





















































 Медицина
МедицинаПохожие презентации:










Генные и хромосомные болезни
1. Генные и хромосомные болезни
2.
Генные болезни- это разнообразная поклинической картине группа
заболеваний , обусловленная мутациями
единичных генов.
В результате мутации гена на
молекулярном уровне возможны
следующие варианты:
1. синтез аномального белка;
2. выработка избыточного количества
генного продукта;
3. отсутствие выработки первичного
продукта;
4. выработка уменьшенного количества
нормального первичного продукта.
3.
По типу наследования генные заболеванияраспределяют на группы:
1)
2)
3)
4)
5)
Аутосомно-доминантные
Аутосомно-рецессивные
Х-сцепленные доминантные
Х-сцепленные рецессивные
Y-сцепленные
Для диагностики генных заболеваний
используют биохимический, генеалогический
методы генетики и метод амниоцентеза.
4. Аутосомно - доминантные заболевания.
1)2)
3)
Синдром Марфана .
Нейрофиброматоз (Болезнь Реклингхаузена ).
Синдром Холт - Орама (Рука-сердце)
5. Синдром Марфана.
Аутосомно-доминантное заболевание из группынаследственных патологий соединительной ткани.
Синдром вызван мутациями генов, кодирующих
синтез гликопротеина фибриллина-1, и является
плейотропным. В классических случаях лица с
синдромом Марфана высоки (долихостеномелия),
имеют удлиненные конечности, вытянутые пальцы
(арахнодактилия) и недоразвитие жировой клетчатки.
Помимо характерных изменений в органах опорнодвигательного аппарата (удлинённые трубчатые кости
скелета, гипермобильность суставов), наблюдается
патология в органах зрения и сердечно-сосудистой
системы, что в классических вариантах составляет
триаду Марфана.
6.
Без лечения продолжительностьжизни лиц с синдромом Марфана
часто ограничивается 30—40 годами,
и смерть наступает вследствие
расслаивающейся аневризмы аорты
или застойной сердечной
недостаточности. В странах с
развитым здравоохранением
больные успешно лечатся и
доживают до преклонного возраста.
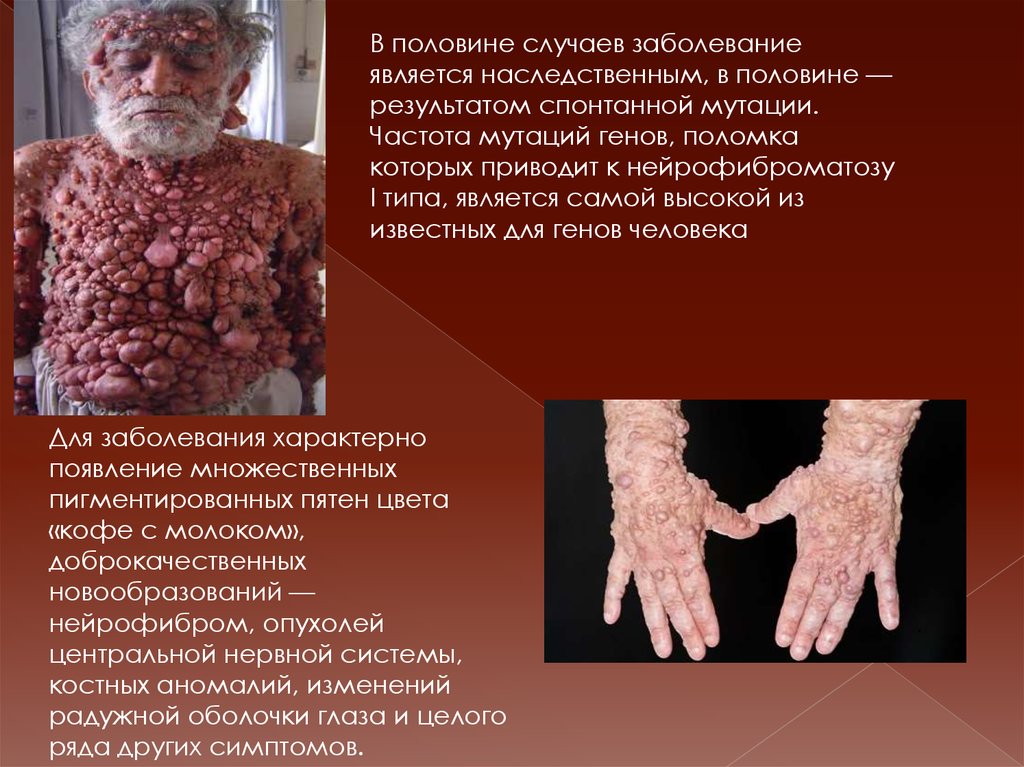
7. Нейрофиброматоз.
Самое распространённоенаследственное заболевание,
предрасполагающее к
возникновению опухолей у человека.
Описан во второй половине XIX века
рядом исследователей, в том числе в
1882 году учеником Рудольфа
Вирхова Фридрихом фон
Реклингхаузеном. Устаревшие
названия — болезнь Реклингхаузена,
периферический
нейрофиброматоз и др. Является
аутосомно-доминантным,
встречается с одинаковой частотой у
мужчин и у женщин, у 1 из 3500
новорождённых
8.
В половине случаев заболеваниеявляется наследственным, в половине —
результатом спонтанной мутации.
Частота мутаций генов, поломка
которых приводит к нейрофиброматозу
I типа, является самой высокой из
известных для генов человека
Для заболевания характерно
появление множественных
пигментированных пятен цвета
«кофе с молоком»,
доброкачественных
новообразований —
нейрофибром, опухолей
центральной нервной системы,
костных аномалий, изменений
радужной оболочки глаза и целого
ряда других симптомов.
9.
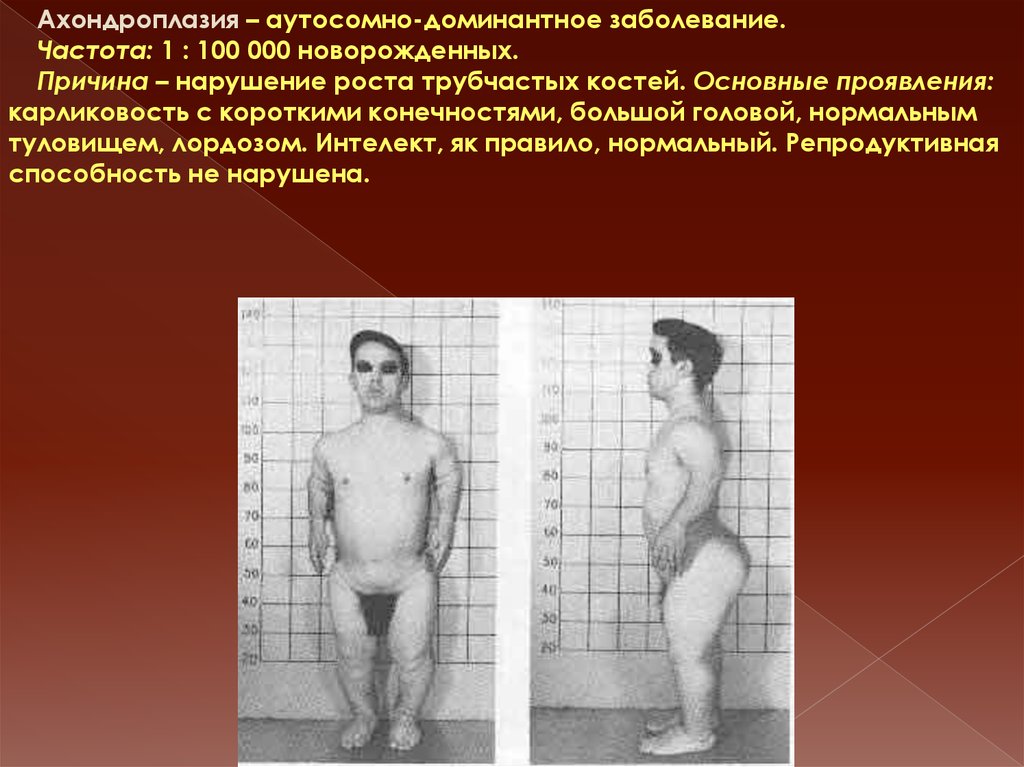
Ахондроплазия – аутосомно-доминантное заболевание.Частота: 1 : 100 000 новорожденных.
Причина – нарушение роста трубчастых костей. Основные проявления:
карликовость с короткими конечностями, большой головой, нормальным
туловищем, лордозом. Интелект, як правило, нормальный. Репродуктивная
способность не нарушена.
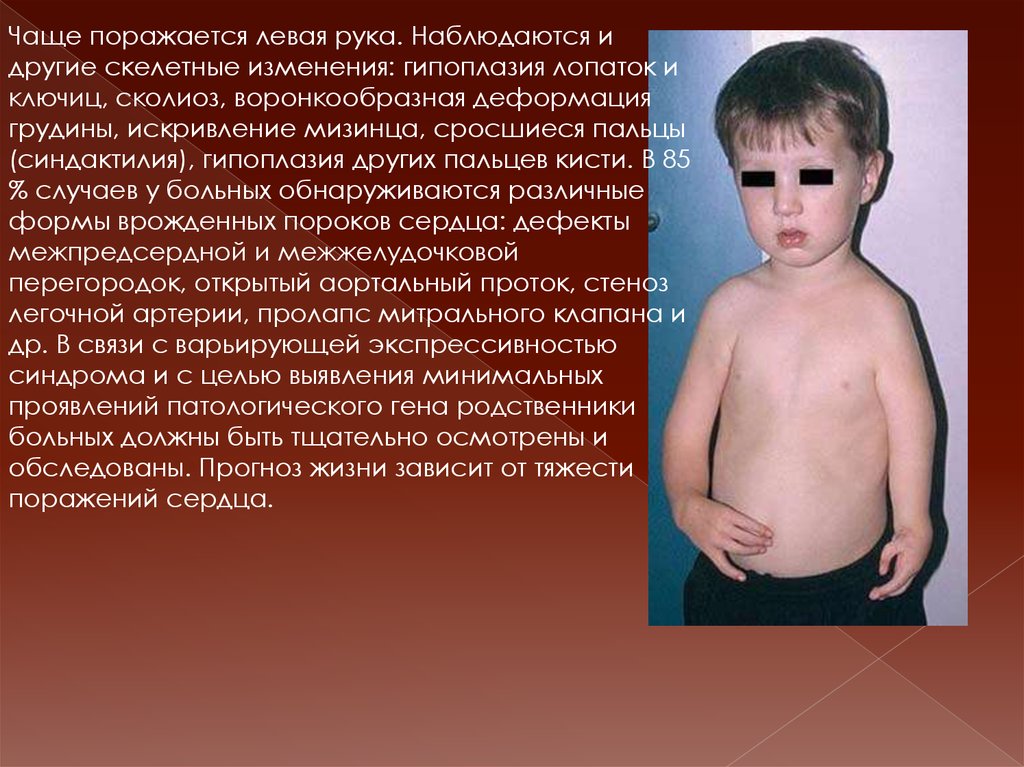
10. Синдром Холт- Орама.
наследственное сочетаниеаномалий больших пальцев рук и
дефекта межпредсердной
перегородки; это моногенный
синдром множественных пороков
развития. Название дано по
фамилиям врачей М. Холта
(педиатра) и С. Орама
(кардиолога). Клиническая картина
характеризуется аномалиями
верхних конечностей и врожденными
пороками сердца. Пороки развития
руки варьируют от недоразвития или
отсутствия 1-го пальца кисти и
трёхфалангового 1-го пальца кисти
до недоразвития или полного
отсутствия лучевой кости с
формированием лучевой
косорукости.
11.
Чаще поражается левая рука. Наблюдаются идругие скелетные изменения: гипоплазия лопаток и
ключиц, сколиоз, воронкообразная деформация
грудины, искривление мизинца, сросшиеся пальцы
(синдактилия), гипоплазия других пальцев кисти. В 85
% случаев у больных обнаруживаются различные
формы врожденных пороков сердца: дефекты
межпредсердной и межжелудочковой
перегородок, открытый аортальный проток, стеноз
легочной артерии, пролапс митрального клапана и
др. В связи с варьирующей экспрессивностью
синдрома и с целью выявления минимальных
проявлений патологического гена родственники
больных должны быть тщательно осмотрены и
обследованы. Прогноз жизни зависит от тяжести
поражений сердца.
12. прогерия
Прогерия (греч.преждевременно
состарившийся) —
патологическое
состояние,
характеризующееся
комплексом
изменений кожи,
внутренних органов,
обусловленных
преждевременным
старением
организма.
Основными
формами является
детская прогерия
(синдром
Гетчинсона
(Хадчинсона) —
Гилфорда) и
прогерия взрослых
(синдром Вернера).
13.
Прогерия (синдром Гетчинсона-Гилфорда)аутосомно-доминантное заболевание. Клинические проявления:
прогрессирующее, быстрое старение организма с 5-6-летнего
возраста. Больные умирают к 12 годам.
14. Аутосомно-рецессивные заболевания.
1)2)
3)
Муковисцидоз(Кистофиброз
поджелудочной железы)
Фенилкетонурия(Фенилпировиноградная
олигофрения)
Адреногенитальный синдром(Врожденная
гиперплазия коры надпочечников)
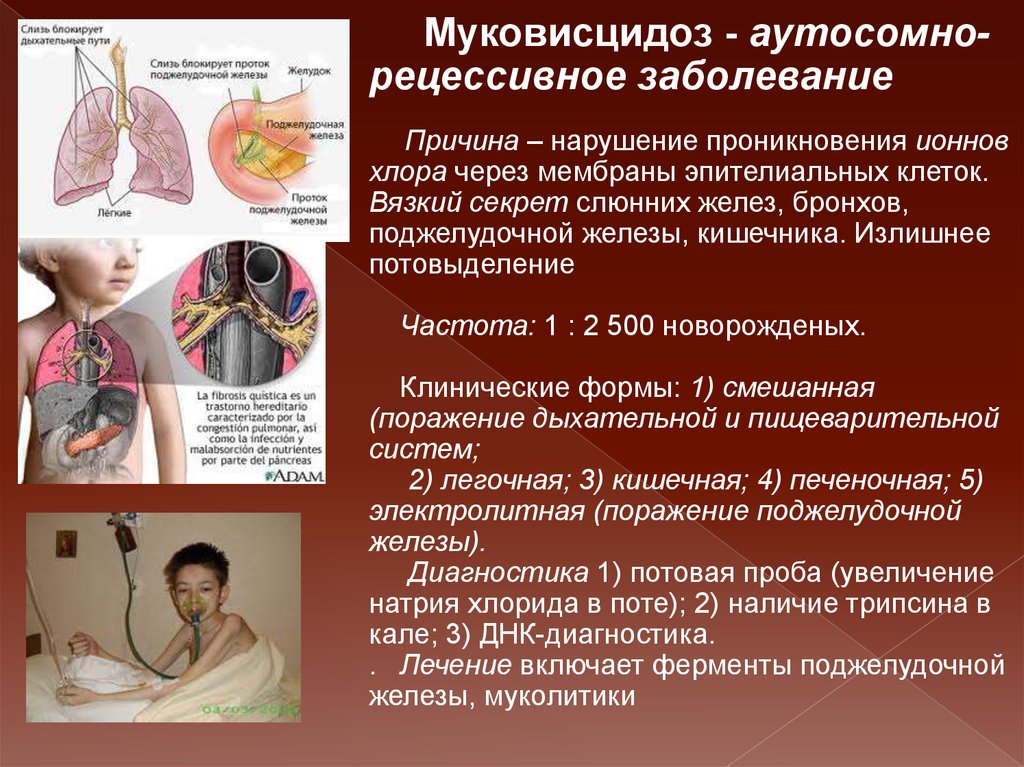
15. Муковисцидоз.
Системное наследственноезаболевание, обусловленное
мутацией гена трансмембранного
регулятора муковисцидоза и
характеризующееся поражением
желёз внешней секреции, тяжёлыми
нарушениями функций органов
дыхания и желудочно-кишечного
тракта.
16.
Патологические изменения в лёгких характеризуются признакамихронического бронхита с развитием бронхоэктазов и диффузного
пневмосклероза. В просвете бронхов находится вязкое содержимое
слизисто-гнойного характера. Нередкой находкой являются участки
эмфиземы. У многих больных течение патологического процесса в
лёгких осложняется наслоением бактериальной инфекции
(патогенный золотистый стафилококк, гемофильная и синегнойная
палочка) и формированием деструкции.
В поджелудочной железе выявляется диффузный фиброз, утолщение
междольковых соединительнотканных прослоек, кистозные изменения
мелких и средних протоков. В печени отмечается очаговая или
диффузная жировая и белковая дистрофия клеток печени, желчные
стазы в междольковых желчных протоках, лимфогистиоцитарные
инфильтраты в междольковых прослойках, фиброзная
трансформация и развитие цирроза.
При мекониевой непроходимости выражена атрофия слизистого
слоя, просвет слизистых желез кишечника расширен, заполнен
эозинофильными массами секрета, местами имеет место отёк
подслизистого слоя, расширение лимфатических щелей. Нередко
муковисцидоз сочетается с различными пороками развития
желудочно-кишечного тракта.
17.
18.
Муковисцидоз - аутосомнорецессивное заболеваниеПричина – нарушение проникновения ионнов
хлора через мембраны эпителиальных клеток.
Вязкий секрет слюнних желез, бронхов,
поджелудочной железы, кишечника. Излишнее
потовыделение
Частота: 1 : 2 500 новорожденых.
Клинические формы: 1) смешанная
(поражение дыхательной и пищеварительной
систем;
2) легочная; 3) кишечная; 4) печеночная; 5)
электролитная (поражение поджелудочной
железы).
Диагностика 1) потовая проба (увеличение
натрия хлорида в поте); 2) наличие трипсина в
кале; 3) ДНК-диагностика.
. Лечение включает ферменты поджелудочной
железы, муколитики
19. Фенилкетонурия.
наследственное заболевание группыферментопатий, связанное с нарушением
метаболизма аминокислот, главным
образом фенилаланина. Сопровождается
накоплением фенилаланина и его
токсических продуктов, что приводит к
тяжёлому поражению ЦНС,
проявляющемуся, в частности, в виде
нарушения умственного развития.
20.
Диагностика производится полуколичественным тестом иликоличественным определением фенилаланина в крови.
При нелеченных случаях возможно выявление продуктов
распада фенилаланина (фенилкетонов) в моче (не ранее 1012 дня жизни ребенка). Также возможно определение
активности фермента фенилаланингидроксилазы в
биоптате печени и поиск мутаций в гене
фенилаланингидроксилазы. Для диагностики 2 и 3 типа,
связанных с мутацией в гене, отвечающем за синтез
кофактора, необходимы дополнительные диагностические
исследования. В возрасте от 2-4 месяцев у больных
появляются такие симптомы, как вялость, судороги,
экзема, мышиный запах.
21.
Фенилкетонурия – аутосомно-рецессивноезаболевание. Причина – недостаток фермента
фенилаланин-4-моноксидазы. Частота: 1 : 20
000 новонарожденных. Характерно
увеличение фенилаланина в крови, судороги,
задержка умственного развития,
гипопигментация кожи, волос. Лечение
состоит в выключении фенилаланина с пищи
до 5-летнего возраста.
22.
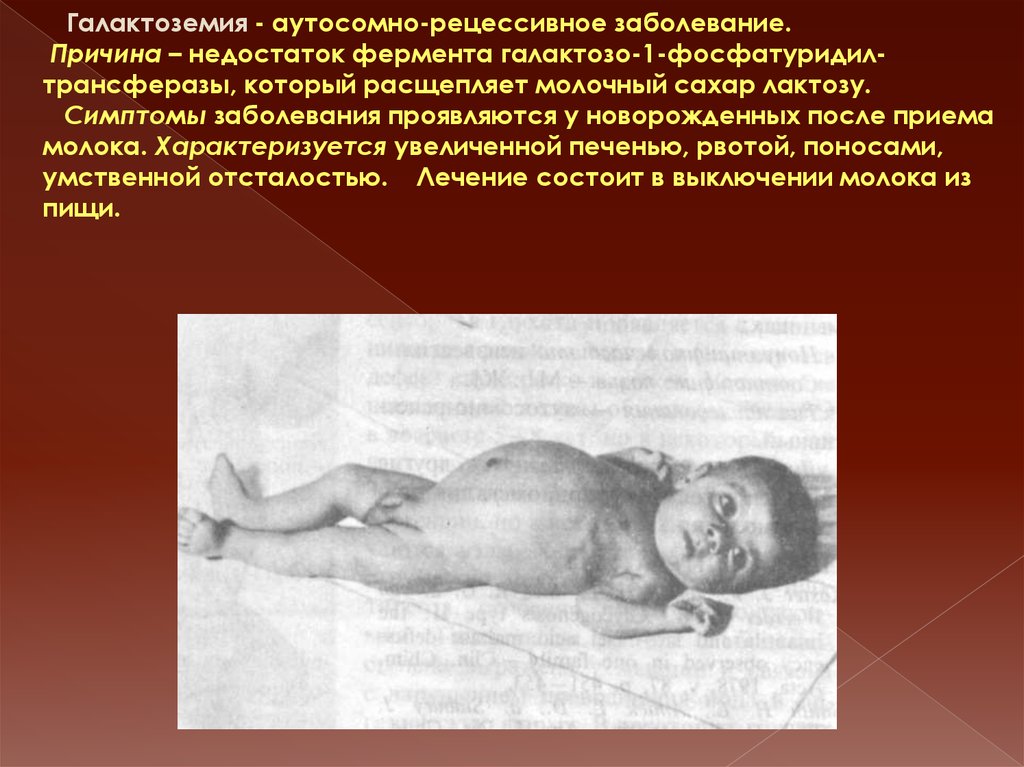
Галактоземия - аутосомно-рецессивное заболевание.Причина – недостаток фермента галактозо-1-фосфатуридилтрансферазы, который расщепляет молочный сахар лактозу.
Симптомы заболевания проявляются у новорожденных после приема
молока. Характеризуется увеличенной печенью, рвотой, поносами,
умственной отсталостью. Лечение состоит в выключении молока из
пищи.
23.
Альбинизм – аутосомно-рецессивноезаболевание. Причина – отсутствие фермента
тирозиназы, необходимого для синтеза
меланина. Проявляется депигментацией кожи,
волос, радужки глаз одинаково для всех рас.
24. Андреногенитальный синдром.
группа заболеваний, наследуемыхпо аутосомно-рецессивному пути,
при которых нарушается выработка
кортизола надпочечниками.
У девочек возникает незначительная
гиперплазия клитора , ускорение
костного возраста, различные
нарушения менструального цикла.
У мальчиков могут быть
преждевременное оволосение
наружных гениталий и снижение
темпов роста за счет раннего
закрытия зон роста.
25. Х- сцепленные рецессивные заболевания.
1)2)
Псевдогипертрофическая мышечная
дистрофия Дюшенна.
Синдром умственной отсталости с ломкой
Х- хромосомой(Синдром Мартина- Белл)
26. Псевдогипертрофическая мышечная дистрофия Дюшенна.
Мышечная дистрофия —врожденноезаболевание, характеризующееся
мышечной слабостью.
Синдром Дюшенна поражает только
мальчиков и является наиболее
распространенным видом
мышечной дистрофии. Это
заболевание отмечается примерно
у одного ребенка из трех тысяч.
27.
Мышечная дистрофия развиваетсяиз-за постепенного разрушения
нервно-мышечных связей.
Это врожденное заболевание,
передающееся по наследству от
родителей к детям. Девочки могут
быть носителями гeна, отвечающего
за наличие мышечной дистрофии,
но обычно сами не подвержены
этому заболеванию.
28. Синдром Мартина- Белл.
Синдром с ломкой Х хромосомы сцепленное с полом доминантноезаболевание.
Главным симптомом синдрома является
интеллектуальное недоразвитие и
своеобразная речь. Такие больные говорят
быстро, сбивчиво, имеются персеверации
(бормочущая речь). Также могут быть
нарушения поведения в виде агрессивности,
двигательной расторможенности. В качестве
одной из частых психопатологических
особенностей отмечена шизофреноподобная
симптоматика, включающая в себя
подпрыгивания, похлопывания руками,
повороты вокруг своей оси, встряхивание
кистями, «манежный» бег, разнообразные
гримасы, монотонное хныканье.
29.
Фенотипические признаки: большая голова свысоким и широким лбом, длинное лицо с
увеличенным подбородком, несколько
уплощённая средняя часть лица, тупой,
слегка клювовидно загнутый кончик носа.
Уши большие, иногда оттопыренные, низко
расположенные. Кисти и стопы широкие,
дистальные фаланги пальцев также
широкие, суставы имеют повышенную
подвижность. Кожа нередко гиперэластична.
Часто встречаются светлоокрашенные
радужные оболочки, светлые волосы. Не
обязательно встречаются все признаки —
могут быть один или несколько.
Неврологическая симптоматика
неспецифична, определяется как и у всех
детей с умственной отсталостью.
Наблюдается некоторая мышечная
гипотония, дискоординация движений.
30.
Гемофилия- нарушениесвёртываемости крови.
Первоначальной
носительницей
мутантного гена
является
английская
королева Виктория
31.
Гемофилия - Х-сцепленное рецессивноезаболевание.
Частота: 1:2500 новорожденных.
Характеризуется кровотечениями,
гемартрозами (кровоизлияния в суставы).
Причина – дефицит VIII или IX факторов
32.
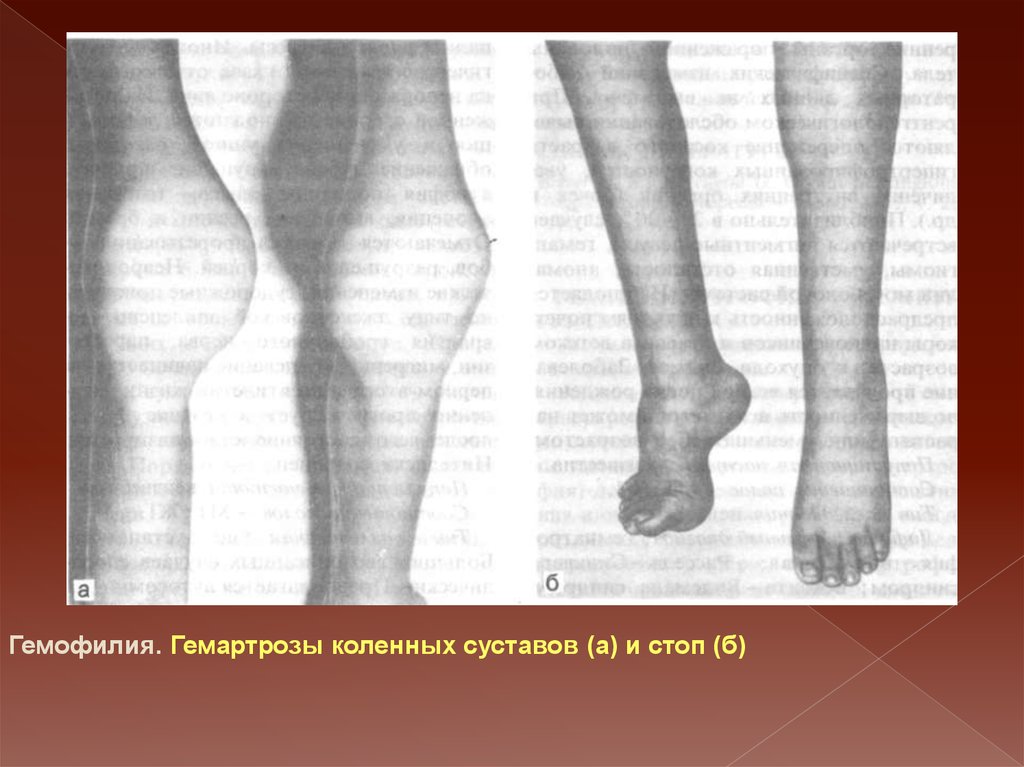
Гемофилия. Гемартрозы коленных суставов (а) и стоп (б)33.
Гидроцефалия - Х-сцепленноерецессивное заболевание. Частота: 1 :
2000 новорожденных.
Причина – нарушение оттока
спинномозковой жидкости.
Характеризуется увеличением размеров
головы, неврологическими
расстройствами, умственной
отсталостью.
34.
Дальтонизмявляется одной из наиболее
распространенных аномалий,
которые наследуются рецессивно,
сцепленно с Х-хромосомой.
Характеризуется нарушением
восприятия красного и зеленого
цветов.
35.
ДальтонизмXN
y
XN XN XN XN y
Xn
XN Xn
Xn y
N = норма
n = дальнонизм
36.
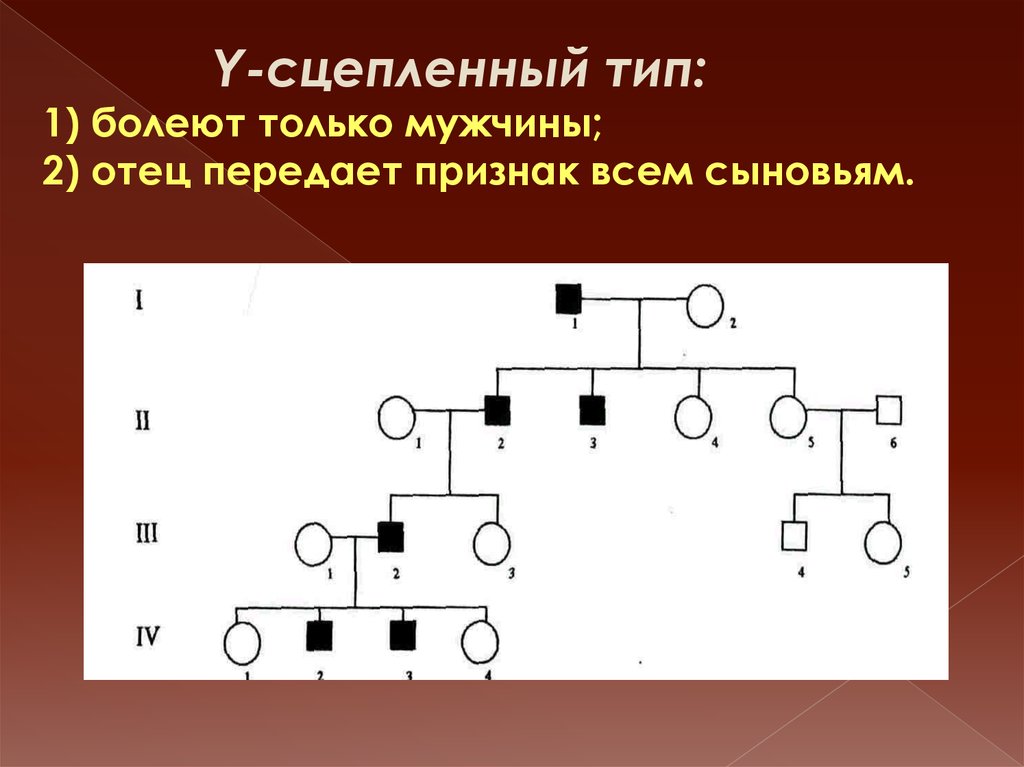
Y-сцепленный тип:1) болеют только мужчины;
2) отец передает признак всем сыновьям.
37.
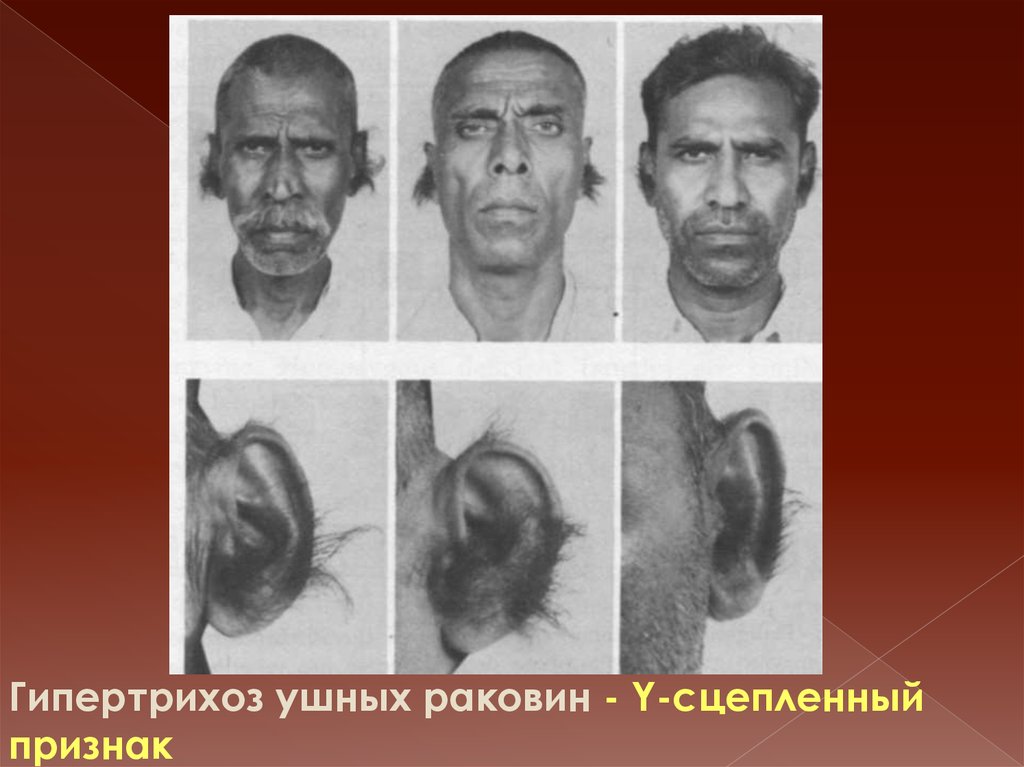
Гипертрихоз ушных раковин - Y-сцепленныйпризнак
38. Сравнение аутосомных и сцепленных с полом генных заболеваний
Автосомныезаболевания
Сцепленные с полом
заболевания
1. Возникают после генной
мутации в аутосомах.
1. Возникают после генной
мутации в половых
хромосомах.
2. Мутантные гены бывают
2. Мутантные гены чаще
доминантные или рецессивные. рецессивные.
3. Встречаются одинаково у
мужчин и женщин.
3. Встречаются чаще у мужчин.
4. Больные гомозиготные по
рецессивным генам;
гомозиготные или
гетерозиготные по
доминантным генам.
4. Больные женщины
гомозиготные по рецессивным
генам и гомозиготные или
гетерозиготные по
доминантным генам Ххромосомы.
39.
Митохондриальные болезниКаждая митохондрия имеет собственную ДНК
кольцевой формы. В этой хромосоме (Мхромосома) содержится 16569 пар нуклеотидов.
Генные мутации в митохондриальной ДНК
наблюдаются при наследственной атрофии
зрительного нерва Лебера, митохондриальных
миопатиях, при миокардиопатиях, атаксии-слепоте.
Митохондрии передаются с цитоплазмой
яйцеклеток, сперматозоиды цитоплазмы почти не
содержат.
Для митохондрального наследования характерны
следующие признаки:
1) болезнь передается только от матери детям;
2) болеют и девочки, и мальчики;
3) больной отец не передает заболевания ни
дочерям, ни сыновьям.
40. Хромосомные болезни.
К хромосомным относятся болезни, обусловленныегеномными мутациями или структурными изменениями
отдельных хромосом. Хромосомные болезни возникают в
результате мутаций в половых клетках одного из родителей.
Из поколения в поколение передаются не более 3—5 % из
них. Хромосомными нарушениями обусловлены примерно
50 % спонтанных абортов и 7 % всех мёртворождений.
Все хромосомные болезни принято делить на две группы:
аномалии числа хромосом и нарушения структуры
хромосом.
41. Аутосомные трисомии.
1)2)
3)
Синдром Дауна(Болезнь Дауна)
Синдром Патау
Синдром Эдвардса
42. Синдром Дауна.
одна из форм геномной патологии,при которой чаще всего кариотип
представлен 47 хромосомами
вместо нормальных 46, поскольку
хромосомы 21-й пары, вместо
нормальных двух, представлены
тремя копиями (трисомия).
Существует ещё две формы
данного синдрома: транслокация
хромосомы 21 на другие
хромосомы (чаще на 15, реже на 14,
ещё реже на 21, 22 и Y-хромосому)
— 4 % случаев, и мозаичный вариант
синдрома — 5 %.
43.
Возраст матери влияет на шансызачатия ребёнка с синдромом Дауна.
Если матери от 20 до 24, вероятность
этого 1 к 1562, если матери от 35 до 39,
то 1 к 214, а в возрасте старше 45,
вероятность 1 к 19. Хоть вероятность и
увеличивается с возрастом матери, 80 %
детей с данным синдромом рождаются
у женщин в возрасте до 35 лет. Это
объясняется более высокой
рождаемостью в данной возрастной
группе. По последним данным
отцовский возраст, особенно если
старше 42 лет, также увеличивает риск
синдрома
44.
«плоское лицо» — 90 %брахицефалия (аномальное укорочение черепа) — 81 %
кожная складка на шее у новорожденных — 81 %
Эпикантус- 80%
гиперподвижность суставов — 80 %
мышечная гипотония — 80 %
плоский затылок — 78 %
короткие конечности — 70 %
брахимезофалангия (укорочение всех пальцев за счёт недоразвития средних фаланг) —
70 %
катаракта в возрасте старше 8 лет — 66 %
открытый рот (в связи с низким тонусом мышц и особым строением нёба) — 65 %
зубные аномалии — 65 %
клинодактилия 5-го пальца (искривлённый мизинец) — 60 %
плоская переносица — 52 %
бороздчатый язык — 50 %
поперечная ладонная складка (называемая также «обезьяньей») — 45 %
короткая широкая шея — 45 %
ВПС (врождённый порок сердца) — 40 %
короткий нос — 40 %
страбизм (косоглазие) — 29 %
деформация грудной клетки, килевидная или воронкообразная — 27 %
стеноз или атрезия двенадцатиперстной кишки — 8 %
врождённый лейкоз — 8 %.
45. Синдром Патау.
Хромосомное заболеваниечеловека, которое характеризуется
наличием в клетках дополнительной
хромосомы 13.
Характерным осложнением
беременности при вынашивании
плода с синдромом Патау является
многоводие: оно встречается почти в
50 % случаев Синдрома Патау.
Встречается с частотой 1:70001:14000. Имеются два
цитогенетических варианта
синдрома Патау: простая трисомия
и робертсоновская транслокация.
46.
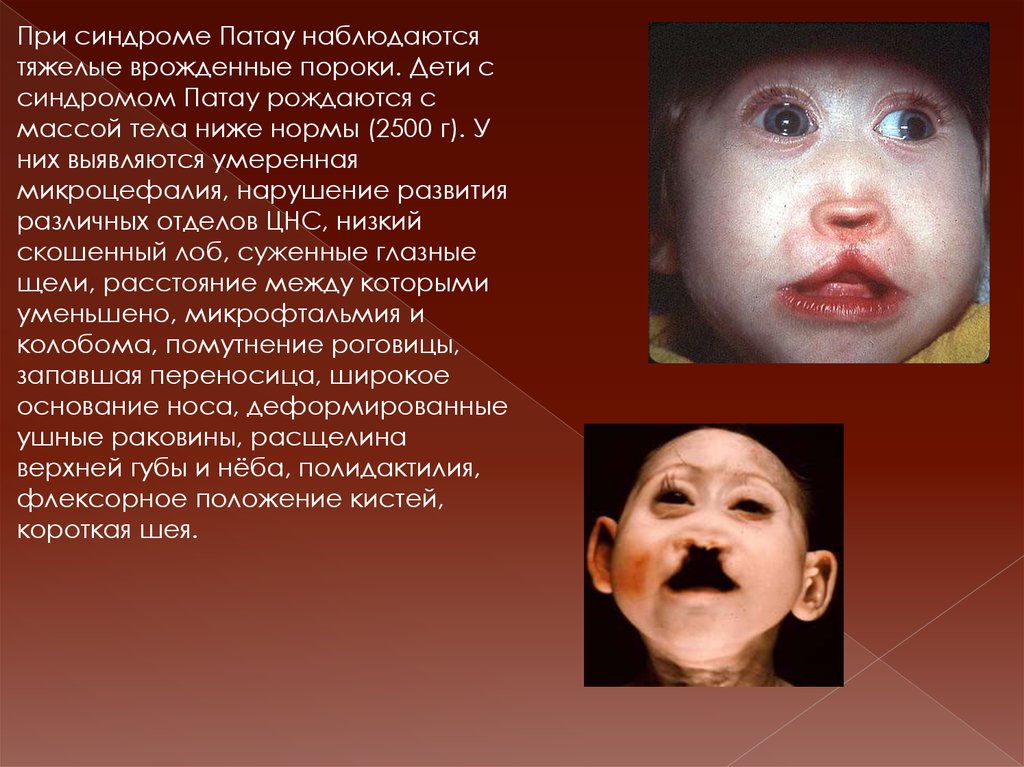
При синдроме Патау наблюдаютсятяжелые врожденные пороки. Дети с
синдромом Патау рождаются с
массой тела ниже нормы (2500 г). У
них выявляются умеренная
микроцефалия, нарушение развития
различных отделов ЦНС, низкий
скошенный лоб, суженные глазные
щели, расстояние между которыми
уменьшено, микрофтальмия и
колобома, помутнение роговицы,
запавшая переносица, широкое
основание носа, деформированные
ушные раковины, расщелина
верхней губы и нёба, полидактилия,
флексорное положение кистей,
короткая шея.
47.
У 80 % новорожденных встречаютсяпороки развития сердца: дефекты
межжелудочковой и
межпредсердной перегородок,
транспозиции сосудов и др.
Наблюдаются фиброкистозные
изменения поджелудочной железы,
добавочные селезенки,
эмбриональная пупочная грыжа.
Почки увеличены, имеют повышенную
дольчатость и кисты в корковом слое,
выявляются пороки развития половых
органов. Для СП характерна
задержка умственного развития.
В связи с тяжелыми врожденными
пороками развития большинство
детей с синдромом Патау умирают в
первые недели или месяцы (95 % —
до 1 года).
48. Синдром Эдвардса.
хромосомное заболевание,характеризуется комплексом
множественных пороков развития и
трисомией 18 хромосомы. Описан в
1960 году Джоном Эдвардсом.
Популяционная частота примерно
1:7000. Дети с трисомией 18 чаще
рождаются у пожилых матерей,
взаимосвязь с возрастом матери
менее выражена, чем в случаях
трисомии хромосомы 21 и 13. Для
женщин старше 45 лет риск родить
больного ребёнка составляет 0,7 %.
Девочки с синдромом Эдвардса
рождаются в три раза чаще
мальчиков.
49.
Фенотипические проявления синдромаЭдвардса многообразны. Чаще всего
возникают аномалии мозгового и лицевого
черепа, мозговой череп имеет
долихоцефалическую форму. Нижняя
челюсть и ротовое отверстие маленькие.
Глазные щели узкие и короткие. Ушные
раковины деформированы и в
подавляющем большинстве случаев
расположены низко, несколько вытянуты в
горизонтальной плоскости. Наружный
слуховой проход сужен, иногда отсутствует.
Грудина короткая, из-за чего межреберные
промежутки уменьшены и грудная клетка
шире и короче нормальной. В 80 % случаев
наблюдается аномальное развитие стопы:
пятка резко выступает, свод провисает
(стопа-качалка), большой палец утолщен и
укорочен.
50.
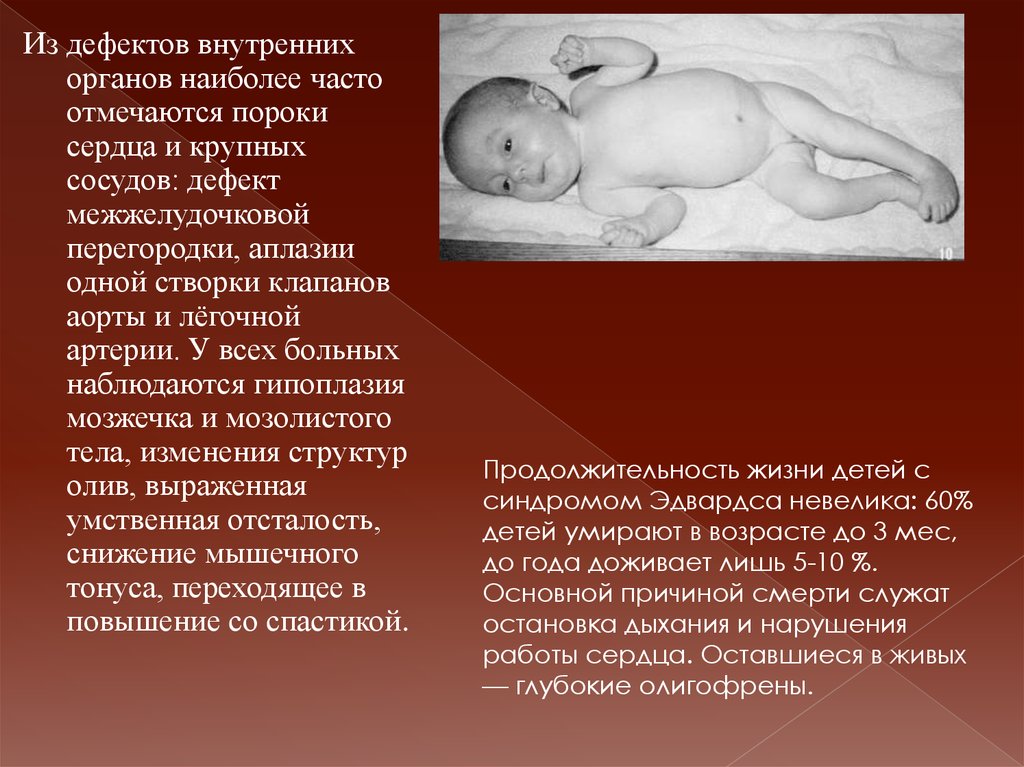
Из дефектов внутреннихорганов наиболее часто
отмечаются пороки
сердца и крупных
сосудов: дефект
межжелудочковой
перегородки, аплазии
одной створки клапанов
аорты и лёгочной
артерии. У всех больных
наблюдаются гипоплазия
мозжечка и мозолистого
тела, изменения структур
олив, выраженная
умственная отсталость,
снижение мышечного
тонуса, переходящее в
повышение со спастикой.
Продолжительность жизни детей с
синдромом Эдвардса невелика: 60%
детей умирают в возрасте до 3 мес,
до года доживает лишь 5-10 %.
Основной причиной смерти служат
остановка дыхания и нарушения
работы сердца. Оставшиеся в живых
— глубокие олигофрены.
51. Полисомии по половым хромосомам.
Синдром трисомии Х2) Синдром Клайнфельтера
3) Синдром дисомии по Y- хромосоме
4) Синдром Шерешевского- Тёрнера.
1)
52. Синдром Трисомии Х.
Заболевание встречается сравнительно редко (1 случай на 700—800 новорожденных девочек).
Часто трисомия-Х выявляется у больных психоза В ряде
случаев могут развиться вторичная аменорея,
преждевременный климакс. У большинства больных
менструальный цикл не нарушен. При нормальной функции
яичников женщины с трисомией-Х способны к
деторождению. По наследству трисомия-Х не передается. ми
и олигофренией.
Прогноз для жизни благоприятный. В
ряде случаев у таких женщин
сохранена детородная функция.
53.
Для заболевания характерно лишь небольшое снижениеинтеллекта. В ряде случаев интеллект сохранен.
Нередко отмечаются сочетание трисомии-Х с
шизофренией, склонность к развитию эпилепсии.
Двигательная активность нередко снижена. У
большинства девочек и женщин соматические
аномалии отсутствуют.
Иногда наблюдаются патологические изменения органа
зрения: двусторонняя атрофия зрительного нерва,
хориоретинит, одностороннее помутнение роговицы.
54. Синдром Клайнфельтера.
Генетической особенностью этогосиндрома является разнообразие
цитогенетических вариантов и их
сочетаний (мозаицизм).
Обнаружено несколько типов
полисомии по хромосомам X и Y у
лиц мужского пола: 47, XXY; 47, XYY;
48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY;
49 XXXYY. Наиболее распространен
синдром Клайнфельтера (47, XXY).
Общая частота его колеблется в
пределах 1 на 500—700
новорождённых мальчиков.
55.
До начала полового развития удаетсяотметить только отдельные
физические признаки, такие как
длинные ноги, высокая талия, высокий
рост. Пик прибавки роста приходится
на период между 5—8 годами и
средний рост взрослых пациентов
составляет приблизительно 179,2 +
6,2 см.
К началу полового созревания формируются
характерные пропорции тела: больные часто
оказываются выше сверстников, но в отличие от
типичного евнухоидизма размах рук у них редко
превышает длину тела, ноги заметно длиннее
туловища. Кроме того, некоторые дети с данным
синдромом могут испытывать трудности в учёбе и в
выражении своих мыслей. В некоторых руководствах
указывается, что у пациентов с синдромом
Клайнфельтера отмечается несколько сниженный
объём яичек до периода полового созревания. Это
утверждение является неверным, поскольку до
периода полового созревания объём яичек у всех
мальчиков небольшой — менее 1 мл
56.
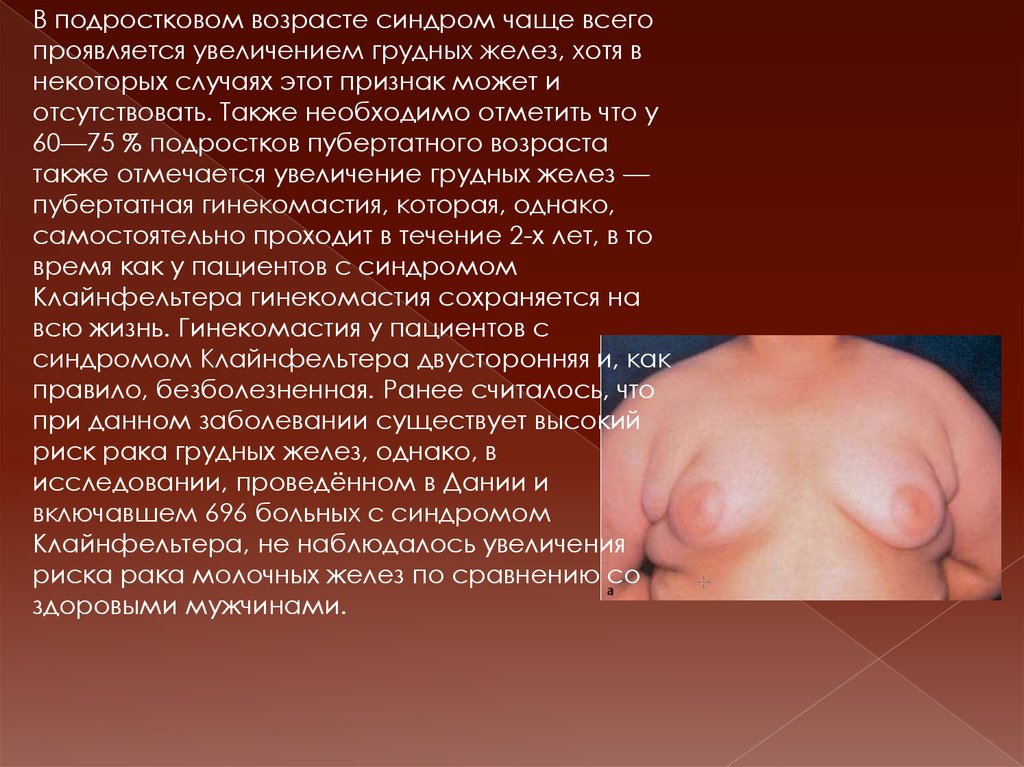
В подростковом возрасте синдром чаще всегопроявляется увеличением грудных желез, хотя в
некоторых случаях этот признак может и
отсутствовать. Также необходимо отметить что у
60—75 % подростков пубертатного возраста
также отмечается увеличение грудных желез —
пубертатная гинекомастия, которая, однако,
самостоятельно проходит в течение 2-х лет, в то
время как у пациентов с синдромом
Клайнфельтера гинекомастия сохраняется на
всю жизнь. Гинекомастия у пациентов с
синдромом Клайнфельтера двусторонняя и, как
правило, безболезненная. Ранее считалось, что
при данном заболевании существует высокий
риск рака грудных желез, однако, в
исследовании, проведённом в Дании и
включавшем 696 больных с синдромом
Клайнфельтера, не наблюдалось увеличения
риска рака молочных желез по сравнению со
здоровыми мужчинами.
57. Синдром дисомии по Y-хромосоме.
Частотой 1 на 1000 новорожденных мальчиков.Мужчины с набором хромосом 47 XYY не отличаются от нормы по
физическому и умственному развитию.
Иногда наблюдается незначительное снижение интеллекта, склонность к
агрессивным и антисоциальным поступкам. По некоторым данным, в
местах заключения мужчин с генотипом XYY в 10 раз больше, чем
мужчин с нормальным генотипом.
58.
Вообще в заболевании нет ничего особенного, Y-хромосома— содержит ген SRY, определяющий мужской пол
организма, а также гены, необходимые для нормального
формирования сперматозоидов.
У большинства больных отмечается ускорение роста в детском
возрасте .
В 30 — 40 % случаев отмечаются определенные симптомы —
грубые черты лица, выступающие надбровные дуги и
переносица, увеличенная нижняя челюсть, высокое нёбо,
аномальный рост зубов с дефектами зубной эмали, большие
ушные раковины, деформация коленных и локтевых суставов.
Интеллект или негрубо снижен, или в норме. Характерны
эмоционально-волевые нарушения: агрессивность,
взрывчатость, импульсивность. В то же время для этого
синдрома характерны подражательность, повышенная
внушаемость, причем больные наиболее легко усваивают
негативные формы поведения.
59. Синдром Шерешевского-Тёрнера.
Хромосомная болезнь,сопровождающаяся характерными
аномалиями физического развития,
низкорослостью и половым
инфантилизмом. Моносомия по Ххромосоме.
При синдроме Тернера патологические признаки по частоте
встречаемости распределяются следующим образом: низкорослость
(98%), общая диспластичность (неправильное телосложение) (92%),
бочкообразная грудная клетка (75%), укорочение шеи (63%), низкий
рост волос на шее (57%), высокое «готическое» нёбо (56%),
крыловидные складки кожи в области шеи (46%), деформация
ушных раковин (46%), укорочение метакарпальных и
метатарзальных костей и аплазия фаланг (46%), деформация
локтевых суставов (36%), множественные пигментные родинки
(35%), лимфостаз (24%), пороки сердца и крупных сосудов (22%),
повышенное артериальное давление (17%).
60.
Половое недоразвитие присиндроме Тернера отличается
определённым своеобразием.
Нередкими признаками являются
геродермия (патологическая
атрофия кожи, напоминающая
старческую) и мошонкообразный
вид больших половых губ, высокая
промежность, недоразвитие малых
половых губ, девственной плевы и
клитора, воронкообразный вход во
влагалище. Молочные железы у
большинства больных не развиты,
соски низко расположены.
Вторичное оволосение появляется
спонтанно и бывает скудным. Матка
недоразвита. Половые железы не
развиты и представлены обычно
соединительной тканью. При
синдроме Тернера отмечается
склонность к повышению
артериального давления у лиц
молодого возраста и к ожирению с
нарушением питания тканей.
61.
Интеллект у большинства больных с синдромом Тернерапрактически сохранен, однако частота олигофрении все
же выше. В психическом статусе больных с синдромом
Тернера главную роль играет своеобразный психический
инфантилизм с эйфорией при хорошей практической
приспособляемости и социальной адаптации.
Прогноз для жизни при синдроме Тернера благоприятный,
исключение составляют больные с тяжёлыми
врождёнными пороками сердца и крупных сосудов и
почечной гипертензией. Лечение женскими половыми
гормонами делает больных способными к семейной
жизни, однако абсолютное большинство из них остаются
бесплодными.
62. Синдромы частичных моносомий.
Синдром «Кошачьего крика»63.
Хромосомно синдром кошачьегокрика объясняется частичной
моносомией; он развивается при
делеции (с утратой от трети до
половины, реже полная утрата)
короткого плеча пятой хромосомы.
Для развития клинической картины
синдрома имеет значение не
величина утраченного участка, а
конкретный незначительный
фрагмент хромосомы. Изредка
отмечается мозаицизм по делеции
или образование кольцевой
хромосомы-5.
64.
При этом синдроме наблюдается:общее отставание в развитии,
низкая масса при рождении и
мышечная гипотония,
лунообразное лицо с широко
расставленными глазами
характерный плач ребёнка,
напоминающий кошачье мяуканье,
причиной которого является
изменение гортани (сужение,
мягкость хрящей, уменьшение
надгортанника, необычная
складчатость слизистой оболочки)
или недоразвитие гортани. Признак
исчезает к концу первого года жизни.
Частота синдрома примерно 1:45000.
Клиническая картина синдрома и
продолжительность жизни людей с этим
синдромом довольно сильно варьирует
по сочетанию врождённых пороков
развития органов.