

")
")



")



")






















")

")













 Медицина
МедицинаПохожие презентации:




. Геморрагические диатезы")





Нарушение системы гемостаза
1. Нарушение системы гемостаза.
Лекция 82. КЛАССИФИКАЦИЯ НАРУШЕНИЙ ГЕМОСТАЗА
по этиологии1) НАСЛЕДСТВЕННЫЕ
2) ПРИОБРЕТЕННЫЕ
по механизму развития
1) НАРУШЕНИЯ СОСУДИСТО-ТРОМБОЦИТАРНОГО
ГЕМОСТАЗА
2) НАРУШЕНИЯ КОАГУЛЯЦИОННОГО ГЕМОСТАЗА
по направленности изменений
1) ГИПОКОАГУЛЯЦИЯ
2) ГИПЕРКОАГУЛЯЦИЯ
3. Нарушение системы гемостаза (по механизму развития)
I. Гемостазиопатии - заболевания связанные с нарушением первичногоили вторичного гемостаза.
• 1) геморрагические
1.1) заболевания связанные с патологией тромбоцитов
(тромбоцитопении, тромбастении)
1.2) с патологией сосудистой стенки (геморрагические вазопатии)
1.3) дефектом плазменных факторов свертывания крови (гемофилии и
другие коагулопатии).
• 2) тромбофилические
тромбозы и тромбоэмболии (дефицит антикоагулянтов, угнетение
плазминной системы крови, атеросклероз, повышенная вязкость
крови).
• 3)смешанного характера (тромбогеморрагические).
приобретенный характер и возникают в результате разбалансирования
системы гемостаза на разных уровнях (ДВС-синдром).
II. Коагулопатии - при нарушениии только процесса свертывания крови.
4. Нарушение системы гемостаза (по направленности изменений)
ГИПОКОАГУЛЯЦИЯГИПЕРКОАГУЛЯЦИЯ
- cнижение способности крови
сворачиваться с появлением
склонности
к
повторным
кровотечениям и кровоизлияниям (спонтанным или после
незначительных травм)
• повышенная
способность
крови образовывать сгустки в
сосудах
1. ТРОМБОЦИТОПЕНИЯ
2. ТРОМБОЦИТОПАТИЯ
1.
2.
3. ВАЗОПАТИЯ
4. КОАГУЛОПАТИЯ
ТРОМБОЗ
ДВС-СИНДРОМ
5. ТРОМБОЦИТОПЕНИЯ
• Патологическое состояниекоторое характеризуется
понижением содержания
тромбоцитов в крови
(меньше 150·109 /л)
6. НАСЛЕДСТВЕННЫЕ ТРОМБОЦИТОПЕНИИ
Врожденные дефекты тромбоцитов или ихобразования ( основная роль принадлежит мутациям).
• синдром Вискотта-Олдрича
Наследственное заболевание, обусловленное генетическими мутациями, в результате которых в
красном костном мозге образуются аномальные, маленькие (менее 1 микрометра в диаметре)
тромбоциты. Из-за нарушенной структуры происходит их чрезмерное разрушение в селезенке, в
результате чего срок их жизни сокращается до нескольких часов.
• синдром Бернара – Сулье
Наследственное аутосомно-рецессивное заболевание, проявляющееся в раннем детском
возрасте. Характеризуется образованием гигантских (6 – 8 микрометров), функционально
несостоятельных тромбоцитов. Они неспособны прикрепляться к стенке поврежденного
сосуда и связываться друг с другом (нарушены процессы адгезии и агрегации) и подвергаются
усиленному разрушению в селезенке.
• врожденная амегакариоцитарная тромбоцитопения
Наследственное аутосомно-рецессивное заболевание, проявляющееся в младенческом возрасте.
Характеризуется мутации гена, отвечающего за чувствительность мегакариоцитов к фактору,
регулирующему их рост и развитие (тромбопоэтину), в результате чего нарушается продукция
тромбоцитов костным мозгом.
7. ПРИОБРЕТЕННЫЕ ТРОМБОЦИТОПЕНИИ
• ПОВРЕЖДЕНИЕ ТРОМБОЦИТОВ- имунными комплексами (Нп, Идиопатическая
тромбоцитопеническая пурпура )
• УГНЕТЕНИЕ ОБРАЗОВАНИЯ ТРОМБОЦИТОВ
(апластическая анемия, химическое и
радиационное повреждение красного костного
мозга, замещение кроветворной ткани опухолью)
• ПОВЫШЕННОЕ ИСПОЛЬЗОВАНИЕ
ТРОМБОЦИТОВ= Тромбоцитопении
потребления
(тромбоз, ДВС-синдром )
8. Идиопатическая тромбоцитопеническая пурпура (ИТП)
ИТП=аутоиммунная тромбоцитопения- характеризуетсяснижением количества тромбоцитов в периферической
крови (состав других клеточных элементов крови не
нарушен) в результате усиленного их разрушения.
Две формы:
• острая, способна прекращаться спонтанно (свойственна
детскому возрасту и не требует специфической терапии)
• хроническая, типична для взрослых, особенно для женщин
в возрасте 20-40 лет.
• Основные клинические симптомы: кровоизлияния в коже,
слизистых оболочках пурпурного цвета. Кровотечения,
кровоизлияния появляются неожиданно, часто после
простуды.
• При лабораторной диагностике количество тромбоцитов
падает, вплоть до полного исчезновения.
9. Идиопатическая тромбоцитопеническая пурпура
10. ТРОМБОЦИТОПАТИИ
• Нарушенние гемостаза вследствиекачественной неполноценности или
дисфункции тромбоцитов, которое
характеризуется нарушением сосудистотромбоцитарного гемостаза, появлением
кровоточивости тканей и органов.
• Тромбоцитопатии – одна из наиболее частых причин
кровоточивости «неясного генеза», при которых количество
тромбоцитов и показатели свертывающей системы крови, как
правило, в норме. Характеризуется микроциркуляторным типом
кровотечения при небольших порезах, после удаления зубов,
опасны кровотечения в сетчатку глаза, головной мозг.
11. Наследственные тромбоцитопатии
• Болезнь Гланцмана:*Причина: дефицит гликопротеинов IIb/IIIa мембран тромбоцитов
Наследование: аутосомно-рецессивное
*Патогенез - тромбоциты не взаимодействуют с фибриногеном и не
агрегируют (фибриноген связывает тромбоциты в агрегаты через гликопротеид
Ilb/IIIa )
*Признаки: петехии, носовые кровотечения, маточные кровотечения
• Болезнь Бернара Сульє (АР)
• Причина – отсутствие гликопротеина Ib на тромбоцитах
• Патогенез – нарушено взаимодействие тромбоцитов с фактором
Виллебранда, фактором V и IX
• Признаки – капиллярные кровотечения (особенно опасны при половом
созревании или родах)
• Сюда же отнесятся тромбоцитопатии, сопровождающиеся тромбоцитопенией
(синдром Вискотта-Олдрича, синдром Бернара-Сулье и
др.)
• Лечение - переливание донорских тромбоцитов.
12. Болезнь Бернара Сульє (АР)
* Наследственное аутосомнорецессивное• гигантские (6 – 8
микрометров),
функционально
несостоятельные
тромбоциты
• усиленному разрушение в
селезенке
*отсутствие или снижение
экспрессии гликопротеинового
комплекса Ib на поверхности
тромбоцитов.
* недостаточное связывание
фактора Виллебранда развивается дефектная
адгезия тромбоцитов
13. Приобретенная тромбоцитопатия
• 1. Лейкозы- мало гранул в тромбоцитахвследствие ускоренного созревания, снижена
адгезия и агрегация
• 2. Иммунные болезни – повреждение рецепторов
иммунными комплексами, нарушение
взаимодействия тромбоцитов с прокоагулянтами
• 3. Гиповитаминоз В12 – нарушено освобождение
гранул
• 4. Медикаментозные влияния (аспирин блокирует
агрегацию тромбоцитов на 4-6 дней)
14. ВАЗОПАТИЯ
• Геморрагический диатез обусловленныйфункциональной и морфологической
неполноценностью сосудистой стенки
- врожденная
- приобретенная
Гемморагические вазопатии могут быть
обусловлены дистрофическими
изменениями и наследственной
патологией сосудистой стенки, ростом
опухолей, инфекционными процессами и
др.. Наиболее распространненная форма
патологии –геморрагический васкулит.
15. Геморрагический васкулит
• Это системное сосудистое заболеваниеиммунного генеза с поражением артериолл,
капилляров, венул комплексами антигенантитело.
• Заболевание встречается в детском и юношеском
возрасте и характеризуется геморрагической
сыпью в основном на конечностях.
• При лабораторном исследовании количество и
качество тромбоцитов не изменено, ретракция
кровяного сгустка в норме, может удлиняться
время капиллярного кровотечения вследствие
нарушения проницаемости сосудистой стенки.
16. Геморрагический васкулит
17. Клиника геморрагического васкулита
18. КОАГУЛОПАТИЯ
• Геморрагический диатез , которыйвозникает в результате патологии
коагуляционной системы гемостаза
** наследственная
** приобретенная
19. НАСЛЕДСТВЕННАЯ КОАГУЛОПАТИЯ
• Генетически обусловленное нарушениесвертывания крови вследствие дефицита
или молекулярной аномалии веществ,
отвечающих за коагуляционный гемостаз
• Среди всех форм коагулопатий страдают:
Гемофилией А
68 – 78%
Болезнь Виллебранда
9 – 18 %
Гемофилией В
6 – 13 %
Гемофилией С, парагемофилией и
гипопроконвертинемией 1 – 2 %
20. НАСЛЕДСТВЕННАЯ КОАГУЛОПАТИЯ
• КЛАССИФИКАЦИЯ1. Коагулопатия вследствие изолированного нарушения
внутреннего механизма формирования протромбиназной
активности (гемофилии А, В, С, Болезнь Виллебранда,
дефицит Хагемана)
2. Коагулопатия вследствие изолированного нарушения
внешнего механизма формирования протромбиназной
активности (гипопроконвертинемия – дефицит VII ф.)
3. Комбинированное нарушение внешнего и внутреннего
механизма формирования протромбиназной активности
(парагемофилия – дефицит V ф., б.Стюарта-Прауэра –
дефи-цит X ф.)
4. Нарушение конечной стадии свертывания крови
(афибриногенемия)
21. Гемофилии
относится к коагулопатиям с непосредственным нарушением гемостаза, для которых
характерна изолированная непостоянность одного какого - либо фактора свертывания
крови.
1. Гемофилия А /классическая гемофилия/
обусловлена дефицитом фактора VIII
составляет 70 - 80 % от всех случаев заболевания,
2. Гемофилия В /болезнь Кристмаса,
рождественская болезнь/
связана с дефицитом фактора IX.
составляет 6 -13% от всех случаев заболевания
На дефицит этих двух факторов свертывания приходится 96 - 98 % всех
наследственных коагулопатий. Гены регулирующие синтез факторов VIII и IX,
локализуются в X – хромосоме.
3. Гемофилия С (Б-нь Розенталя)
связана с дефицитом ХI фактора.
Гемофилия С наследуеся аутосомно, ею болеют мужчины и женщины.
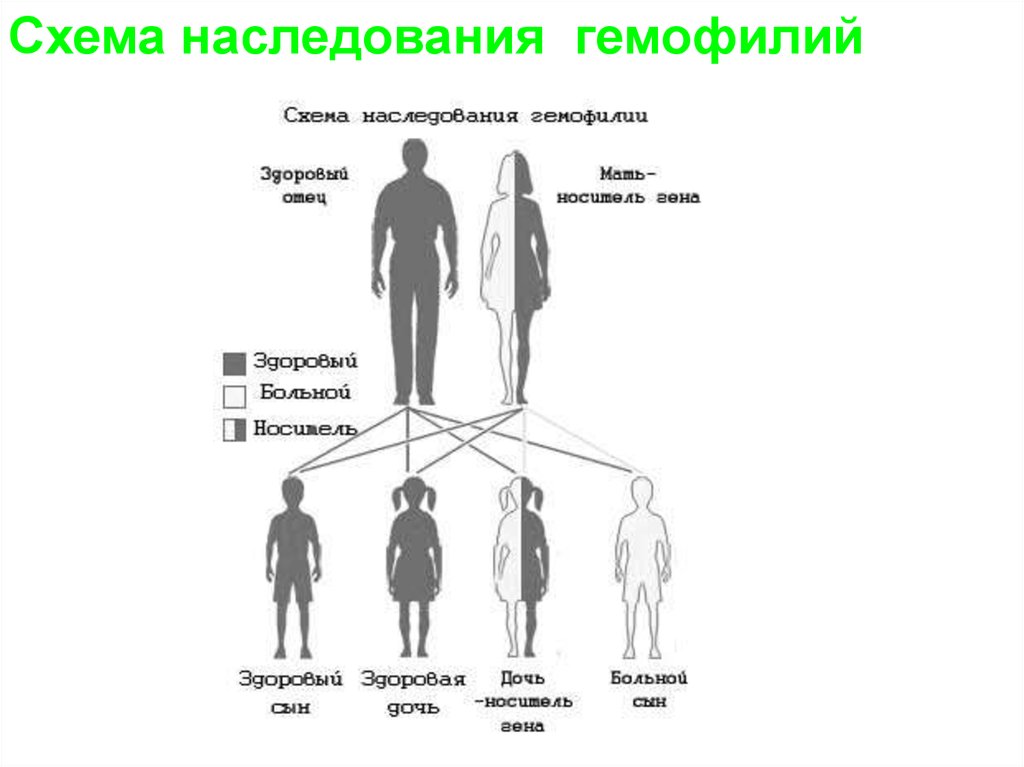
22.
Схема наследования гемофилийзаболевания.
23.
Потомки королевы ВикторииИмператрица Александра Федоровна и цесаревич
Алексей. У Алексея была наследственная
гемофилия, которой страдали мужчины в роду его
матери. 1913 г.
Потомки королевы Виктории страдали
гемофилией. Считается, что болезнь
была передана династии Викторией
(1819-1901), которая произвела на свет
девятерых детей.
24. Клинические проявления гемофилии
• Клинически отдельные формы гемофилии протекают одинаково,проявляясь обильными и продолжительными кровотечениями при
порезах и травмах, кровоизлияниями в суставы, возникновением
мышечных и внутримышечных гематом. Выраженность кровоточивости
зависит от формы гемофилии: тяжелые формы заболевания чаще
наблюдаются при гемофилии А, Гемофилия В обычно протекает легче.
Заболевание выявляется в детском возрасте. Течение заболевания
характеризуется периодами повышенной кровоточивости,
сменяющимися промежутками относительного клинического
благополучия. Одно из характерных проявлений гемофилии –
кровоизлияния в крупные суставы конечностей: коленные, реже –
локтевые, голеностопные, плечевые. Малейшее движение сустава
резко болезненно. Как следствие повторных кровоизлияний
развиваются хронические артрозы: сустав увеличен в обьеме,
деформирован, развивается атрофия и слабость мышц конечностей.
Для гемофилии характерно также образование обширных подкожных,
внутримышечных и межмышечных гематом, которые могут возникать в
любом участке тела.
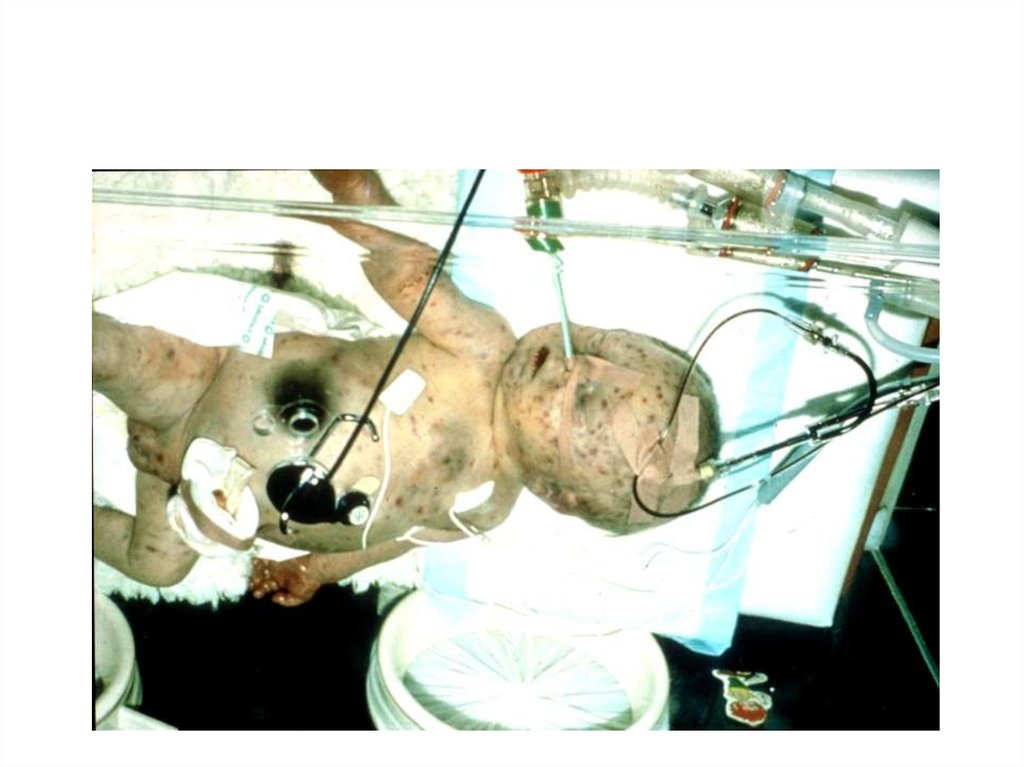
25. Гематомный тип кровоточивости
Гематома в месте инъекцииГематома языка
Гематома глаза
26. Гемофилия
27. Гемофилия
Гематома уноворожденного
ребенка
Гематома у ребенка
после выполнения
иньекции
28. Диагностика гемофилии
• Диагноз гемофилии устанавливают на основаниихарактерных клинических симптомов и данных
лабораторных исследований.
• Основными лабораторными показателями,
характерными для всех гемофилии, являются
увеличение времени свертывания крови,
снижение потребления протромбина в процессе
свертывания крови и соотвественно, снижение
концентрации VIII, IX, X, XI плазменных факторов.
• Основной принцип лечения кровоточивости при
гемофилии – введение в организм больного
недостающего фактора свертывания крови в
достаточном количестве.
29.
БОЛЕЗНЬ ВИЛЛЕБРАНДАБолезнь Виллебранда-Диана — наследственное заболевание крови,
характеризующееся возникновением эпизодических спонтанных
кровотечений, которые схожи с кровотечениями при гемофилии.
• Причина кровотечений — нарушение свертываемости крови из-за
недостаточной активности фактора Виллебранда, который участвует
в адгезии тромбоцитов на коллагене и защищает VIII фактор
от протеолиза.
• При дефиците Фактора Виллебранда VIII фактор подвергается
протеолизу, и его содержание в плазме снижается.
Заболевание обычно наследуется по аутосомно-доминантному типу, но
может отмечаться и рецессивное наследование.
30. БОЛЕЗНЬ ВИЛЛЕБРАНДА
• Характер кровоточивости – микроциркуляторногематомный. Характерны кровотечения изслизистых оболочек, из носа. Одно из характерных
проявлений болезни – обильные, продолжительные
кровотечения из ран, после травм, операций.
• У больных болезнью Виллибранда кровотечение
начинается, как правило, сразу после травмы, у
больных гемофилией – через некоторое время.
• Болезни Виллебранда свойственно резкое
увеличение времени кровотечения при нормальном
количестве тромбоцитов и неизменной ретракции
сгустка крови, снижении адгезии тромбоцитов к
коллагену и соединительной ткани, снижение
коагуляционной активности 8 фактора.
• Лечение. В период кровотечений вводят препараты,
содержащие комплекс VIII фактора.
31. Дефицит XIII фактора
• наследственный и приобретенный.• Наблюдается при тяжелой патологии печени, XIII
фактор, фибриназа, переводит растворимый
фибрин в нерастворимый, т.е. обеспечивает
поддержание окончательного тромба. При
дефиците фактора тромб непрочен и возможны
повторные кровотечения.
• Первый признак наследственной патологии –
медленное заживление пупочной ранки и
кровотечение из нее.
32. ПРИОБРЕТЕННАЯ КОАГУЛОПАТИЯ
• Особенность – полидефицитная• 1. Иммунная ингибиция прокоагулянтов (резус конфликт)
2. Дефицит вит. К–зависимых факторов коагуляции (7, 10, 9, 2)
а) нарушение синтеза в кишечнике (дисбактериоз, понос)
б) нарушение всасывания вит. К (дефицит желчи)
в) тяжелое повреждение печени
3. Передозировка гепарина
• При тяжелой патологии печени, лейкозах, тяжелых инфекциях,
гемолитической анемии может развиваться коагулопатия
связанная с дефицитом фибриногена.
• Дефицит ионов Са2+ дает клинические проявления в виде судорог.
Наблюдается при поражении паращитовидной железы, при
уремии.
33. ТИПЫ КРОВОТЕЧЕНИЯ
• 1. Микроциркуляторный.• Характеризуется мелкоточечными кровоизлияниями на коже и
слизистых.
• Могут быть десневые, носовые, маточные кровотечения.
• Такой тип кровотечения бывает при тромбоцитопении,
тромбоцитопатии, дефиците II, V, VII, X факторов.
• 2. Гематомный (макроциркуляторный)
• Характеризуется крупными кровоизлияниями в мышцы, суставы,
ткани.
• При этом нарушается питание тканей, сдавливаются нервы,
нарушается функция суставов, что приводит к инвалидности. Могут
быть профузные кровотечения – носовые, желудочно-кишечные.
• Развиваются при дефиците факторов VIII, IX, XI.
• 3. Cмешанный
• характеризуется сочетанием кровоточивости слизистых и
гематомного типа.
• Наблюдается при болезни Виллибранда, дефиците VII и XIII
факторов, при ДВС-синдроме.
34. ТРОМБОФИЛИИ
• Заболевания характеризующиеся повышеннойсвертываемостью крови, прижизненным образованием
тромбов в венах и артериях.
• Это частые осложнения сердечно-сосудистых заболеваний,
опухолей, инфекционно-воспалительных процессов,
обширных травм.
• Факторами высокого риска являются атеросклероз,
гипертоническая болезнь. Сахарный диабет, ожирение,
переломы костей нижних конечностей и др..
• Непосредственными факторами тромогенеза являются:
повреждение сосудистой стенки, активация тромбоцитов,
активация системы свертывания крови.
35. ТРОМБОФИЛИИ
• Клинические проявления:• рецидивирующие множественные тромбозы разной
локализации, тромбоэмболиями в бассейне легочной артерии,
инфарктами органов, развивающимися, как правило, у
больных сравнительно молодого возраста.
• Диагноз основывается на выявлении склонности к тромбозам
в сравнительно молодом возрасте, непереносимости
внутривенных манипуляций, выявлении семейной
предрасположенности к тромбозам ("тромбоэмболическиа
семьи") и на характерных лабораторных признаках того или
иного вида тромбофилии.
• При исследовании гемостазиограммы отмечается активация
адгезии и агрегации тромбоцитов, активация плазменных
факторов, снижение антикоагулянтной и фибринолитической
активности крови.
36. ДВС-СИНДРОМ (СИНДРОМ ДЕСИМИНОРОВАННОГО ВНУТРИСОСУДИСТОГО СВЕРТЫВАНИЯ КРОВИ )
• – это патологический процесс в основекоторого лежит массивное свертывание
крови, ведущее к блокаде
микроциркуляторного русла жизненно
важных органов рыхлыми массами фибрина
и агрегатами клеток и сопровождающееся
избыточным потреблением факторов
коагуляции, тромбоцитов и других клеток.
• - нарушенная свёртываемость крови по причине
массивного освобождения из тканей
тромбопластических веществ.
• - обширное отложение фибрина в мелких
кровеносных сосудах, что приводит к повреждению
ткани или органа в результате ишемии
37. ЭТИОЛОГИЯ ДВС-СИНДРОМА
Инфекции, септические состояния
Шок (при септическом – смертность 100 %)
Хирургические вмешательства, ожеги
Все терминальные состояния, остановка сердца
Острый внутрисосудистый гемолиз
Акушерская патология (20-25 %) (эмболия околоплодными водами,
преждевременная отслойка плаценты, внутриутробная гибель плода при длительной его
ретенции в матке, при тяжелых поздних токсикозах (эклампсия).
Гемобластозы (при о. лейкозе – 33-45 %)
Деструктивные процессы в паренхиматозных органах
Аллергические реакции
Воздействие гемокоагулирующих змеиных ядов (гадюковые:
гюрза, эфа и др; гремучие змеи: кроталиды, ботропсы и т.д.);
38.
39. Абу - Ибрахим- Джурджани (1110 год нашей эры)
«- люди, укушенные, ядовитой змеей,погибают от свертывания крови в
полостях сердца, сосудах и истечения
крови из всех отверстий…»
40. Аксиомы ДВС- синдрома:
• ДВС-синдром всегда вторичен, т.е.сопровождает или осложняет течение
основного заболевания – индуктора.
• ДВС- синдром имеет «дозовую»
зависимость от выраженности и тяжести
основного заболевания.
• ДВС- синдром всегда является
полиорганной патологией.
41. Патогенез ДВС-синдрома:
• Он возникает вследствие попадания в кровотокактиваторов свертывания крови. Например, при гемолизе
эритроцитов, на обломках их мембран активируется VII, XII
факторы.
• В отличие от физиологического гемостаза, когда
тромбоциты активируются в месте повреждения сосуда, при
ДВС-синдроме тромбоциты активируются во всем
кровотоке. Они приобретают способность к адгезии и
агрегации, приклеиваются к эндотелию сосудистой стенки,
образуя тромбоэмболы.
• Эти процессы приводят к образованию множественных
рыхлых сгустков фибрина, закупоривающих мелкие
сосуды различных органов. В связи с повышенным
потреблением на эти процессы плазменных факторов
свертывания и фибрина, наступает их дефицит. На этом
фоне развивается кровоточивость, возникают профузные
неконтролируемые кровотечения.
42. Стадии ДВС-синдрома
• 1) Гиперкоагуляция (образование множественныхтромбов вследствие активации системы коагуляции)
• 2) Коагулопатия потребления (истощение системы
каогулянтов, чрезмерное использование тромбоцитов для образования тромбов)
• 3) Гипокоагуляция (понижение активности коагулянтов, активация антикоагулянтов, активация
фибринолиза)
• 4) Завершение (выздоровление, осложнения,
смерть)
43. Клиника ДВС-синдрома
1. Гемокоагуляционный шокпричина
* нарушения микроциркуляции (вызывают развитие
гипоксии тканей)
* накопление токсических продуктов протеолиза
проявления
* понижение артериального давления
* понижение центрального венозного давления
* кровотечения (провоцируют геморрагический шок)
44. Клиника ДВС-синдрома
2. Нарушения гемостазаа) гиперкоагуляция
Главное проявление – тромбоз
Кровь сворачивается в пробирке
б) гипокоагуляция
Главное проявление – кровотечения
(одновременно истощается система
фибринолиза)
45. Клиника ДВС-синдрома
• 3. Тромбоцитопения• Возникает вследствие образова-ния
большого количества тромбов в
сосудах (тромбоцитопения
потребления)
46. Клиника ДВС-синдрома
4. Блокада микроциркуляторного русла-
-
(вызывает повреждение органов-мишеней)
Легкие (тромбы заносятся из венозной системы) – острая
дыхательная недостаточность
Почки - острая почечная недостаточность
Желудок и кишечник - глубокая дистрофия слизистой,
интоксикация, аутолиз кишечника, профузные кровотече-ния,
шок (високая летальность)
Надпочечники
Печень
Гипофиз
47. Клинические проявления ДВС-синдрома
Клинические проявления ДВСсиндрома48. Органопатология ДВС-синдрома
ПОЧКИ• окклюзии микроциркуляторного русла,
• повреждения стенок сосудов,
• поражения эпителия канальцев.
КОЖА
петехиальные, мелко- и крупнопятнистая
иногда сливная геморрагическая сыпь,
множественные кровоизлияния в местах
инъекций, операционных разрезов
49. Лабораторная диагностика ДВС-синдрома.
Лабораторная диагностика ДВСсиндрома.• В зависимости от течения ДВС-синдрома выделяют
острую и хроническую формы.
• При остром ДВС-синдроме обычно увеличено
протромбиновое время, снижено количество
тромбоцитов, увеличено количество продуктов
деградации продуктов фибрина. Снижена
концентрация многих факторов свертывания крови,
особенно фибриногена и факторов 5, 8, 13.
• При хроническом ДВС-синдроме количество
тромбоцитов и уровень факторов свертывания могут
быть в пределах нормы, но концентрация продуктов
деградации фибрина будет повышена. В мазке
крови в 50% случаев могут присутствовать
фрагментированные эритроциты.
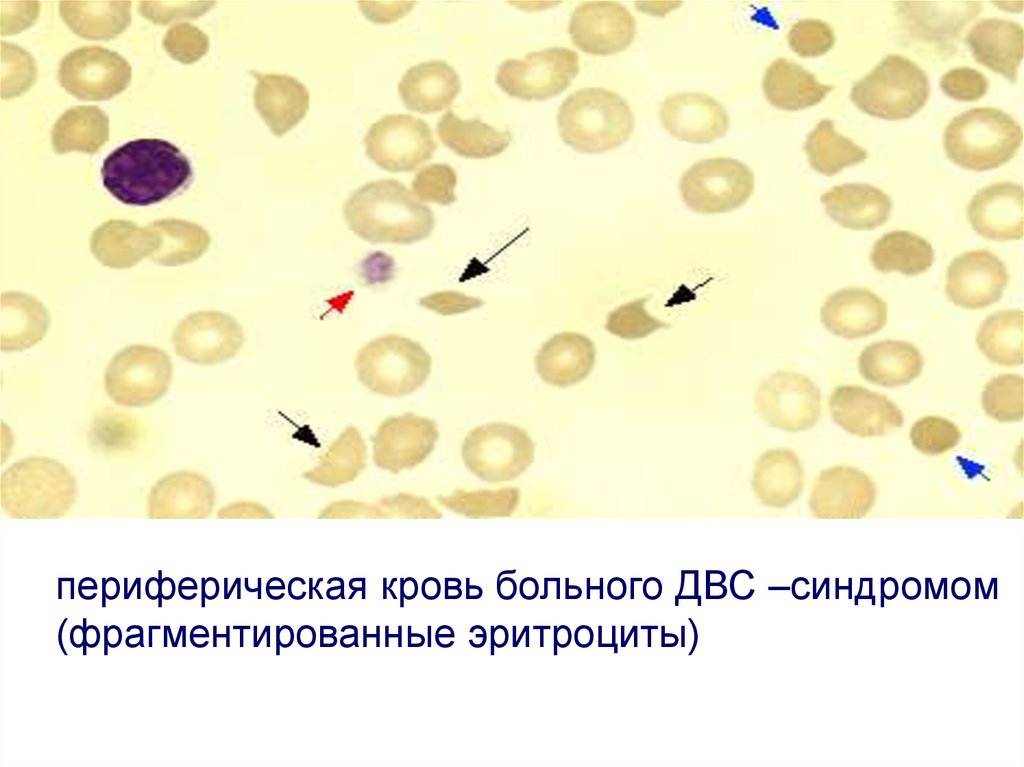
50.
периферическая кровь больного ДВС –синдромом(фрагментированные эритроциты)
51.
Дополнения52.
ТИПОВЫЕ ФОРМЫИЗМЕНЕНИЙ
В СИСТЕМЕ ТРОМБОЦИТОВ
ТРОМБОЦИТОЗЫ
ТРОМБОЦИТОПЕНИ
И
ТРОМБОЦИТОПАТИ
И
52
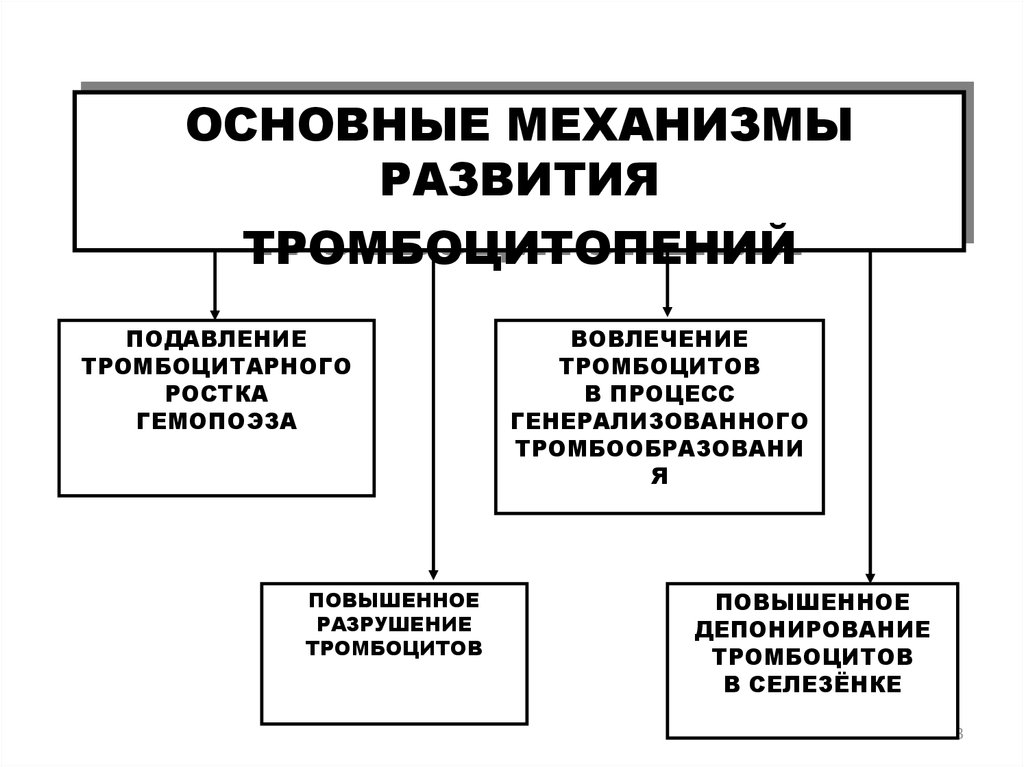
53.
ОСНОВНЫЕ МЕХАНИЗМЫРАЗВИТИЯ
ТРОМБОЦИТОПЕНИЙ
ПОДАВЛЕНИЕ
ТРОМБОЦИТАРНОГО
РОСТКА
ГЕМОПОЭЗА
ПОВЫШЕННОЕ
РАЗРУШЕНИЕ
ТРОМБОЦИТОВ
ВОВЛЕЧЕНИЕ
ТРОМБОЦИТОВ
В ПРОЦЕСС
ГЕНЕРАЛИЗОВАННОГО
ТРОМБООБРАЗОВАНИ
Я
ПОВЫШЕННОЕ
ДЕПОНИРОВАНИЕ
ТРОМБОЦИТОВ
В СЕЛЕЗЁНКЕ
53
54.
ИЗМЕНЕНИЯ В СИСТЕМЕГЕМОСТАЗА ПРИ
ТРОМБОЦИТОПЕНИЯХ
СНИЖЕНИЕ
КОНЦЕНТРАЦИИ
И/ИЛИ АКТИВНОСТИ
ТРОМБОЦИТАРНЫХ
ФАКТОРОВ
СВЕРТЫВАЮЩЕЙ
СИСТЕМЫ
КРОВИ
УВЕЛИЧЕНИЕ
ДЛИТЕЛЬНОСТИ
КРОВОТЕЧЕНИЯ
СНИЖЕНИЕ
СТЕПЕНИ
РЕТРАКЦИИ
СГУСТКА
КРОВИ
ГЕМОРРАГИЧЕСКИЙ
СИНДРОМ
54
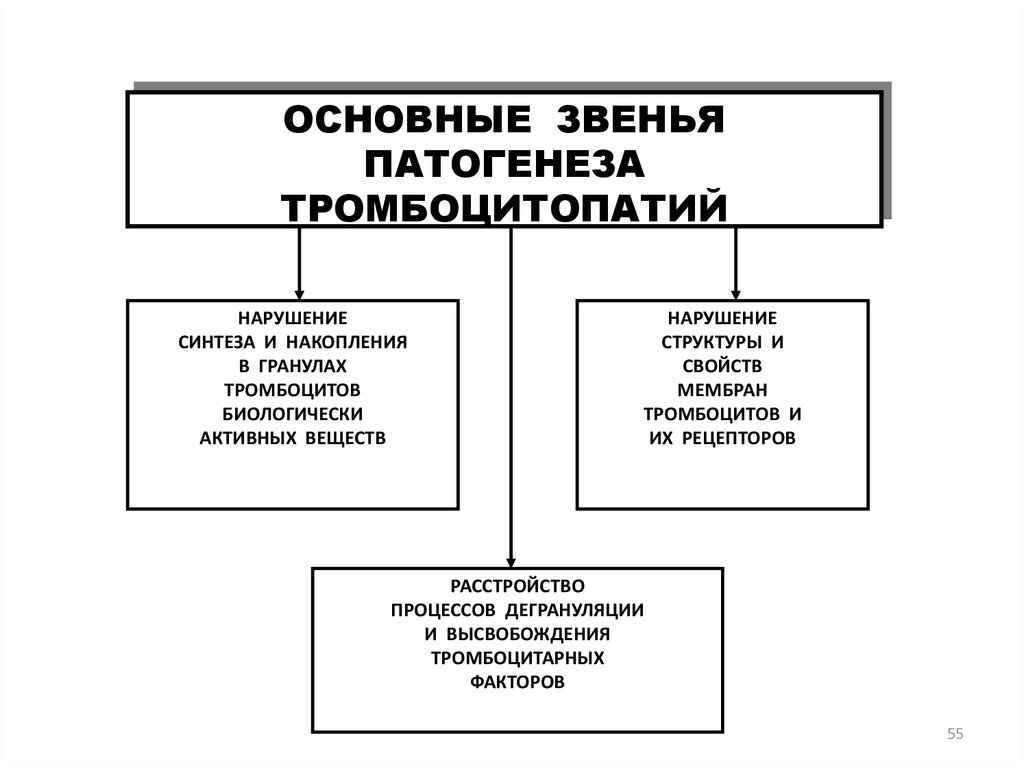
55.
ОСНОВНЫЕ ЗВЕНЬЯПАТОГЕНЕЗА
ТРОМБОЦИТОПАТИЙ
НАРУШЕНИЕ
СИНТЕЗА И НАКОПЛЕНИЯ
В ГРАНУЛАХ
ТРОМБОЦИТОВ
БИОЛОГИЧЕСКИ
АКТИВНЫХ ВЕЩЕСТВ
НАРУШЕНИЕ
СТРУКТУРЫ И
СВОЙСТВ
МЕМБРАН
ТРОМБОЦИТОВ И
ИХ РЕЦЕПТОРОВ
РАССТРОЙСТВО
ПРОЦЕССОВ ДЕГРАНУЛЯЦИИ
И ВЫСВОБОЖДЕНИЯ
ТРОМБОЦИТАРНЫХ
ФАКТОРОВ
55