
































")












 аминокислот")















")
")











")
")














































")







 анемия")



")
")




































































")






































































 Биология
БиологияПохожие презентации:
")
")





")


Метаболизм простых и сложных белков
1. МЕТАБОЛИЗМ ПРОСТЫХ И СЛОЖНЫХ БЕЛКОВ
UNIVERSITATEA DE STAT DE MEDICINĂ ȘI FARMACIE«NICOLAE TESTEMIȚANU»
CATEDRA BIOCHIMIE ȘI BIOCHIMIE CLINICĂ
МЕТАБОЛИЗМ
ПРОСТЫХ И СЛОЖНЫХ
БЕЛКОВ
Svetlana Protopop
doctor în științe medicale,
conferențiar universitar
2. ПЕРЕВАРИВАНИЕ И ВСАСЫВАНИЕ БЕЛКОВ
3. НОРМЫ БЕЛКА В ПИТАНИИ
• 0,8 г/кг массы – взрослый человек(умственный труд или средняя физическая
нагрузка).
• 130–150 г белка в сутки – при тяжелой
физической работе.
• Суточные потребности в белке возрастают
при беременности и лактации, а также при
патологических состояниях, когда организм
теряет белок (асцит, нефриты, тяжелые
инфекционные заболевания, ожоги, травмы).
4. Биологическая ценность пищевых белков
зависит от:• аминокислотного состава (чем ближе
аминокислотный состав пищевого белка к
составу белков тела, тем выше его
биологическая ценность).
• степени усвоения пищевого белка
(эффективности распада под влиянием
ферментов желудочно-кишечного тракта).
5. Незаменимые аминокислоты
Val, Leu, Ile, Met, Thr, Lys, Phe, TrpПолузаменимые аминокислоты
Arg, His
Если белок содержит все незаменимые
аминокислоты в необходимых пропорциях
и он легко подвергается действию протеаз,
то он является полноценным (яйца, молоко,
мясо).
6. Динамическое состояние белкового обмена
Белковый состав организма постоянен, чтоявляется результатом равновесия между
скоростями синтеза и распада белков.
Аминокислоты
Vсинтеза
Белки
Vраспада
Взрослый здоровый человек –
Vсинтеза=Vраспада
7. Азотистый баланс
разница между количеством азота,поступающего с пищей, и количеством
выделяемого азота (мочевина, аммонийные
соли).
• Азотистое равновесие – здоровый взрослый
человек при полноценном питании.
• Положительный азотистый баланс – дети,
пациенты, выздоравливающие после тяжелых
болезней.
• Отрицательный азотистый баланс – старение,
голодание, тяжелые заболевания.
8. Белковая недостаточность
• Исключение даже одной незаменимойаминокислоты сопровождается отрицательным
азотистым балансом, истощением, остановкой
роста, нарушениями функций нервной
системы.
• Отсутствие цистеина – некроз печени.
• Отсутствие гистидина – катаракта.
• Отсутствие метионина – анемия, ожирение,
цирроз печени, облысение, геморрагии в
почках.
• Отсутствие лизина – анемия, внезапная
смерть.
9. Белковая недостаточность
• Белковая недостаточность – «квашиоркор»– «золотой (или красный) мальчик» –
Центральная Африка.
• Развивается у детей, которые не получают
животные белки, а питаются растительной
пищей (бананы, просо, кукуруза).
• Задержка роста, анемия, отеки
(гипопротеинемия), жировое перерождение
печени.
10. Белковая недостаточность
• Из-за атрофии клеток поджелудочной железыне усваивается даже то небольшое количество
белков, которое поступает с пищей.
• Из-за поражения почек увеличивается
экскреция аминокислот с мочой.
• Длительная белковая недостаточность
приводит к необратимым нарушениям
физиологических и умственных функций.
• Лечение – перевод больного на богатую
белком пищу, добавление в пищу лизина.
11. Парентеральное белковое питание
• Введение белков парентерально приводит кразвитию сенсибилизации, а повторное введение
белков может вызвать анафилактический шок.
• Для белкового питания используют гидролизаты
белков, что не вызывает аллергических реакций
(свободные АК не обладают видовой и тканевой
специфичностью).
• Показания – непроходимость пищевода (ожоги,
отравления), тяжелые раковые поражения
пищевода и желудка, после операций на желудке
и кишечнике.
12. Переваривание белков в ЖКТ
• Происходит в желудке и кишечнике поддействием протеолитических ферментов
(протеазы, пептидазы) желудка,
поджелудочной железы и кишечника.
• Протеолитические ферменты синтезируются
в неактивной форме – профермент (зимоген).
• Пепсиноген – пепсин.
• Прокарбоксипептидаза – карбоксипептидаза.
• Механизм активации –
частичный (ограниченный) протеолиз.
13. Переваривание белков в ЖКТ
Пептидазы делятся на:• Экзопептидазы – катализируют отщепление
концевых аминокислот. Делятся на аминопептидазы и
карбоксипептидазы.
• Эндопептидазы – гидролизуют пептидные связи
внутри полипептидной цепи.
Аминопептидаза
Карбоксипептидаза
Эндопептидазы
14. Переваривание белков в ЖКТ
• Пептидазы обладают разной субстратнойспецифичностью (расщепляют пептидные
связи, в образовании которых участвуют
определенные аминокислоты).
• Это свойство пептидаз применяется при
определении первичной структуры белков.
15. Переваривание белков в желудке
Условия для переваривания белков:Наличие протеолитических ферментов:
пепсин – у взрослых,
гастриксин, реннин – у детей.
Оптимальная кислая среда – создается
соляной кислотой.
16. Роль соляной кислоты в переваривании белков
• Превращает неактивный пепсиноген вактивный пепсин.
• Создает оптимальную среду для действия
пепсина.
• Осуществляет набухание белков, частичную
денатурацию, гидролиз сложных белков.
• Стимулирует выработку секретина в
двенадцатиперстной кишке.
• Ускоряет всасывание железа.
• Оказывает бактерицидное действие.
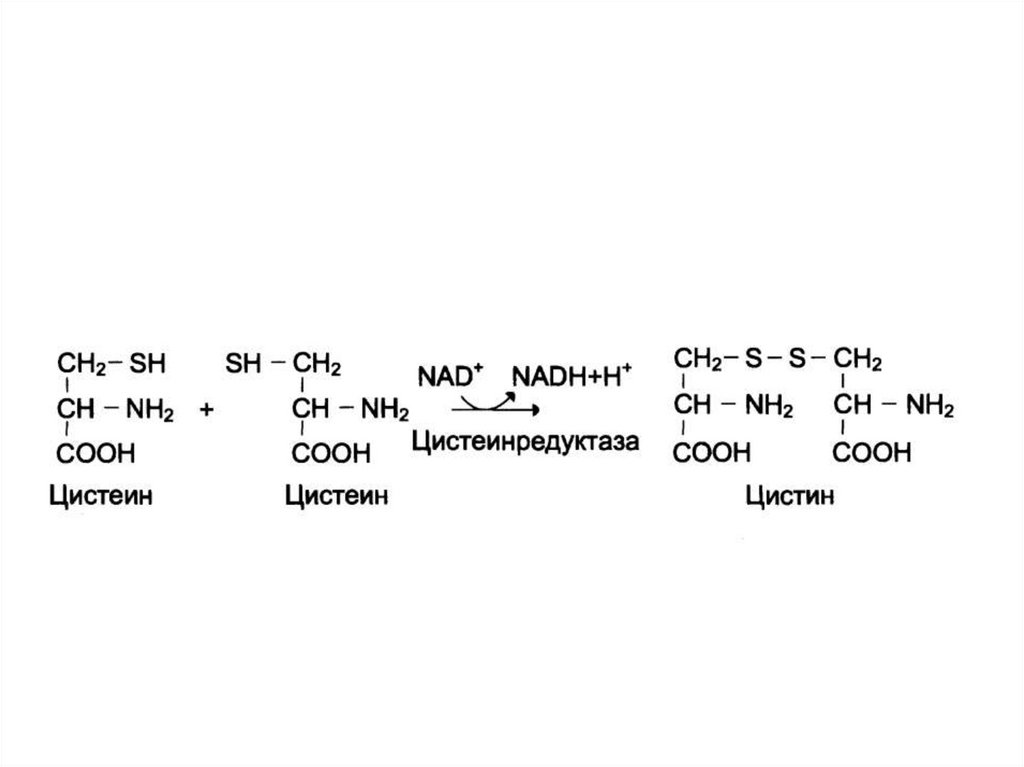
17. Пепсин
• Пепсин синтезируется в главных клеткахслизистой оболочки желудка в неактивной
форме – в виде пепсиногена.
• Превращение пепсиногена в активный
пепсин происходит в полости желудка.
• Механизм активации – частичный
протеолиз (отщепление от N-конца
пепсиногена 42-х аминокислот) под
действием HCl и аутокаталитически.
18. Пепсин
• Специфичность действия – эндопептидаза.Расщепляет пептидные связи, в
образовании которых участвуют
аминогруппы Phe, Tyr, Trp.
• Под действием пепсина в желудке
образуются короткие пептиды.
• Оптимальное рН действия – 1,5-2,5.
19. Переваривание белков в желудке у детей
• Пепсиноподобный фермент гастриксин,(образуется из пепсиногена), оптимум рН
действия – 3,0.
• Реннин – катализирует свертывание молока
(превращение растворимого казеиногена в
нерастворимый казеин, кальциевая соль
которого нерастворима, и он выпадает в
осадок).
• При этом резко замедляется продвижение
нерастворимого казеина через
пищеварительный канал, в результате чего он
дольше подвергается действию протеиназ.
20. Образование соляной кислоты
Источником H+ является H2CO3, котораяобразуется в обкладочных клетках желудка из
CO2, диффундирующего из крови, и H2O.
Фермент – карбангидраза.
H2O + CO2 → H2CO3 →HCO3- + H+
HCO3- выделяется в плазму в обмен на ионы Cl-,
а H+ поступают в просвет желудка в обмен на К+
путем активного транспорта(Н+/K+-ATP-аза).
Ионы Cl- поступают в просвет желудка через
хлоридный канал.
21. Образование соляной кислоты
1 - карбангидраза3- анионный канал
2 - H+/K+-ATP-аза
4 - хлоридный канал
22. Регуляция секреции желудочного сока
• Гистамин стимулирует секрециюсоляной кислоты.
• Гастрин стимулирует секрецию
пепсиногена и соляной кислоты.
23. Определение кислотности желудочного сока
• Повышенная кислотность – изжога, диарея,симптом язвы желудка или 12-перстной
кишки.
• Пониженная кислотность – гипоацидные
гастриты.
• Анацидность (желудочная ахилия) –
атрофические гастриты, сопровождается
пернициозной анемией вследствие
недостаточности выработки фактора Кастла и
нарушения всасывания витамина В12.
Предрасполагает к раку желудка.
24.
25. Ингибиторы желудочной секреции
• Циметидин – блокирует гистаминовые Н2рецепторы обкладочных клеток желудка,
ингибируя секрецию соляной кислоты.
• Омепразол – ингибирует H+/K+-ATP-азу.
26. Протеиназы поджелудочного сока
ПроферментАктивация Активный
фермент
Специфичность
действия
Трипсиноген
энтеротрипсин
киназа,
аутокатализ
трипсин,
химотрипсин
аутокатализ
трипсин
эластаза
Эндопептидаза
Lys, Arg
Химотрипсиноген
Проэластаза
Прокарбоксипептидаза
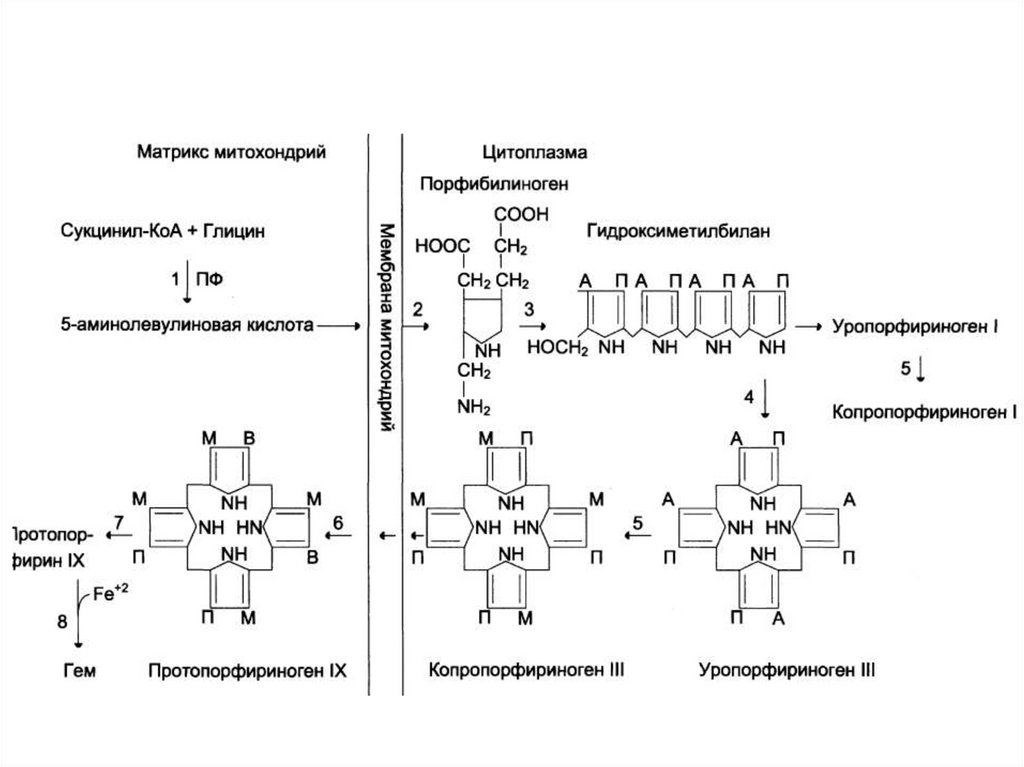
трипсин
карбоксипептидаза A
карбоксипептидаза В
Эндопептидаза
Phe, Tyr, Trp
Эндопептидаза
Gly, Ala, Ser
Экзопептидаза
Phe, Tyr, Trp
Экзопептидаза
Lys, Arg
27. Кишечные протеолитические ферменты
Аминопептидазы:• Аланинаминопептидаза – пептидные связи,
в образовании которых участвует Nконцевой аланин.
• Лейцинаминопептидаза – не обладает
строгой субстратной специфичностью
(гидролизует пептидные связи,
образованные любой N-концевой
аминокислотой).
28. Кишечные протеолитические ферменты
• Дипептидазы:• глицилглицин-дипептидаза (гидролиз
соответствующего дипептида до двух
молекул глицина).
• пролил-дипептидаза (пролиназа) –
катализирует гидролиз пептидной связи, в
образовании которой участвует СООНгруппа пролина.
• пролин-дипептидаза (пролидаза) – гидролиз
дипептида, в образовании которой
участвует имино-группа пролина.
29. Регуляция переваривания белков в кишечнике
• Секретин стимулирует секрециюподжелудочного сока, содержащего НСО3-,
что приводит к нейтрализации соляной
кислоты желудочного сока. рН возрастает
до 7.
• Холецистокинин стимулирует выделение
панкреатического сока, содержащего
протеолитические ферменты.
30. Всасывание аминокислот в кишечнике
• Происходит в проксимальных отделахтонкого кишечника.
• Механизм – вторично-активный транспорт
(ATP и Na+) с участием транслоказ:
1. Для нейтральных АК с короткой боковой
цепью.
2. Для нейтральных АК с длинной боковой
цепью (ароматических аминокислот).
3. Для щелочных АК и цистеина.
4. Для кислых АК.
5. Для пролина и гидроксипролина.
31.
32. Всасывание пептидов
Некоторые растительные токсины (абрин ирицин), а также токсины ботулизма, холеры и
дифтерии всасываются непосредственно в
кровь.
Дифтерийный токсин тормозит
внутриклеточный синтез белка.
У новорожденных всасываются антитела
молозива (секрет молочных желез,
обогащенный антителами и антитоксинами).
33. Гниение аминокислот в толстом кишечнике
превращения аминокислот под действиемферментов микроорганизмов кишечника.
При гниении аминокислот образуются
токсичные и нетоксичные продукты.
34. Гниение аминокислот в толстом кишечнике (токсичные вещества)
• Цистин, цистеин → сероводород H2S.• Метионин → метилмеркаптан CH3SH.
• Орнитин → путресцин
(декарбоксилирование).
• Лизин → кадаверин
(декарбоксилирование).
• Тирозин → крезол и фенол.
• Триптофан → скатол и индол.
35. Гниение аминокислот в толстом кишечнике
CH2 CH COOHNH2
HO
Tyr
NH
CH2 CH COOH
NH2
Trp
HO
CH3
Крезол
HO
Фенол
CH3
NH
NH
Скатол
Индол
36. Обезвреживание токсичных продуктов в печени
происходит путем химического связывания
с серной или глюкуроновой кислотой.
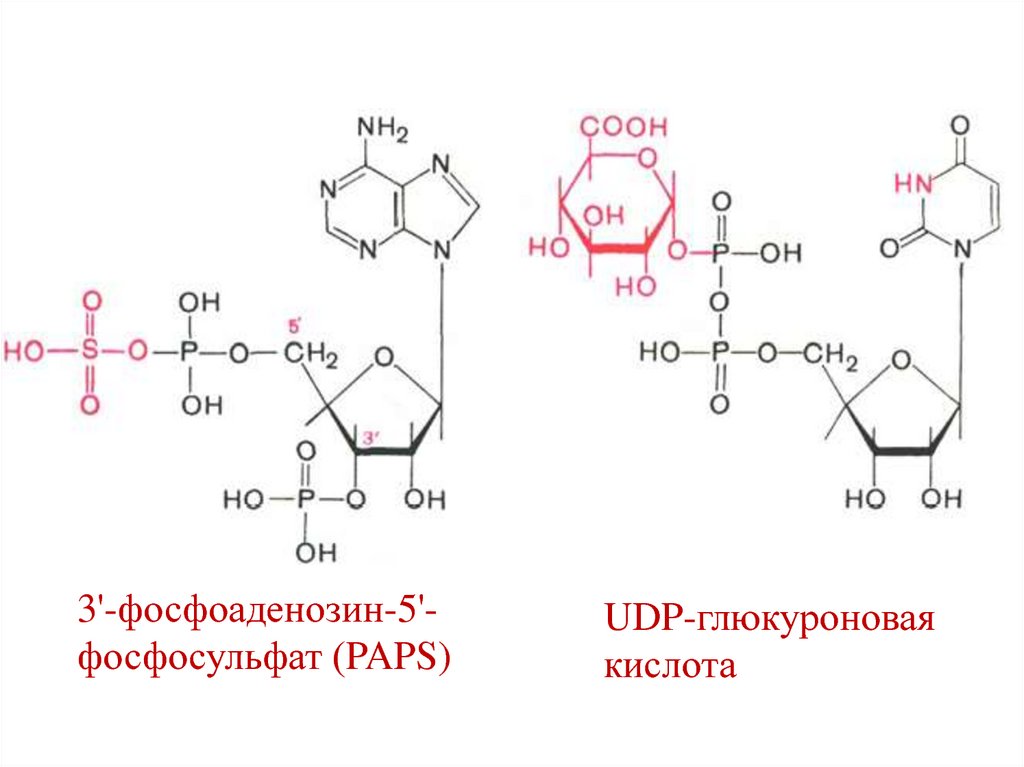
Активная форма серной кислоты –
3'-фосфоаденозин-5'-фосфосульфат (PAPS);
фермент – арилсульфотрансфераза.
Активная форма глюкуроновой кислоты –
UDP-глюкуронат;
фермент – UDP-глюкоронилтрансфераза.
37.
3'-фосфоаденозин-5'фосфосульфат (PAPS)UDP-глюкуроновая
кислота
38. Обезвреживание токсичных продуктов в печени
O2NH
индол
NADPH+H+
OH
NADP+
H2 O
NH
индоксил
O SO3H
PAPS
PAP
NH
индоксилсульфат
O SO3K
NH
индикан
39. Обезвреживание токсичных продуктов в печени
• Калиевая соль индоксилсульфата –животный индикан, выводится с мочой.
Диагностическое значение:
• Повышение количества индикана в
моче – усиленное гниение белков в
кишечнике.
• Снижение количества индикана в моче
– нарушение обезвреживающей
функции печени.
40. Оценка функционального состояния печени
определение скорости образования ивыделения гиппуровой кислоты с мочой
после приема бензойной кислоты.
Бензойная
кислота
Глицин
Гиппуровая
кислота
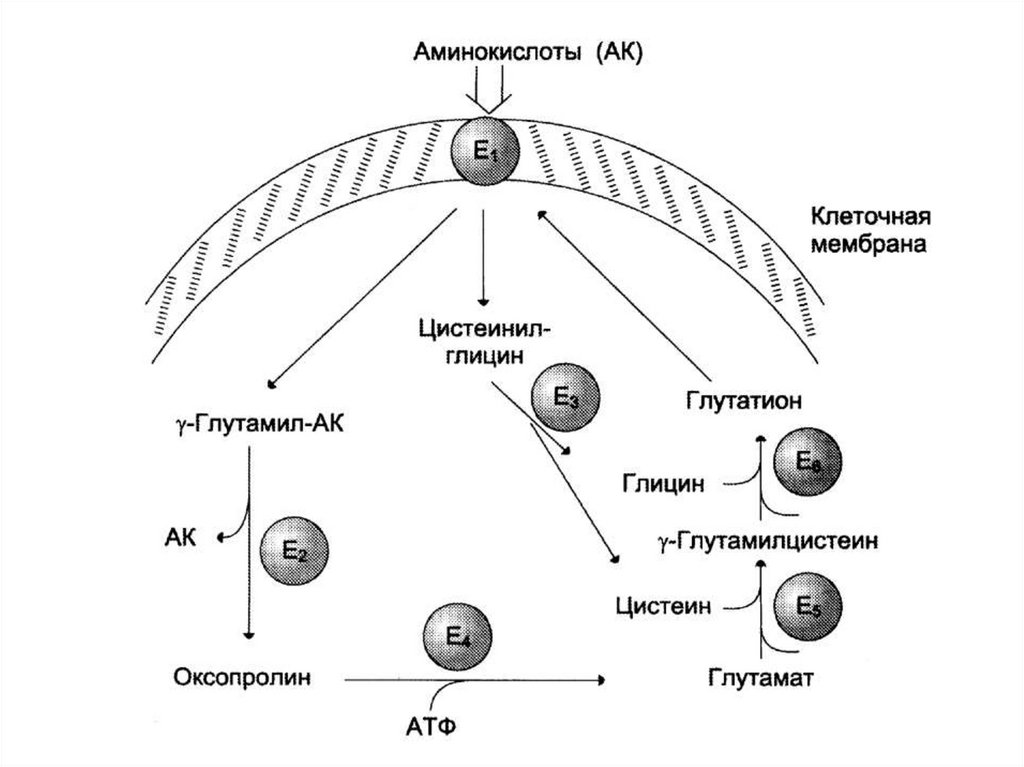
41. Транспорт аминокислот через клеточные мембраны
• γ-глутамильный цикл• Кишечник, мозг, почки.
• Состоит из 6-и реакций.
• Центральный фермент – мембранносвязанная γ-глутамилтрансфераза.
• Кофактор – глутатион
(γ-глутамил-цистеинил-глицин).
42.
43. Реакции γ-глутамильного цикла
1. перенос γ-глутамильной группы отглутатиона на аминокислоту – образуется
γ-глутамил-аминокислота и цистеинилглицин.
2. γ-глутамил–аминокислота распадается на
свободную аминокислоту и 5-оксопролин.
3. Цистеинил-глицин расщепляется до
свободных аминокислот.
Следующие три реакции направлены на
ресинтез глутатиона, для чего
используются 3 молекулы АТФ.
44. Первая реакция γ-глутамильного цикла
45. Вторая реакция γ-глутамильного цикла
46. ОБЩИЕ ПУТИ ОБМЕНА АМИНОКИСЛОТ В ТКАНЯХ. ОБЕЗВРЕЖИВАНИЕ АММИАКА
47. Использование аминокислот в тканях
Синтез заменимых аминокислот.
Синтез белков.
Синтез пептидов.
Образование биогенных аминов.
Синтез гормонов.
Синтез пуриновых и пиримидиновых
нуклеотидов, порфиринов.
• Синтез углеводов.
• Синтез липидов.
• Окисление (энергия).
48. ОБЩИЕ ПУТИ ОБМЕНА АМИНОКИСЛОТ
• Трансаминирование• Дезаминирование
• Декарбоксилирование
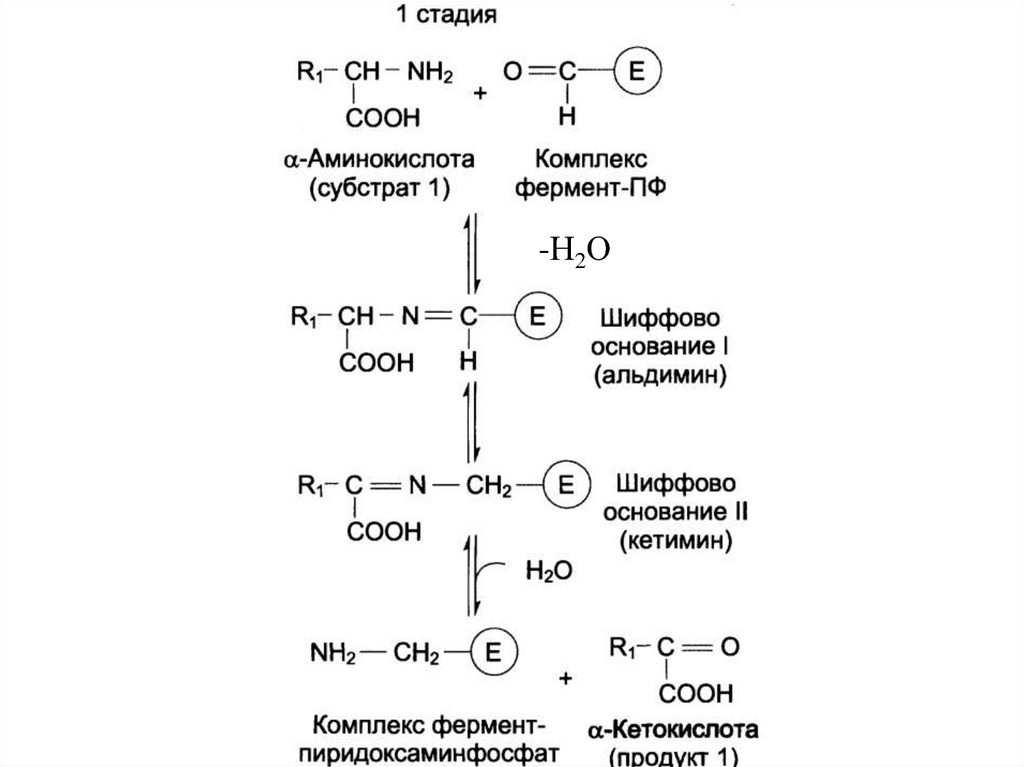
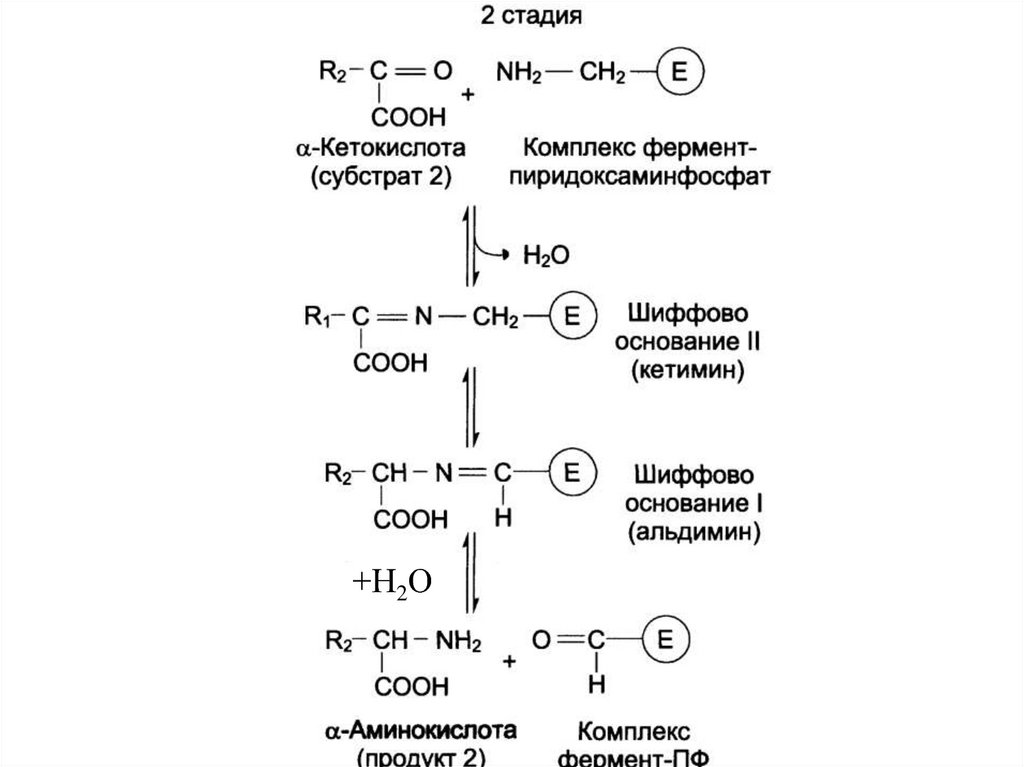
49. Трансаминирование (ТА) аминокислот
• реакция переноса аминогруппы (NH2—)с α-аминокислоты на α-кетокислоту и
перенос кетогруппы с α-кетокислоты на
α-аминокислоту, в результате образуется
новая аминокислота и новая кетокислота.
• Реакции ТА являются обратимыми.
• Ферменты – аминотрансферазы
(трансаминазы).
• Кофермент – пиридоксальфосфат
(производное витамина В6).
50. Активный центр аминотрансфераз
Альдиминнаясвязь
51. Трансаминирование аминокислот
COOHCH NH2
R1
+
COOH
C O
R2
COOH
COOH
C O + CH NH2
R2
R1
Механизм трансаминирования
Реакция протекает по типу «пинг-понг».
аминокислота1
кетокислота1
пиридоксальфосфат
аминокислота2
пиридоксаминфосфат
кетокислота2
52.
ПиридоксальфосфатO
H
C
PO3H2
HO
H3C
N
O
C E
H
Пиридоксаминфосфат
HO
H3C
CH2 NH2
PO3H2
N
H2N H2C E
53. Механизм трансаминирования
Первый этапCOOH
CH NH2
R1
+
O
C E
H
COOH
C O + H2N H2C E
R1
Второй этап
COOH
C O + H2N H2C E
R2
COOH
CH NH2
R2
+
O
C E
H
54.
-Н2О55.
+Н2О56. БИОЛОГИЧЕСКАЯ РОЛЬ ТА
Трансаминирование аминокислотиграет центральную роль в
метаболизме аминокислот:
является начальным этапом
в катаболизме аминокислот и
конечным этапом
в анаболизме аминокислот!!!
57. Основные реакции трансаминирования
COOHCH NH2
CH3
Аланин
+
COOH
C O
CH2
АЛТ COOH
CH2
COOH
α-кетоглутарат
C O
CH3
Пируват
+
COOH
CH NH2
CH2
CH2
COOH
Глутамат
АЛТ – аланин аминотрансфераза
ГПТ – глутамат пируват аминотрансфераза
Цитоплазматический фермент
58. Основные реакции трансаминирования
COOHCH NH2
CH2
COOH
Аспартат
+
COOH
C O
CH2
АСТ COOH
CH2
COOH
α-кетоглутарат
C O
CH2
+
COOH
Оксалоацетат
COOH
CH NH2
CH2
CH2
COOH
Глутамат
АСТ – аспартат аминотрансфераза
ГОТ – глутамат оксалоацетат аминотрансфераза
Цитоплазма, митохондрии
59. Диагностическое значение определения трансаминаз
• В норме активность АЛТ и АСТ в крови низкая.• АЛТ и АСТ активны в печени, сердце,
скелетных мышцах.
• При повреждении этих органов повышается
активность АЛТ и АСТ в крови.
• ↑АЛТ – гепатиты, циррозы.
• ↑АСТ – инфаркт миокарда, циррозы.
• АСТ/АЛТ – коэффициент де Ритиса (1,33±0,42):
˃ инфаркт, цирроз, ˂ гепатиты.
60. Дезаминирование аминокислот
реакция отщепления аминогруппы в
виде аммиака.
Типы дезаминирования:
Окислительное;
Неокислительное;
Внутримолекулярное;
Непрямое (трансдезаминирование).
61. Окислительное дезаминирование глутамата
COOH NAD+ NADH+H+ COOHCOOH
CH NH2
C NH +H2O C O
+NH
3
CH2
CH2
CH2
CH2
NADP+ NADPH+H+CH2
COOH
Глутамат
-H2O
COOH
α-иминоглутарат
CH2
COOH
α-кетоглутарат
Фермент – глутаматдегидрогеназа
62. Окислительное дезаминирование глутамата
Глутаматдегидрогеназа – регуляторныйфермент.
Ингибиторы – ATP, GTP, NADH.
Активатор – ADP.
Глюкокортикоиды индуцируют фермент.
63. Окисление L-аминокислот
Оксидазы L-аминокислот имеет оптимальноеpH = 10,0.
При физиологических значениях pH активность
фермента низкая.
Протекает в печени и в почках.
64.
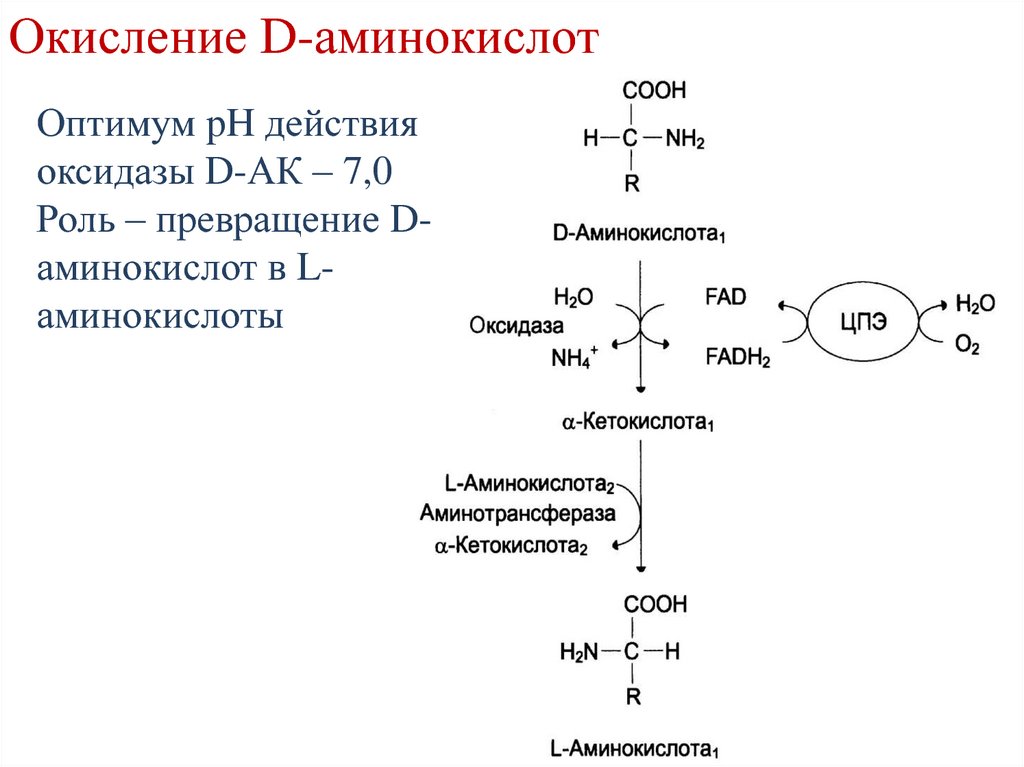
Окисление D-аминокислотОптимум pH действия
оксидазы D-АК – 7,0
Роль – превращение Dаминокислот в Lаминокислоты
65. Неокислительное дезаминирование
Кофермент – пиридоксальфосфатПротекает в печени
66. Неокислительное дезаминирование
цистатионин-γ-лиазаH2O
H2S
NH3
Cys
Пируват
Кофермент – пиридоксальфосфат
67. Внутримолекулярное дезаминирование
Реакция протекает только в печени и коже.68. Непрямое дезаминирование (трансдезаминирование)
Протекает в 2 стадии:1.Трансаминирование аминокислоты с
α-кетоглутаратом → образуется
глутамат.
2. Окислительное дезаминирование
глутамата.
69. Непрямое дезаминирование (трансдезаминирование)
70. Трандезаминирование является начальным этапом катаболизма большинства АК
71. Трансреаминирование является основным путем синтеза заменимых АК
72. Судьба продуктов дезаминирования аминокислот
АминокислотаАммиак
Мочевина
Аммонийные
соли
α-кетокислота
Глюкоза
ЖК
Кетоновые
Заменимые
тела
АК
Окисление
до СО2 и Н2О
73. Источники аммиака
Дезаминирование аминокислот.
Дезаминирование нуклеотидов.
Обезвреживание биогенных аминов.
Гниение белков в кишечнике.
74. Механизм токсичности аммиака
Аммиак легко проникает через мембраны и вмитохондриях сдвигает реакцию,
катализируемую глутаматдегидрогеназой в
сторону образования глутамата.
NH3 + α-кг + NADPH+H+→Glu + NADP+ +H2O
Уменьшение концентрации α-кетоглутарата
вызывает:
• Снижение скорости цикла Кребса → ↓ АТФ.
• Угнетение синтеза АК → ↓ синтеза
нейромедиаторов.
75. Механизм токсичности аммиака
Увеличение концентрации аммиака в крови:• Сдвигает рН в щелочную сторону (алкалоз) →
увеличение сродства гемоглобина к кислороду
→ гипоксия тканей.
• Стимулирует синтез глутамина.
NH3 + Glu + ATP → Gln + ADP + H3PO4
Накопление глутамина → повышение
осмотического давления → набухание
астроцитов → отек мозга.
Уменьшение концентрации глутамата →
нарушение синтеза нейромедиаторов (ГАМК).
76. Механизмы обезвреживания аммиака
Временные механизмы:
Восстановительное аминирование
α-кетоглутарата.
Синтез глутамина и аспарагина
(периферические ткани).
Окончательные механизмы:
Образование аммонийных солей (почки).
Синтез мочевины (печень).
77. Восстановительное аминирование α-кетоглутарата
NH3 + α-кг + NADPH+H+→Glu + NADP+ +H2OФермент – глутаматдегидрогеназа.
Интенсивно протекает в головном мозге.
78. Синтез глутамина
Глутамин является основной транспортной формойаммиака от мышц, мозга, печени.
Транспортируется в почки и кишечник.
79. Освобождение аммиака из глутамина
Почки – аммиак идет на синтез аммонийных солей.Кишечник – аммиак транспортируется через
воротную вену к печени → синтез мочевины????
80. Образование аммонийных солей в почках
NH3 + H+ + Cl-→ NH4ClОбразование аммонийных солей в почках
является одним из механизмов регуляции
кислотно-щелочного равновесия и
сохранения основных катионов (Na+, K+)
для поддержания осмотического давления.
Глутаминаза почек индуцируется при
ацидозах (аммиак нейтрализует кислые
продукты, которые выводятся с мочой).
При алкалозах активность фермента
снижается.
81. Транспорт аммиака в виде аланина (глюкозо-аланиновый цикл)
82. Синтез мочевины (орнитиновый цикл, цикл Кребса-Гензелейта)
• Происходит в печени.• Мочевина является основным
конечным продуктом азотистого
обмена (90% всего азота).
• За сутки с мочой выделяется 20-35 г
мочевины.
83. Синтез мочевины
84. Синтез мочевины
85. Синтез мочевины
86. Синтез мочевины
87. Суммарное уравнение синтеза мочевины
NH3 + CO2 + Asp + 3ATP + 2H2O →NH2-CO-NH2 + фумарат + 2 ADP +
AMP + 4H3PO4
Затраты энергии – 3ATP
(4 макроэргические связи).
Связь с циклом Кребса: фумарат →малат
→ оксалоацетат → аспартат.
88. Связь орнитинового цикла с циклом Кребса
89. Связь орнитинового цикла с циклом Кребса
90. Гипераммониемия
- повышение содержания аммиака в крови.6000 мкмоль/л
Концентрация аммиака в крови в норме –
60 мкмоль/л
Причины гипераммониемии:
• Наследственные дефекты ферментов
орнитинового цикла.
• Поражение печени – циррозы, гепатиты.
91. Гипераммониемия
Тошнота, рвота.
Головокружение.
Судорожные припадки.
Потеря сознания.
Отставание физического развития (при
хронических врожденных формах).
• Кома.
• Летальный исход.
92. Врожденные гипераммониемии
ЗаболеваниеГипераммониемия,
тип I
Гипераммониемия,
тип II
Цитрулинемия
Аргининосукцинатурия
Гипераргининемия
Дефект фермента
Карбамоилфосфатсинтетаза I
Орнитинкарбамоилтрансфераза
Аргининосукцинатсинтетаза
Аргининосукцинатлиаза
Аргиназа
93. Принципы лечения гипераммониемии
• Малобелковая диета.• Введение кетоаналогов аминокислот.
• Стимуляция выведения аммиака в обход
нарушенных реакций:
1. Путем связывания и выведения аммиака.
2. Повышением концентрации
промежуточных продуктов орнитинового
цикла, образующихся вне блокируемых
реакций.
94. Принципы лечения гипераммониемии I типа
• Примеры:• Фенилацетат + глутамин → фенилацетилглутамин → выводится с мочой →
происходит стимуляция синтеза глутамина
→ снижение концентрации аммиака.
• Бензойная кислота + глицин →гиппуровая
кислота →выводится с мочой. Недостаток
глицина компенсируется синтезом из
серина или из NH3 и CO2 → снижение
концентрации аммиака.
95. Принципы лечения гипераммониемий
Гипераммониемия II типа:• Введение больших доз цитруллина
стимулирует синтез мочевины из аспартата
→ выведение азота из организма.
Аргининосукцинатурия:
• Введение больших доз аргинина
стимулирует регенерацию орнитина и
выведение азота в виде цитруллина и
аргининосукцината.
96. Гликогенные и кетогенные аминокислоты
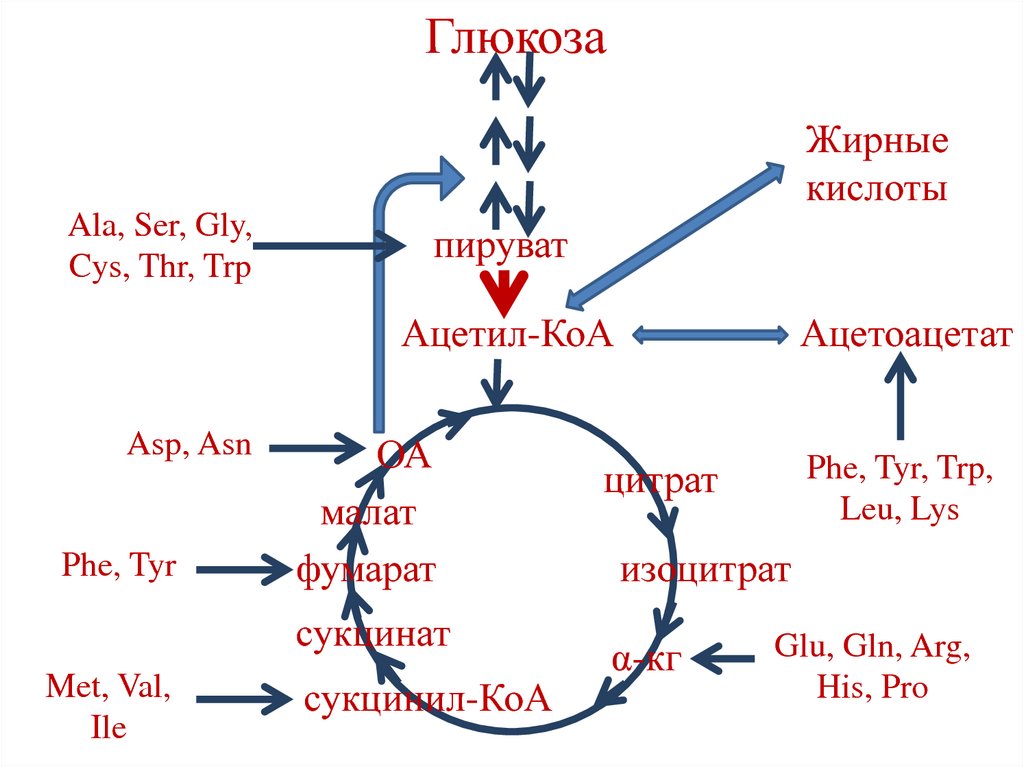
• Гликогенные аминокислоты – АК, которыепревращаются в промежуточные продукты
цикла Кребса или пируват.
• Кетогенные аминокислоты – АК, которые
превращаются в ацетоацетат или ацетилКоА (Leu).
• Смешанные аминокислоты – гликокетогенные АК (Phe, Tyr, Trp, Lys).
97.
ГлюкозаЖирные
кислоты
Ala, Ser, Gly,
Cys, Thr, Trp
пируват
Ацетил-КоА
Asp, Asn
Phe, Tyr
ОА
малат
фумарат
сукцинат
Met, Val,
Ile
сукцинил-КоА
Ацетоацетат
Phe, Tyr, Trp,
Leu, Lys
цитрат
изоцитрат
α-кг
Glu, Gln, Arg,
His, Pro
98. Синтез заменимых аминокислот
1. Трансреаминирование.2. Из незаменимых аминокислот:
Phe →Tyr
Met + Ser →Cys
99. Синтез заменимых аминокислот
100. Синтез заменимых аминокислот
101. Синтез заменимых аминокислот
102. Синтез заменимых аминокислот
103. Синтез заменимых аминокислот
104. Синтез заменимых аминокислот
105. Синтез заменимых аминокислот
106. Декарбоксилирование аминокислот
отщепление карбоксильной группыаминокислот в виде СО2.
• 4 типа декарбоксилирования аминокислот:
1. α-Декарбоксилирование.
2. ω-Декарбоксилирование.
3. Декарбоксилирование, связанное с
реакцией трансаминирования.
4. Декарбоксилирование, связанное с
реакцией конденсации двух молекул.
107. α-декарбоксилирование аминокислот
• Локализация – печень, почки, мозг, кишечник.• приводит к образованию биогенных аминов
(биологически активные вещества).
• Ферменты – декарбоксилазы аминокислот.
• Простетическая группа – пиридоксальфосфат
(производное витамина В6).
COOH
R CH NH2
Аминокислота
-СО2
R CH2 NH2
Биогенный амин
108.
NNH
CH2 CH NH2
COOH
Гистидин
Гистидин
декарбоксилаза N
-СО2
CH2 CH2 NH2
NH
Гистамин
Биологическая роль гистамина:
•Сосудорасширяющее действие.
•Участвует в воспалении (вызывая расширение
сосудов в очаге воспаления, ускоряет приток
лейкоцитов).
•Участвует в аллергических реакциях (явления
сенсибилизации и десенсибилизации).
•Является медиатором боли.
•Участвует в секреции соляной кислоты в желудке.
109. Декарбоксилирование ароматических аминокислот
Происходит под действиемдекарбоксилазы ароматических
аминокислот (не обладает строгой
субстратной специфичностью и
катализирует декарбоксилирование
триптофана, 5-окситриптофана и 3,4диоксифенилаланина (ДОФА)).
110.
HONH
CH2 CH COOH
NH2
Гидрокситриптофан
-СО2
HO
CH2 CH2 NH2
NH
Серотонин
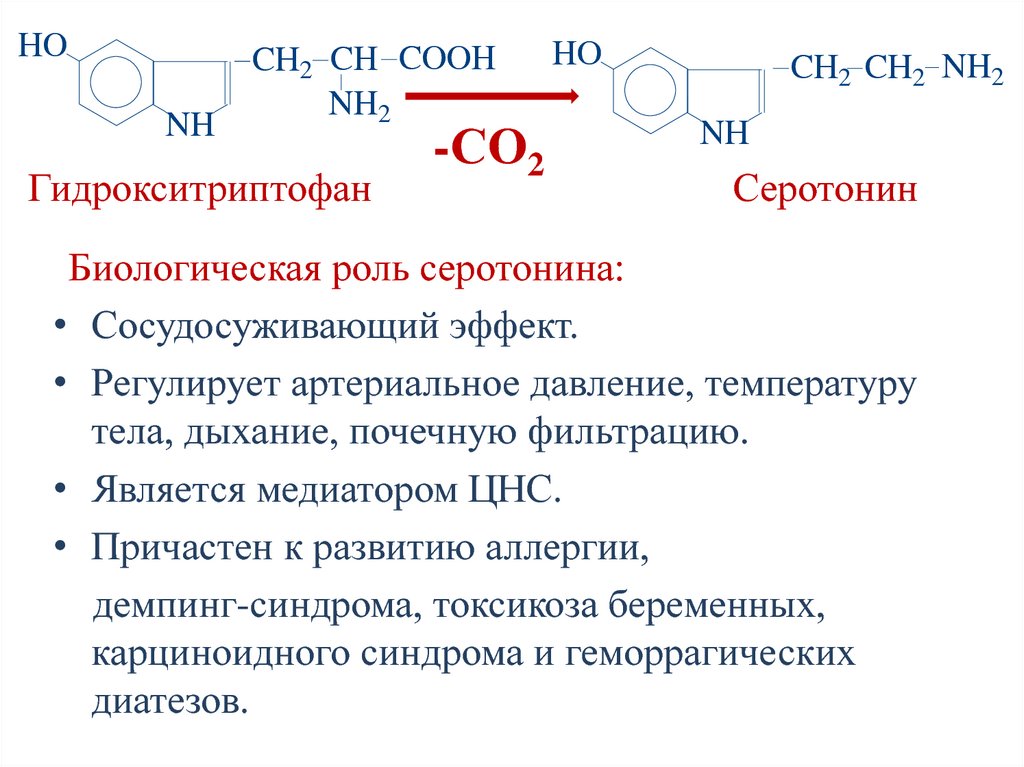
Биологическая роль серотонина:
• Сосудосуживающий эффект.
• Регулирует артериальное давление, температуру
тела, дыхание, почечную фильтрацию.
• Является медиатором ЦНС.
• Причастен к развитию аллергии,
демпинг-синдрома, токсикоза беременных,
карциноидного синдрома и геморрагических
диатезов.
111.
HOHO
CH2 CH COOH
NH2
Диоксифенилаланин
(ДОФА)
HO
-СО2
CH2 CH2 NH2
HO
Дофамин
Биологическая роль дофамина:
•Предшественник катехоламинов
(норадреналина и адреналина).
•Медиатор ЦНС.
•Ингибиторы декарбоксилазы ароматических
аминокислот вызывают снижение артериального
давления
α-метилдофа (альдомет)
112.
α-декарбоксилирование глутаминовой кислотыCOOH
CH NH2
CH2
CH2
COOH
Глутамат
Глутамат
декарбоксилаза
-СО2
CH NH2
CH2
CH2
COOH
γ-аминомасляная
кислота (ГАМК)
Биологическая роль ГАМК:
•Тормозящие действие на ЦНС.
•ГАМК и глутаматдекарбоксилаза – в сером веществе
коры большого мозга.
•ГАМК и Glu используются при заболеваниях ЦНС,
связанных с возбуждением коры большого мозга
(эпилепсия).
113. Нейтрализация биогенных аминов
MAO (моноаминоксидаза)R-CH2-NH2 + E-FAD + H2O →R-CHO + NH3 + E-FADH2
1/2O2
R-COOH
мочевина
E-FADH2 + О2 → E-FAD + H2O2
каталаза
H2O +1/2O2
114. Обмен отдельных аминокислот
115. Обмен фенилаланина и тирозина
• Фенилаланин – незаменимая аминокислота.• Тирозин – заменимая аминокислота,
синтезируется из фенилаланина.
• Фенилаланин → Тирозин.
• Фермент – фенилаланингидроксилаза
• Коферменты –
тетрагидробиоптерин, НАДФН.
116. Синтез тирозина из фенилаланина
117. Роль фенилаланина и тирозина
• Синтез катехоламинов (дофамин,норадреналин, адреналин) –
надпочечники, нервная ткань.
• Синтез гормонов щитовидной
железы (Т3, Т4) – щитовидная
железа.
• Синтез меланинов – меланоциты.
118. Синтез катехоламинов
119. Гормоны щитовидной железы
120. Синтез меланинов
121. Катаболизм фенилаланина и тирозина
122. Наследственные нарушения обмена фенилаланина и тирозина
фенилаланинтирозин
1
фенилпируват
меланины
2
гидроксифенилпируват
фениллактат
гомогентизат
3
фенилацетат
123. 1. Фенилкетонурия
Причины:• Мутации гена фенилаланингидроксилазы
(частота – 1на 10 000).
• Мутации генов, контролирующих обмен
тетрагидробиоптерина.
124. Фенилкетонурия
Клинические проявления:• Нарушение умственного и физического
развития.
• Причины – высокие концентрации
фенилаланина, фенилпирувата,
фениллактата ограничивают транспорт
тирозина и триптофана через ГЭБ →
нарушается синтез нейромедиаторов.
125. Метаболические нарушения при фенилкетонурии
126. 2. Альбинизм
• Причина – врожденный дефекттирозиназы (катализирует
превращение тирозина в ДОФА в
меланоцитах).
• Частота – 1:20 000.
• Клинические проявления:
• Отсутствие пигментации кожи и волос.
• Предрасположенность к раку кожи.
127. 3. Алкаптонурия
• Причина – дефект гомогентизатдиоксигеназы.
• Клинические проявления:
• «черная моча» – гомогентизиновая кислота
окисляется, образуя темные пигменты –
алкаптоны.
• Охроноз – пигментация соединительной
ткани.
• Артрит.
128. Тирозинемии
• Тирозинемия I типа (тирозиноз) – дефектфумарилацетоацетатгидролазы.
• Тирозинемия II типа (синдром РихнераХанхорта) – дефект
тирозинаминотрансферазы.
• Тирозинемия новорожденных
(кратковременная) –дефект парагидроксифенилпируватдиоксигеназы.
129. Фолиевая кислота
• Является витамином• Состоит из 3-х компонентов:
1. Птеридин
2. Пара-аминобензойная кислота
3. Глутамат
130. Фолиевая кислота
131. Тетрагидрофолиевая кислота (ТГФК)
• Является коферментом фолиевой кислоты.• Образуется из фолиевой кислоты в ходе 2-х
реакций восстановления (печень).
1. Ферменты – фолатредуктаза и
дигидрофолатредуктаза.
2. Кофермент – НАДФН.
1
2
ФК
НАДФН+Н+
ДГФК
НАДФ+ НАДФН+Н+
ТГФК
НАДФ+
132. Тетрагидрофолиевая кислота
НН
Н
Н
133. Тетрагидрофолиевая кислота
134. Роль ТГФК
ТГФК является переносчикомодноуглеродных фрагментов:
–СН3 (метил)
– СОН
(формил)
–СН2– (метилен) – СН=NH (формимино)
–СН= (метенил)
Одноуглеродные фрагменты связываются к
N5 и/или N10 ТГФК.
135. Роль ТГФК
136. Роль ТГФК
ТГФК участвует в обмене:
серина
глицина
метионина
пуриновых нуклеотидов
пиримидиновых нуклеотидов
(тимидилата)
137. Роль ТГФК в обмене серина и глицина
138. Роль ТГФК в обмене метионина
139. Мегалобластная (макроцитарная) анемия
Причина – недостаточность фолиевойкислоты и/или витамина В12.
Проявления:
• Уменьшение количества эритроцитов.
• Снижение количества гемоглобина в них.
• Увеличение размера эритроцитов.
Причина симптомов – нарушение синтеза
ДНК и РНК (уменьшение синтеза
тимидилата и пуриновых нуклеотидов).
140. Механизм антибактериального действия сульфаниламидных препаратов
• Сульфаниламидные препараты похожипо строению на парааминобензойную
кислоту.
141. Механизм антибактериального действия сульфаниламидных препаратов
Подавление синтеза фолиевой кислоты:• Конкурентно ингибируют бактериальные
ферменты синтеза фолата.
• Могут использоваться как
псевдосубстраты из-за относительной
субстратной специфичности ферментов.
В результате нарушается синтез
нуклеиновых кислот, что вызывает
прекращение размножения бактерий.
142. Обмен метионина
Незаменимая аминокислота.
Роль:
Синтез белков.
Образование S-аденозилметионина
(активная форма метионина).
• Синтез цистеина (Мет является источником
серы).
• Гликогенная аминокислота.
143. Роль S-аденозилметионина (SАМ)
Является донором метильной группы:• Синтез адреналина из норадреналина.
• Синтез фосфатидилхолина из
фосфатидилэтаноламина.
• Синтез холина из этаноламина → ацетилхолин
• Синтез креатина.
• Синтез меланина.
• Синтез карнитина.
• Синтез анзерина из карнозина.
• Обезвреживание токсичных веществ, лекарств.
• Метилирование азотистых оснований.
144. Синтез S-аденозилметионина (SАМ)
145. Синтез фосфатидилхолина из фосфатидилэтаноламина
146. Синтез карнитина
147. SАМ + R → SАГЦ +R-CH3
Метилкобаламин(В12)
148. Синтез цистеина
Кофермент – пиридоксальфосфат (В6)149. Обмен метионина
150. Синтез креатина
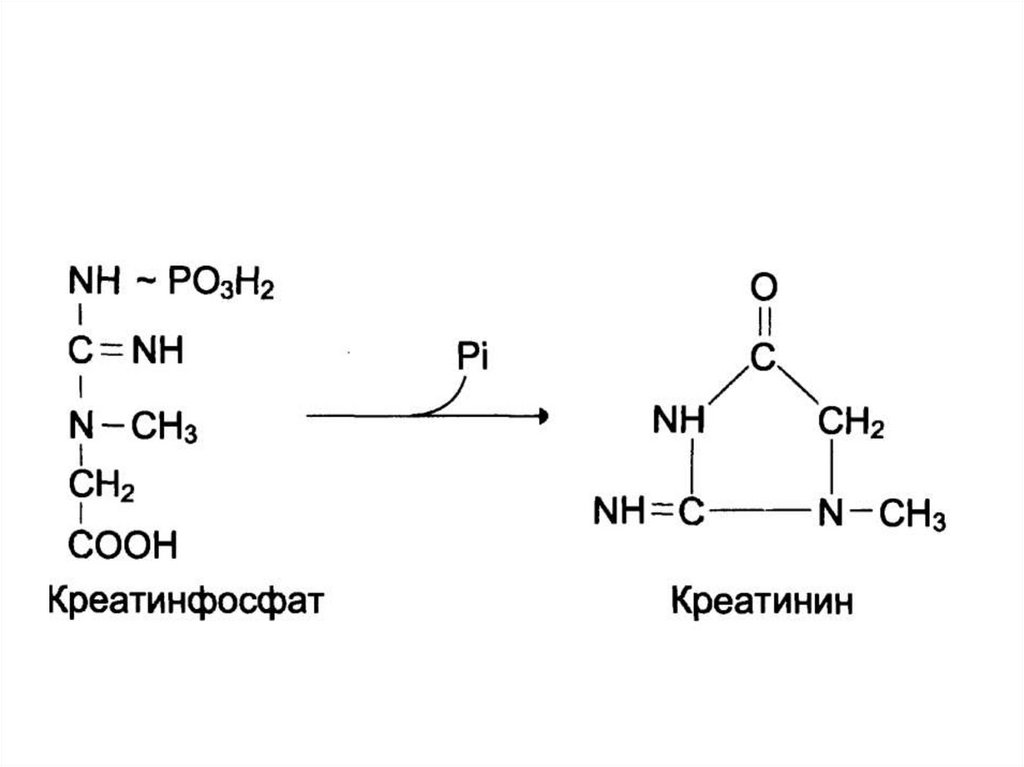
151. Синтез креатина
152. Синтез креатина
153.
154. Гипергомоцистеинемия
• Причины – дефект цистатионин синтазы,• недостаточность витаминов В6, В12 и
фолевой кислоты
• Гипергомоцистеинемия – фактор риска
атеросклероза.
155. Гомоцистинурия нарушение использования гомоцистеина, он восстанавливается в гомоцистин. Причина – насcледсвенное нарушение
обменагомоцистеина или гиповитаминозы фолиевой
кислоты, витаминов В12 и В6
156. Обмен цистеина
• Заменимая аминокислота, синтезируется изсерина (источник углеродного скелета) и
метионина (источник серы).
157. Роль цистеина
• Синтез цистина (образование дисульфидныхсвязей между 2-мя молекулами цистеина) –
стабилизация третичной структуры белков.
• НS- группы цистеина входят в состав.
активных центров многих ферментов.
• Входит в состав глутатиона.
• Синтез таурина (конъюгирование желчных
кислот).
• Синтез тиоэтиламина (НS-КоА).
• Синтез белков.
• Синтез глюкозы.
158. Катаболизм цистеина
Сульфиты превращаются в сульфаты,которые выводятся с мочой.
Пируват → ацетил-КоА → цикл Кребса
159. Цистинурия
• Причина – нарушение транспорта цистеинаи щелочных аминокислот в кишечнике и
почках.
• Цистеин восстанавливается до цистина,
который малорастворим.
• Образуются почечные камни.
160.
161.
162. Глутатион
• Трипептид γ-Glu-Cys-Gly• Синтез – из соответствующих аминокислот:
Glu + Cys → γ-Glu-Cys (АТФ)
γ-Glu-Cys + Gly → γ-Glu-Cys-Gly (АТФ)
• Ферменты – γ-Glu-Cys-синтетаза и
глутатион-синтетаза.
• Роль:
• Восстанавливающий агент – является
коферментом многих ферментов.
163. Ферменты, коферментом которых является глутатион
Глутатион пероксидаза
Гамма-глутамил трансфераза
Глутатион-инсулин трансгидрогеназа
Малеил-ацетоацетат изомераза
Формальдегид дегидрогеназа
Глиоксилаза
Предотвращает окисление тиогрупп цистеинсодержащих ферментов.
• Глутатион восстанавливается ферментом
глутатион редуктаза
164. Обмен серина
• Серин – заменимая аминокислота.• Синтезируется из 3-фосфоглицерата.
165. Синтез серина
166. Пути использования серина
Синтез глицина.
Синтез цистеина.
Синтез сфингозина.
Синтез фосфолипидов.
Синтез белков.
Синтез глюкозы.
Синтез липидов.
Окисление в цикле Кребса.
167. Пути использования серина
• Источник одноуглеродныхфрагментов для ТГФК.
• Входит в состав активных центров
многих ферментов
(серинпротеиназы).
• По остаткам серина происходит
фосфорилирование многих
ферментов.
168. Катаболизм серина
1Ацетил-КоА
2
Серин
Цикл Кребса
Глицин
СО2 + NH3
169. Обмен глицина
Глицин – заменимая аминокислота,
Синтезируется из:
Серина.
Треонина.
Аммиака и СО2 с участием ТГФК.
170. Синтез глицина
171. Пути использования глицина
Синтез серина.
Синтез гема.
Синтез глутатиона.
Синтез креатина.
Синтез пуриновых нуклеотидов.
Синтез формиата и оксалата.
Конъюгация желчных кислот.
172. Пути использования глицина
• Источник одноуглеродныхфрагментов для ТГФК.
• Тормозной медиатор ЦНС.
• Синтез белков.
• Синтез глюкозы.
• Синтез липидов.
• Окисление в цикле Кребса.
173. Катаболизм глицина
12
Глицин
Ацетил-КоА
Серин
Пируват
Цикл Кребса
174. Обмен глицина – образование глиоксилата
175. Обмен глицина – образование оксалата и одноуглеродных фрагментов для ТГФК
176. Наследсвенные нарушения обмена глицина
Гиперглицинемия – недостаточностьглицинрасщепляющей ферментной
системы.
• Клинические проявления – судороги,
гипотония, нарушение дыхания.
177. Наследсвенные нарушения обмена глицина
• Глицинурия – повышение выделенияглицина с мочой из-за нарушения
реабсорбции глицина в почках; в крови
содержание глицина нормальное.
• Клинические проявления – образование
оксалатных камней в почках при
нормальном содержании оксалатов в моче.
• Оксалат образуется из глиоксилата (продукт
дезаминирования глицина).
178. Наследсвенные нарушения обмена глицина
• Первичная гипероксалатурия – высокоевыделение оксалата с мочой, независимо от
его содержания в пище.
• Клинические проявления – образование
оксалатных камней в мочевыводящих
путях, нефрокальциноз и инфекция
мочевыводящих путей.
• Смерть в раннем возрасте из-за почечной
недостаточности или гипертонии.
179. Обмен аргинина
Полузаменимая аминокислота
Синтезируются из α-кетоглутарата
Роль:
Синтез белков
Гликогенная аминокислота
Синтез мочевины
Синтез креатин-фосфата
Синтез NO
180. Синтез NO
NH2NH2
CH COOH
CH2
CH2
O2
CH2
CH2
CH2
CH2
+
NH NADPH+H
C
NH
NH2
Arg
NADP+
H2O
NH2
CH COOH
NH
C
CH2
O2
½NADPH+H+
N OH
NH2
Гидроксиаргинин
CH COOH
CH2
½NADР+
H2O
+ NO
CH2
NH
C O
NH2
Цитруллин
Фермент – NO-синтаза
Коферменты – NADPH, FMN, FAD,
гем-Fe3+, тетрагидробиоптерин
181. Изоформы NO-синтазы
ИзоформаЛокализация
Эндотелиальная Эндотелий
NO-синтаза
Миокард
Тромбоциты
Неурональная
Нервная система
NO-синтаза
Скелетные
мышцы
Индуцируемая Иммунная
NO-синтаза
система
Макрофаги
Функции
Сосудорасширяющий эффект
Межклеточное
взаимодействие
Иммунный
ответ
Бактерицидный
ответ
182. Биологическая роль NO
Расслабляетгладкую
мускулатуру
Сосудорасширяющий
эффект
Предотвращает
агрегацию
тромбоцитов
NO
Регулирует
апоптоз
Антиканцерогенное
действие
Является
медиатором
183. Обмен триптофана
• Незаменимая аминокислота• Глюко- и кетогенная аминокислота
Роль:
• Синтез белков
• Синтез глюкозы
• Синтез триптамина
• Синтез серотонина
• Синтез меланина
• Синтез никотинамида
184. Синтез серотонина
NHО2
CH2 CH COOH
NH2
NH
NADPH+H+
Trp
HO
-CО2
HO
NADP+
Н2О
CH2 CH COOH
NH2
Гидрокситриптофан
CH2 CH2 NH2
NH
Серотонин
Ферменты: 1.Триптофан гидроксилаза
2.Декарбоксилаза ароматических аминокислот
185. Синтез мелатонина
HOHO
CH2 CH2 NH2
NH
NH
Серотонин СН3-СО-SCoA
HSCoA
CH3 O
NH
SАМ
SАНС
CH2 CH2 NH C CH3
O
N-ацетилсеротонин
CH2 CH2 NH C CH3
O
Мелатонин
Ферменты: 1.Серотонин N-ацетилаза
2.N-ацетилсеротонин О-метил-трансфераза
186. Мелатонин
• Является гормоном• Синтезируется в эпифизе (максимальная
концентрация наблюдается ночью).
• Синтез и секреция мелатонина зависит от
освещенности – избыток света понижает его
синтез, а снижение освещенности увеличивает
синтез и секрецию мелатонина.
Роль:
• Регуляция циркадного ритма (цикл сонбодрствование).
187. Обмен глутамата и глутамина
• Заменимые аминокислоты.• Синтезируются из α-кетоглутарата:
• α-кетоглутарат → глутамат → глутамин.
188. Использование глутамата
• Занимает центральное место в синтезе икатаболизме аминокислот
(трансдезаминирование и
трансреаминирование).
• Медиатор ЦНС.
• Предшественник ГАМК.
• Входит в состав глутатиона.
• Входит в состав фолиевой кислоты.
189. Использование глутамина
• Основная форма транспорта аммиака.• Участвует в синтезе пуринового кольца.
• Участвует в синтезе пиримидинового
кольца.
• Синтез пуриновых и пиримидиновых
нуклеотидов в качестве донора
аминогруппы.
190. Катаболизм глутамата и глутамина
• Глутамин → глутамат → α-кетоглутарат →цикл Кребса.
• Гликогенные аминокислоты.
191. Обмен аспартата и аспарагина
• Заменимые аминокислоты.• Синтезируются из оксалоацетата и аммиака
(трансреамиенирование):
• Оксалоацетат → аспартат → аспарагин.
192. Использование аспартата
Синтез мочевины.
Синтез пуринового кольца.
Синтез пиримидинового кольца.
Синтез пуриновых нуклеотидов.
NMDA – медиатор ЦНС.
193. Катаболизм аспартата и аспарагина
• Аспарагин → аспартат → оксалоацетат →цикл Кребса.
• Гликогенные аминокислоты.
194.
Обмен пуриновыхи пиримидиновых
нуклеотидов
195. Переваривание нуклеопротеинов в ЖКТ
• В желудке под действием HClпроисходит распад нуклеопротеинов и
денатурация белка.
• Расщепление нуклеиновых кислот
происходит в тонком кишечнике.
196. Переваривание нуклеиновых кислот в ЖКТ
Нуклеиновые кислотыДНК-азы и РНК-азы
Олигонуклеотиды
Фосфодиэстеразы
Мононуклеотиды
Нуклеотидазы
Нуклеозиды + фосфат
Нуклеозидфосфорилазы
Азотистые основания + пентоза-1-фосфат
197. Переваривание нуклеопротеинов в ЖКТ
• В энтероцитах основная часть пуриновыхоснований под действием ксантиноксидазы
превращается в мочевую кислоту, которая
выделяется с мочой.
• Большая часть пиримидиновых оснований
не всасывается, а в полости кишечника под
действием микрофлоры расщепляется до
СО2, Н2О, -аланина и -аминоизобутирата.
198. Синтез пуриновых нуклеотидов de novo
• Происходит в основном в печени, откудапуриновые нуклеотиды поступают в ткани,
не способные к их синтезу (эритроциты,
головной мозг).
• Пуриновый цикл синтезируется на остатке
рибозо-5-фосфата при участии доноров
углерода и азота.
199. Источники атомов пуринового кольца
200. Синтез пуриновых нуклеотидов de novo
201. Синтез пуриновых нуклеотидов de novo
202. Синтез пуриновых нуклеотидов de novo
В следующих реакциях участвуютглицин, глутамин, аспартат, СО2,
N5,N10-метенил- THF, N10формилTHF, АТФ (для энергии), в
результате образуется ИМФ
(инозин-монофосфат), который
является предшественником АМФ
и ГМФ.
203. Синтез АМФ и ГМФ
Asp, GTPGDP, Pi
аденилосукцинат
ИМФ
NAD+, H2O
NADH+H+
КМФ
Gln, ATP
фумарат
АМФ
ГМФ
Glu
AMP, PPi
204. Синтез нуклеозиддифосфатов и нуклеозидтрифосфатов
GMP + ATP → GDP + ADPФермент – гуанилаткиназа
GDP + ATP → GTP + ADP
Фермент – нуклеозиддифосфаткиназа
AMP + ATP → 2 ADP
Фермент – аденилаткиназа
ADP + H3PO4 → ATP
Окислительное фосфорилирование
205. «Запасной» путь синтеза пуриновых нуклеотидов
Аденозин + АТФ → АМФ + АДФФермент – аденозинкиназа
206. «Запасной» путь синтеза пуриновых нуклеотидов
207. Регуляция синтеза пуриновых нуклеотидов
208. Регуляция синтеза пуриновых нуклеотидов
• ФРПФ синтетаза – аллостерическийфермент:
• Активатор – неорганический фосфат
• Ингибиторы – пуриновые нуклеотиды
• Амидофосфорибозилтрансфераза:
• Активатор – ФРПФ
• Ингибиторы – пуриновые нуклеотиды
209. Катаболизм пуриновых нуклеотидов
210. Катаболизм пуриновых нуклеотидов
АМФаденозин
инозин
Н2О
Н3РО4
Н2О
NН3
Н2О
Н3РО4
Н3РО4
Рибозо-1-Р
Н3РО4
Рибозо-1-Р
гипоксантин
О2 Н2О
Н2О
NН3
Н2О2
ГМФ
гуанозин
гуанин
ксантин
О2
Н2О
Н2О2
Мочевая кислота
211. Катаболизм пуриновых нуклеотидов
ON
HN
N
N
H
Гипоксантин
Ксантиноксидаза
O
O
О2 Н2О
N
HN
N
H
Н2О2
N
H
Ксантин
О2 Н2О
Ксантиноксидаза
O
H
N
HN
O
Н2О2
O
N
H
N
H
Мочевая кислота
212. Катаболизм пуриновых нуклеотидов
• Конечный продукт обмена пуринов учеловека – мочевая кислота.
• Концентрация мочевой кислоты в крови –
0,15-0,47 ммоль/л (0,3-0,7 г/л).
• Мочевая кислота выделяется с мочой – 0,40,6 г/сутки.
• Повышение концентрации мочевой кислоты
– гиперурикемия.
213. Подагра
• Заболевание, при котором происходитотложение уратов в суставных хрящах,
синовиальной оболочке, подкожной
клетчатке.
• Клиническая картина:
• Подагрический артрит – приступы острых
болей в мелких суставах.
• Нефролитиаз – уратные камни в
мочевыводящих путях.
• Подагрические тофусы (узлы).
214. Подагра
• Концентрация мочевой кислоты приподагре – 2-4 г/л (при легкой форме, без
тофусов), до 30 г (при тяжелых формах).
• Распространенность – 0,3-1,7%.
• Мужчины болеют в 20 раз чаще, чем
женщины.
• Подагра носит семейный характер
(генетически детерминирована).
215. Причины подагры (генетические дефекты )
• Высокая активность ФРПФ синтетазы (неподается контролю со стороны
нуклеотидов).
• Недостаточность глюкозо-6-фосфатазы
(накапливается глюкозо-6-фосфат →
пентозо-фосфатный путь → много рибозы).
• Недостаточность гипоксантин-гуанинфосфорибозил-трансферазы.
216. Синдром Lesch-Nyhan
• Тяжелая форма гиперурикемии.• Рециссивный признак, сцепленный с Ххромосомой, болеют только мальчики. Причина –
полное отсутствие активности гипоксантингуанин-фосфорибозил-трансферазы.
Клинические проявления:
• Тофусы, уратные камни.
• Неврологические отклонения (нарушение речи,
церебральный паралич, снижение интеллекта,
склонность к нанесению себе увечий).
• Ранняя смерть (до 10 лет).
217. Лечение подагры
OКсантиноксидаза
N
HN
N
H
N
Гипоксантин
O
O
О2 Н2О
N
HN
N
H
Н2О2
Ксантин
HN
O
N
N
О2 Н2О
Ксантиноксидаза
O
N
H
Аллопуринол – структурный
Аналог гипоксантина!
N
H
H
N
HN
O
Н2О2
O
N
H
N
H
Мочевая кислота
218. Лечение подагры
Механизм действия аллопуринола:• Конкурентно ингибирует ксантиноксидазу,
останавливая катаболизм пуриновых
нуклеотидов на стадии гипоксантина,
растворимость которого в 10 раз выше
мочевой кислоты.
• Являясь псевдосубстратом, используется на
синтез нуклеотидов по «запасному пути»,
ингибируются регуляторные ферменты
синтеза нуклеотидов de novo.
219. Синтез пиримидиновых нуклеотидов
220. Синтез пиримидиновых нуклеотидов
221. Синтез пиримидиновых нуклеотидов
222.
223. Синтез дезоксирибонуклеотидов
224. Синтез дезоксирибонуклеотидов
Ферменты:
Рибонуклеотидредуктаза
Тиоредоксинредуктаза
Синтезируются dАДФ, dГДФ, dЦДФ,
dУДФ.
• dНДФ + АТФ→ dНТФ + АДФ
• Ферменты – НДФ-киназы
225. Синтез тимидиловых нуклеотидов
226. Синтез тимидиловых нуклеотидов
Ферменты:2 – тимидилатсинтаза
4 – дигидрофолат редуктаза
5 - серингидроксиметилтрансфераза
227. Катаболизм пиримидиновых нуклеотидов
ЦитозинУрацил
Тимин
-аланин
-аминоизобутират
NH3 СО2
NH3 СО2
228. Регуляция синтеза пиримидиновых нуклеотидов
• Карбамоилфосфатсинтетаза II (КАДфермент):• Активатор – ФРПФ
• Ингибиторы – пиримидиновые нуклеотиды.
• Рибонуклеотидредуктаза и
тимидилатсинтаза – индуцируемые
ферменты (их синтез достигает максимума
во время активного синтеза ДНК)
• Рибонуклеотидредуктаза – ингибируется
дезоксирибонуклеозид-фосфатами.
229. Нарушения обмена пиримидиновых нуклеотидов
Оротацидурия
Причина – врожденный дефект УМФ-синтазы.
Клинические проявления:
мегалобластная анемия, которая не подается
лечению фолиевой кислотой
Неврологические нарушения
Повышенная чувствительность к инфекциям
Образование камней
Ранняя смерть
Лечение – уридин
230. Противоопухолевые препараты
Является аналогом урацилаПродукт его превращения
в организме – 5-фтор-дУМФ
является конкурентным
ингибитором
тимидилатсинтазы.
Используется в качестве
противоопухолевого
средства.
231. Противоопухолевые препараты
Нуклеозид, в которомрибоза замещена на
арабинозу.
В организме
превращается в дНТФ,
который ингибирует
ДНК-полимеразу и
снижает скорость
репликации.
232. Противоопухолевые препараты
Метотрексат и аминоптерин – структурныеаналоги фолиевой кислоты.
Ингибируют дигидрофолатредуктазу,
снижая синтез ДНК.
233. Противовирусные препараты
Азидотимидин (АЗТ)фосфорилируется с
образованием нуклеотидов,
которые используются
вирусной ДНК-полимеразой
для синтеза ДНК.
Прекращается синтез
ДНК.
Используется при
ВИЧ-инфекции.
234. Противовирусные препараты
5-йод-2-дезоксиуридиниспользуется при лечении
кератитов и поражении
роговицы вирусом герпеса.
Азатиоприн в организме
превращается в
6-меркаптопурин, который
оказывает выраженное
иммуносупрессорное
действие. Используется
в трансплантологии.
235.
ОБМЕНГЕМОПРОТЕИНОВ
236. ГЕМОПРОТЕИНЫ
Гемоглобин
Миоглобин
Цитохромы дыхательной цепи
Цитохром Р450
Каталаза
Пероксидаза
237. Синтез гема
• Происходит во всех тканях, но снаибольшей скоростью в костном
мозге (для синтеза гемоглобина) и в
печени (для синтеза цитохрома Р450).
• Протекает в митохондриях (первая
реакция и конечные этапы – от
копропорфириногена до гема) и в
цитоплазме.
238.
239. Первая реакция синтеза гема
240. Вторая реакция синтеза гема
241. Регуляция синтеза гема
242. Регуляция синтеза гема
1. Аминолевулинатсинтаза:• Аллостерическая регуляция –
ингибируется гемом
• Регуляция на уровне трансляции –
увеличение концентрации железа
стимулирует синтез фермента.
1. Аминолевулинатдегидратаза
аллостерически ингибируется гемом
2. Гем регулирует синтез глобина.
243. Нарушения синтеза гема
• Порфирии – наследственные иприобретенные нарушения синтеза гема,
сопровождающиеся повышением
содержания порфириногенов и продуктов
их окисления в тканях и в крови, а также
появлением их в моче.
• «Порфирин» в переводе с греческого
означает пурпурный.
244. Наследсвенные порфирии
• Причины – генетические дефектыферментов, участвующих в синтезе гема, за
исключением аминолевулинатсинтазы.
• Снижается синтез гема.
• Накапливаются промежуточные продукты
синтеза гема – аминолевулинат и
порфириногены.
245. Наследсвенные порфирии
• Печеночные порфирии –накопление порфиринов в
гепатоцитах.
• Эритропоэтические порфирии –
накопление порфиринов в
эритроцитах.
246. Наследсвенные порфирии
Клинические проявления тяжелых форм:• Нейропсихические расстройства
(аминолевулинат и порфириногены
являются нейротоксинами)
• Повреждения кожи (фотосенсибилизация и
изъязвления открытых участков кожи)
• Нарушения функции РЭС
247. Наследсвенные порфирии
Легкие формы могут протекать
бессимптомно, но прием лекарств,
являющихся индукторами
аминолевулинатсинтазы, вызывают
обострение.
Сульфаниламиды
Барбитураты
Диклофенак, вольтарен
Стероиды
248. Приобретенные порфирии
• Отравления солями свинца (свинецингибирует
аминолевулинатдегидратазу и
феррохелатазу).
• Некоторые галогенсодержащие
гербициды и инсектициды
(индуцируют
аминолевулинатсинтазу).
249. Обмен железа
• Содержание железа в организме – 3-4 г/л, вплазме – 3,5 мг.
• Гемоглобин – 68%
• Ферритин – 27%
• Миоглобин – 4%
• Трансферрин – 0,1%
• Ферменты – 0,6%
250. Всасывание железа
• В пище железо находится в окисленнойформе (Fe3+) – в составе белков и солей
органических кислот.
• Кислая среда желудочного сока
способствует освобождению железа из
солей органических кислот.
• Аскорбиновая кислота восстанавливает
железо и улучшает его всасывание
(всасывается только двухвалентное железо).
• Железо всасывается в основном в
двенадцатиперстной кишке.
251. Всасывание железа
• Поступление железа из энтероцитов в кровьзависит от скорости синтеза в них
апоферритина.
• Апоферритин связывает железо и
превращается в ферритин, который
остается в энтероцитах → снижается
поступление железа в кровь.
• При недостатке железа в организме
апоферритин почти не синтезируется →
железо поступает в кровь.
252. Транспорт железа в плазме крови
• Поступившее в кровь Fe2+ окисляется поддействием церулоплазмина в Fe3+ и
связывается трансферином.
• Трансферрин – гликопротеин,
синтезируется в печени, связывает только
Fe3+.
• Одна молекула трансферрина может связать
1 или 2 иона Fe3+.
• В норме трансферрин насыщен железом на
33%.
253. Поступление железа в клетки
• Трансферрин взаимодействует с мембраннымирецепторами
• → в клетке образуется комплекс Са2+кальмодулин-ПКС
• → фосфорилирование рецептора
• → образование эндосомы
• → в кислой среде эндосомы железо
освобождается от трансферрина
• → комплекс рецептор-трансферрин
возвращается на поверхность мембраны
• → апотрансферрин поступает в кровь.
254. Использование железа в тканях
1. Синтез железосодержащих белков2. Депонируется в составе ферритина.
• Ферритин – олигомерный белок
• Состоит из 24-х протомеров
• Образует полую сферу, в которой может
содержатся до 4500 ионов железа.
• Высокое содержание ферритина – печень,
селезенка, костный мозг.
255. Ферритин
• Концентрация ферритина вкрови является важным
диагностическим
показателем запасов железа в
тканях при
железодефицитной анемии.
256. Обмен железа
257. Железодефицитная анемия
Причины:
Кровотечения
Беременность
Частые роды
Язвы и опухоли ЖКТ
Операции на ЖКТ
258. Железодефицитная анемия
• Уменьшение размера и пигментацииэритроцитов (гипохромная анемия)
• Уменьшение содержания гемоглобина
в эритроцитах
• Понижение насыщения железом
трансферрина
• Снижение концентрации в тканях и в
крови ферритина.
259. Гемохроматоз
• Избыток железа в клетках(превышение ферритинового депо)
приводит к образованию гемосидерина
(железо откладывается в виде
аморфных отложений в белковой части
молекулы ферритина).
• Гемосидерин плохо растворим в воде и
содержит до 37% железа.
260. Гемохроматоз
• накопление гранул гемосидерина вклетках печени, селезенки,
поджелудочной железы с
повреждением этих органов.
• Причины – наследственно
обусловленное увеличение всасывания
железа в кишечнике.
• Содержание железа в организме
достигает 100 г.
261. Гемохроматоз
• Клинические проявления:• Сахарный диабет (разрушение клеток поджелудочной железы)
• Цирроз печени
• Сердечная недостаточность
(отложение гемосидерина в
кардиомиоцитах).
• Лечение – кровопускания.
262. Катаболизм гемоглобина
• Продолжительность жизниэритроцитов –120 дней.
• Катаболизм происходит в
клетках РЭС селезенки, печени,
лимфатических узлов, костного
мозга.
263. Катаболизм гемоглобина
• Первая реакция – расщеплениеметенового мостика между 1-м и 2-м
пиррольными кольцами –
раскрывается структура кольца –
образуется вердоглобин.
• От вердоглобина отщепляется
белковая часть и железо – образуется
биливердин.
• Фермент – гемоксигеназа.
264. Катаболизм гемоглобина
265. Катаболизм гемоглобина
• Биливердин восстанавливается вбилирубин под действием фермента
биливердин редуктаза.
266. Катаболизм гемоглобина
267. Катаболизм гемоглобина
• Билирубин из клеток РЭС выделяетсяв кровь, связывается и
транспортируется альбуминами.
• Связанный с альбумином билирубин
называется неконъюгированный
билирубин.
• На поверхности гепатоцитов комплекс
альбумин-билирубин диссоциирует.
268. Катаболизм гемоглобина
• Транспорт билирубина в клеткипечени происходит облегченной
диффузией с участием 2-х белков:
лигандина (основная часть) и белка Z.
• В гепатоцитах (гладкий ЭР)
происходит конъюгация билирубина с
1-м или 2-мя остатками глюкуроновой
кислоты – образуется
конъюгированный билирубин.
269. Катаболизм гемоглобина
• Донором глюкуроновойкислоты является UDPглюкуронат. Фермент –
UDP-глюкуронилтрансфераза.
• Фенобарбитал индуцирует
фермент.
270. Катаболизм гемоглобина
271. Структура конъюгированного билирубина
272. Катаболизм гемоглобина
• Конъюгированный билирубинсекретируется в желчь
(активный транспорт).
• Небольшая часть
конъюгированного билирубина
диффундирует в кровь.
273. Катаболизм гемоглобина
• В кишечнике конъюгированныйбилирубин гидролизуется под
действием бактериальных
ферментов -глюкуронидаз
(отщепление глюкуроновой
кислоты).
• Освободившийся билирубин
восстанавливается под действием
кишечной микрофлоры до
уробилиногенов.
274. Катаболизм гемоглобина
• В подвздошной и толстой кишкахнебольшая часть уробилиногенов
всасывается, с кровью воротной
вены поступают в печень.
• Большая часть уробилиногенов в
толстом кишечнике
восстанавливается до
стеркобилиногена, который
окисляется до стеркобилина
(пигмент коричневого цвета).
275. Диагностическое значение определения билирубина
• Нормальная концентрация билирубинав крови – 1,7-17 мкмоль/л.
• 75% – неконъюгированный (непрямой)
билирубин.
• Повышение концентрации билирубина
в крови – гипербилирубинемия.
• При превышении верхних пределов
нормы более чем в 2,5 раза возникает
желтуха.
276. Желтухи
• Гемолитическая(надпеченочная)
• Паренхиматозная (печеночная)
• Механическая, обтурационная
(подпеченочная)
• Желтуха новорожденных
277. Гемолитическая желтуха
• Причина – интенсивный гемолиз(наследственные или
приобретенные анемии).
• Чрезмерное образование
билирубина, превышающее
способность печени конъюгировать
билирубин.
278. Гемолитическая желтуха
• Повышение в крови концентрациинеконъюгированного билирубина.
• Большое количество пигментов в
кишечнике (интенсивно
окрашенный кал).
• Большое содержание
уробилиногенов в моче
(интенсивная окраска мочи).
279. Желтуха новорожденных
• Причины:• Усиленный гемолиз
• Недостаточность UDPглюкуронилтрансферазы.
• Лечение – большие дозы
фенобарбитала.
• Неконъюгированный билирубин
проходит ГЭБ – билирубиновая
энцефалопатия.
280. Токсичность билирубина
• Разобщение окисления ифосфорилирования
• Нарушение синтеза белка
• Нарушение транспорта калия
через мембраны
281. Печеночная желтуха
• Повреждение гепатоцитов и желчныхкапилляров (вирусные и токсические
гепатиты, цирроз печени).
• Нарушается:
• Захват билирубина гепатоцитами
• Конъюгирование билирубина
• Экскреция билирубина в желчные
капилляры
• Проницаемость мембран – билирубин
диффундирует в кровь
282. Печеночная желтуха
• Повышение в крови билирубиназа счет обеих фракций.
• В кишечник попадает меньше
билирубина, меньше образуется
уробилиногенов (гипохоличный
кал).
• Интенсивно окрашенная моча
(прямой билирубин).
283. Механическая желтуха
• Закупорка желчных протоков:• Желчнокаменная болезнь
• Опухоли поджелудочной
железы, желчного пузыря,
печени
• Послеоперационное сужение
общего желчного протока
284. Механическая желтуха
• В крови повышаетсяконцентрация
конъюгированного билирубина.
• Обесцвеченный кал (пигменты
не поступают в кишечник).
• Интенсивно окрашенная моча
(прямой билирубин).
285. Наследственные нарушения обмена билирубина
Синдром Crigler-Najjar I
Полное отсутствие UDPглюкуронилтрансферазы.
Аутосомно-рециссивный тип
наследования.
Повышение непрямого билирубина.
Введение фенобарбитала не приводит
к снижению билирубина.
Дети умирают в раннем возрасте от
билирубиновой энцефалопатии.
286. Наследственные нарушения обмена билирубина
Синдром Crigler-Najjar II
Снижение активности UDPглюкуронилтрансферазы.
Аутосомно-доминантный тип
наследования.
Умеренное повышение непрямого
билирубина.
Введение фенобарбитала снижает
билирубин.
Клинически проявляется в юности или у
взрослых.
287. Наследственные нарушения обмена билирубина
Синдром Dubin-Johnson• Нарушение транспорта
конъюгированного билирубина из
гепатоцитов в желчные капилляры.
• Аутосомно-доминантный тип
наследования.
• Повышение прямого билирубина.
• Клинически проявляется у
взрослых.