Подход 1 + 1 = клинический диагноз
Основной критерий: потеря ПЖК по типу парциальной илигенерализованной ЛД (с уточнением для СПЛ)
Дополнительные критерии:
СД с выраженной ИР
Выраженная гипертриглицеридемия
НАЖБП
Семейный анамнез
Выраженность скелетной мускулатуры и флебомегалия
на конечностях
Гиперфагия
Вторичный гипогонадизм у мужчин/аменорея у женщин
Handelsman Y. et al., an AACE consensus statement, 2016
34. Дифференциальная диагностика
ИстощениеНервная анорексия
Декомпенсированный СД
Тиреотоксикоз
Надпочечниковая недостаточность
Хронические инфекции
Гиперинсулинизм вследствие мутации INSR
Акромегалия
Синдром Кушинга
Абдоминальное ожирение
Brown R.J., Araujo-Vilar D., Pik To Cheung., Dunger D., Garg A., 2016
35. Метаболические осложнения
Липоатрофический диабетГиперфагия
Дислипидемия
НАЖБП
Репродуктивные нарушения
Сердечно-сосудистые заболевания
Патология почек
Brown R.J., Araujo-Vilar D., Pik To Cheung., Dunger D., Garg A., 2016
36. Основные причины смерти
Заболевания сердца
Кардиомиопатии
Аритмии
Сердечная недостаточность
Инфаркт миокарда
Заболевания печени
Печеночная недостаточность
Желудочно-кишечные кровотечения
Гепатоцелюллярная карцинома
Почечная недостаточность
Острый панкреатит
Сепсис
Brown R.J., Araujo-Vilar D., Pik To Cheung., Dunger D., Garg A., 2016
37. Принципы лечения
Строгая антиатерогенная диетаРегулярные физические нагрузки
Заместительная терапия адипокинами
(метрелептин)
Терапия СД с использованием
тиазолиндионов
Плазмаферез
Бариатрическая хирургия
Пластическая хирургия
Brown R.J., Araujo-Vilar D., Pik To Cheung., Dunger D., Garg A., 2016
38.
ФриденштейнАлександр
Яковлевич
(1924 – 1997)