




























































 Биология
БиологияПохожие презентации:





")


")

Изменчивость
1.
Крымский федеральный университетимени В.И. Вернадского
Медицинская академия имени С.И. Георгиевского
Кафедра биологии медицинской
ИЗМЕНЧИВОСТЬ
Лекция №4 для фармацевтического
факультета
Агеева Елизавета Сергеевна
доктор медицинских наук,
доцент по кафедре патофизиологии,
Заведующий кафедрой биологии медицинской
Стоматологического факультета
2.
Изменчивость – фундаментальное свойство живыхорганизмов приобретать новые признаки и
свойства в процессе онтогенеза.
Выделяют модификационную, комбинативную и
мутационную изменчивость
3.
Фенотипическая ненаследственнаяПризнаки для которых характерен этот вид
изменчивости: рост, масса , окраска
Механизм: условия среды воздействуют на реакции и
приводят к их изменению
Факторы:
1. определяются влиянием среды. Имеют
направленный характер
2. изменения не наследуются потомками, связаны
только с фенотипом
3. изменения появляются массово
4. изменения носят адаптивный характер
(приспособительный)
4.
Предел, в котором изменяется фенотипическоепроявление генотипа, в зависимости от условий
внешней среды
Узкая - изменения признака незначительны
Широкая
Графическое выражение изменчивости признака,
отражает размах вариации и частоты
встречаемости - вариационным рядом
5.
Генотипическая наследственнаяПолучение новых сочетаний генов в генотипе
Механизм:
1. независимое расхождение хромосом в анафазе
мейоза-1, хроматид в анафазе мейоза-2
2. перекомбинация генов при кроссинговере
3. случайное сочетание гамет при оплодотворении
6.
Генотипическая наследственнаяМутации генов в генотипе
Свойства мутаций:
1. возникают внезапно
2. наследуются
3. ненаправлены
4. могут возникать повторно
NB! В ходе репликации и рекомбинации постоянно возникают
различные нарушения в структуре ДНК и хромосом, которые
распознаются и исправляются системами репарации.
Нарушения во время реализации этого процесса может
приводить к мутациям.
7.
Наследственная патология связана с мутациями(т.е. с изменением генетического материала).
-6
Средняя частота мутаций 10 на 1 гамету в
поколение.
Причины мутаций - мутагены.
8.
физические - ионизирующее излучение,температурный фактор,
химические – АФК, ароматические
углеводороды, цитостатики, органические
растворители, пестициды, препараты ртути,
биологические – вирусы кори, краснухи,
гриппа, антигены некоторых
микроорганизмов
9.
Если по происхождению мутагеныотносятся к факторам окружающей
среды, то они называются
экзогенные.
Если они образуются во время
жизнедеятельности организма, то эндогенные.
Мутагенез - это, соответственно,
процесс.
10.
в соматических клеткахзлокачественные опухоли
врожденные пороки развития
в половых (генеративные)
наследственные болезни
болезни с наследственной
предрасположенностью ( МФЗ)
врожденные пороки развития (мутации во
время органогенеза)
11.
Спонтанные (самопроизвольные)Под действием естественных мутагенных
факторов внешней среды без вмешательства
человека
Индуцированные
Появляются в результате направленного
воздействия определенных мутагенных
факторов
12.
ЛетальныеВызывают гибель организма
Сублетальные
Снижают жизнедеятельность
Нейтральные
Не влияют жизнедеятельность
13.
ДоминантныеСубдоминантные (проявляющиеся частично)
Рецессивные
14.
Генные – изменение молекулярной структурыгена (последовательности нуклеотидов) в
пределах одного гена
Хромосомные – изменение структуры хромосом
Геномные – изменение количества хромосом
15.
ГеныСтруктурные
Регуляторные
в кодирующей части –может качественно
измениться синтез белка
в регуляторной части – например, в промоторе –
количественно измениться
в интронах – ничего не будет – нейтральная
(сайлент) мутация
16.
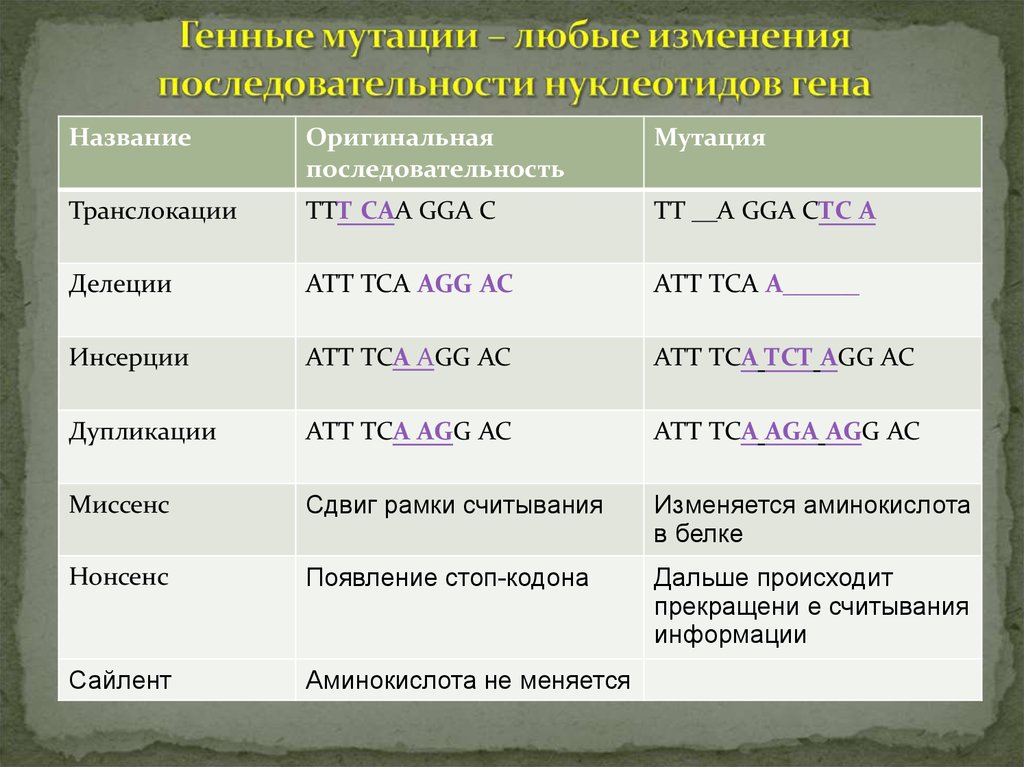
НазваниеОригинальная
последовательность
Мутация
Транслокации
ТТТ САА GGA C
ТТ __А GGA CТС А
Делеции
АТТ ТСА АGG AC
АТТ ТСА А______
Инсерции
АТТ ТСА АGG AC
АТТ ТСА ТСТ АGG AC
Дупликации
АТТ ТСА АGG AC
АТТ ТСА АGА АGG AC
Миссенс
Сдвиг рамки считывания
Изменяется аминокислота
в белке
Нонсенс
Появление стоп-кодона
Дальше происходит
прекращени е считывания
информации
Сайлент
Аминокислота не меняется
17.
Заболевания связанные с генными мутациямимогут быть – моногенными (1 ген – 1 заболевание)
Пример – фенилкетонурия – аутосомнорецессивное заболевание, обусловленное
нарушением обмена незаменимой аминокислоты
фенилаланина, поступающей в организм человека
с белковой пищей
ФКУ 1 типа – дефицит фенилаланин-4-
гидроксилазы
ФКУ 2 и 3 типа – дефект птеринового кофактора (13%)
18.
Эпидемиология фенилкетонурияПричина – рецессивная мутация
Частота ФКУ составляет 1 на 10 000 новорожденных
(1 : 4370 в Турции, 1 : 80500 в Японии, 1 : 12280
Италия)
(1 : 4735 Курская область, 1 : 18000 Республика
Тыва, 1 : 7600 Санкт-Петербург, 1 : 6772 Москва)
Частота носителей гена (гетерозиготы) 1 на 50
19.
Фенилкетонурия1 тип - дефицит фермента фенилаланингидроксилазы
ведет к накоплению фенилаланина и продуктов его
распада в биологических жидкостях – 12q22-q24.1
2 тип – дефицит цитозольной дигидроптеринредуктазы,
который приводит к блоку на пути превращения
фенилаланина в тирозин, а также предшественников
образованиякатехоламинов и серотонина – 4р15.3
3 тип – недостаточность цитозольной 6пирувоилтетрагидроптеринсинтетазы в печени и
эритроцитах, участвующей в процессе синтеза
тетрогидробиоптерина из дигидронеоптерина трифосфата
– q22.3-23.3
20.
ЗаболеваниеНа первом году жизни (2-6 месяцев) – вялость,
отсутствие интереса к окружающему, иногда
повышенная раздражительность, беспокойство,
срыгивание, гипотония, судороги, явления
дерматита
Задержка мышечного и психического развития
Микроцефалия и гидроцефалия
Гипопигментация кожи
Эпилептические приступы
Течение прогрессирующее, приводящее к смерти
на 2-3 году жизни
21.
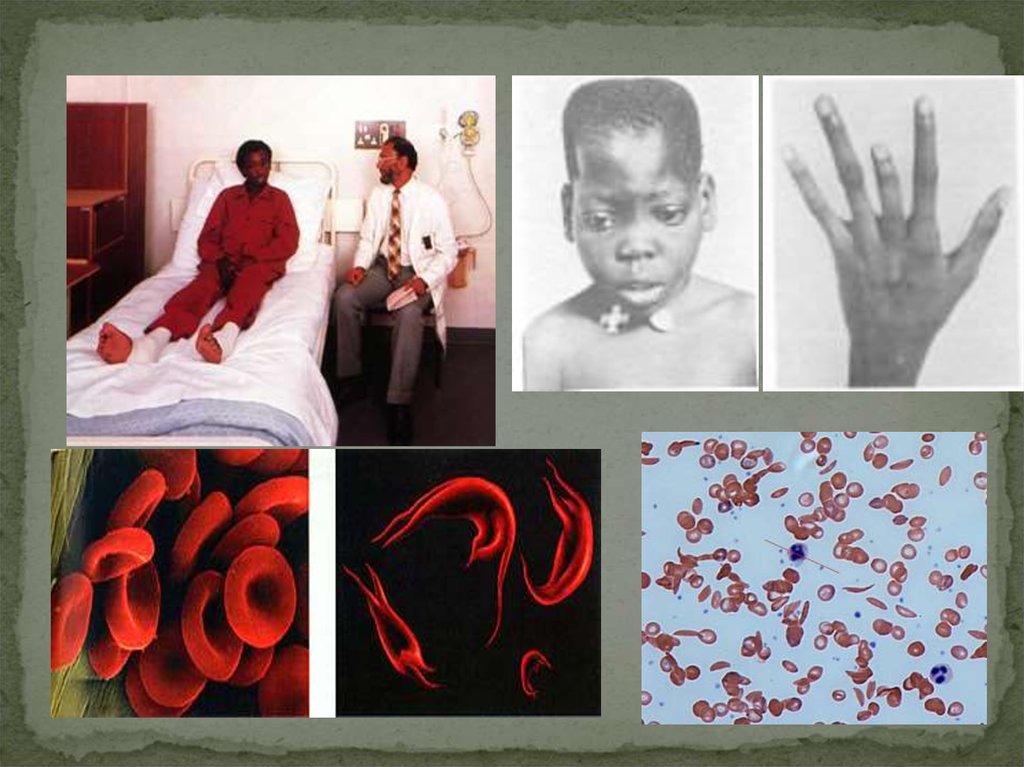
Серповидноклеточная анемияМутация в гене β-субъединицы гемоглобина
Дефект гена HBB - 11p15.5.
Дефектный гемоглобин HbS образуется в результате замены
валина на глутаминовую кислоту
Отдавая тканям кислород гемоглобин полимеризуется с
образованием волокон, которые деформируют эритроциты с
формированием длинных цепей, эритроциты становятся
серповидными
Это вызывает увеличение вязкости крови, стаз; создается
механическая преграда в мелких артериолах и капиллярах, что
приводит к тканевой ишемии (с чем связаны болевые кризы),
образуются тромбы, возникает анемия.
22.
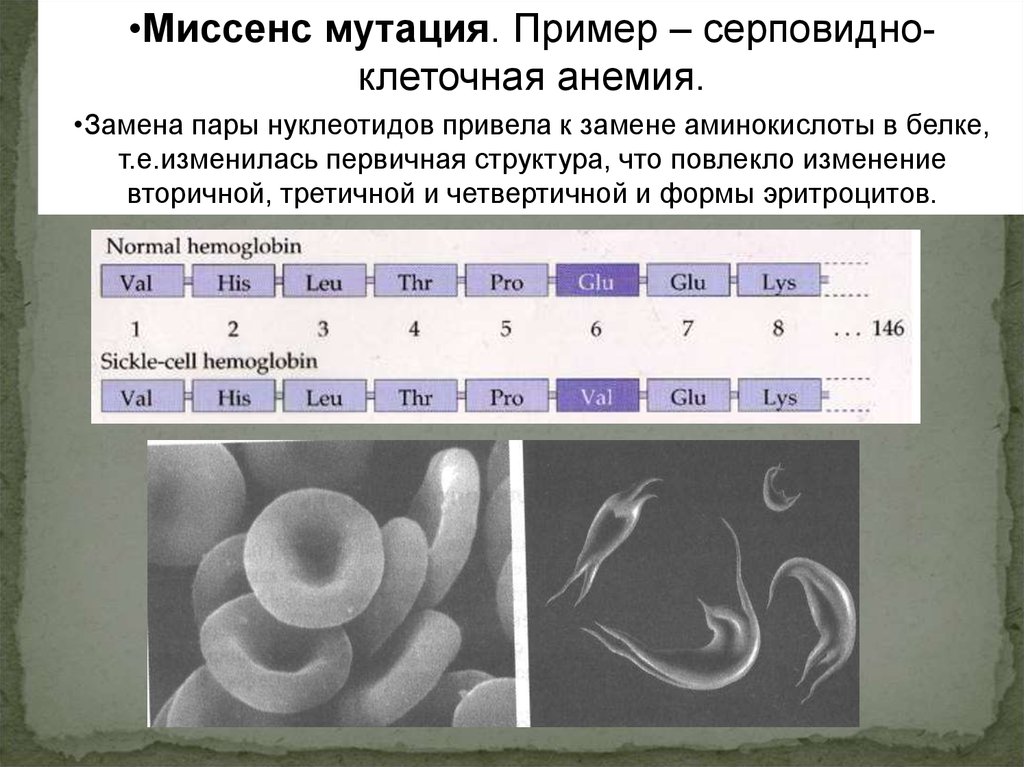
•Миссенс мутация. Пример – серповидноклеточная анемия.•Замена пары нуклеотидов привела к замене аминокислоты в белке,
т.е.изменилась первичная структура, что повлекло изменение
вторичной, третичной и четвертичной и формы эритроцитов.
ЦТТ в ДНК
ГАА в РНК
ЦАТ в ДНК
ГУА в РНК
23.
24.
Нонсенс мутация может возникнуть как в
результате замены нуклеотида, так и при
сдвиге рамки считывания. Пример: группа
крови 0.
У людей с данной группой крови в гене
произошло выпадение (делеция) одного
нуклеотида – в результате возник стоп-кодон.
Синтезируется короткий и неактивный белокфермент.
25.
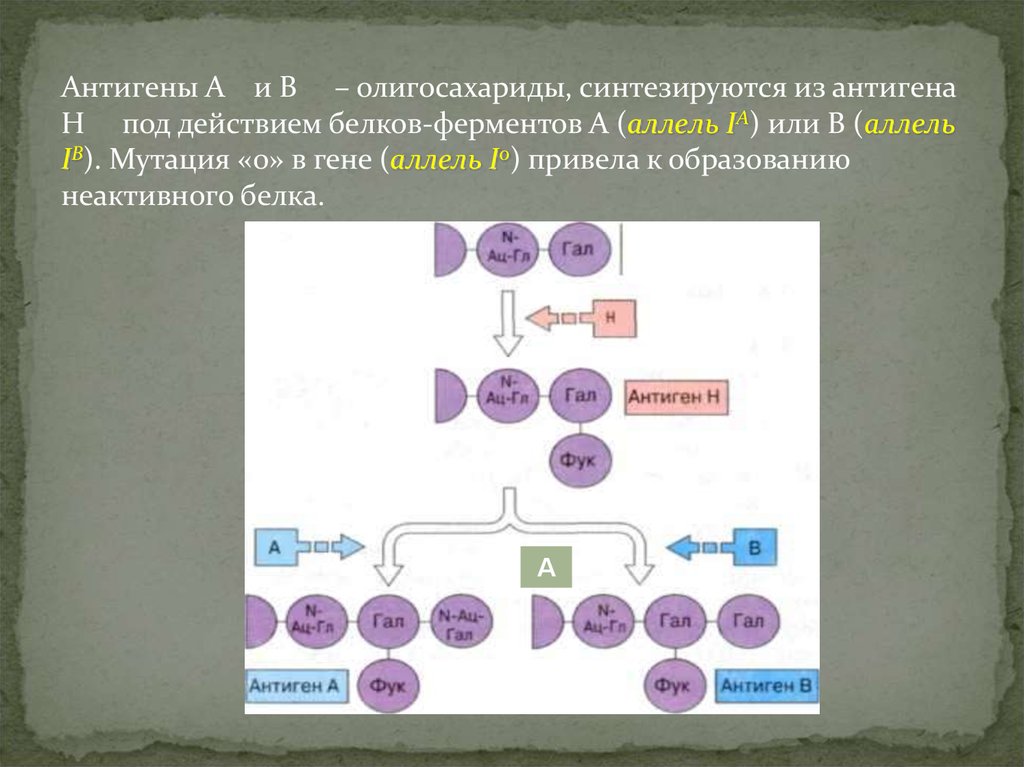
Антигены А и В – олигосахариды, синтезируются из антигенаН под действием белков-ферментов А (аллель IA) или В (аллель
IВ). Мутация «0» в гене (аллель I0) привела к образованию
неактивного белка.
А
26.
27.
28.
• Соматические мутации в специфических генах,участвующих в контроле клеточного деления,
дифференцировки, пролиферации, являются
причиной развития многих онкологических
заболеваний.
• Эти гены делятся на два класса: доминантные и
рецессивные онкогены.
• Таким образом, рак – это болезнь генов,
обусловленная мутациями, чаще всего
возникающими de novo в тех соматических клетках
и тканях.
29.
Основная сложность состоит в понимании генетических основмультифакториальных заболеваний, к которым относится
большинство распространенных болезней человека, в том
числе артериальная гипертония, коронарный атеросклероз,
сердечная недостаточность, сахарный диабет и т.д.
Моногенные болезни
Мутации
дефект (мутация) одного гена, в силу
особенностей его участия в
метаболических процессах
организма, вызывает заболевание
Мультфакториальные болезни
Полиморфизмы
Распространенные
генетические варианты
(частота >1%)
•Участвует множество генов
•Факторы внешней среды
•Межгенные взаимодействия
30.
Нарушение структуры хромосомымежхромосомные, изохромосомные
31.
Большинство мутаций летальны.Патологический кариотип характерен для всех
клеток.
Если мутации во время органогенеза- мозаицизм
32.
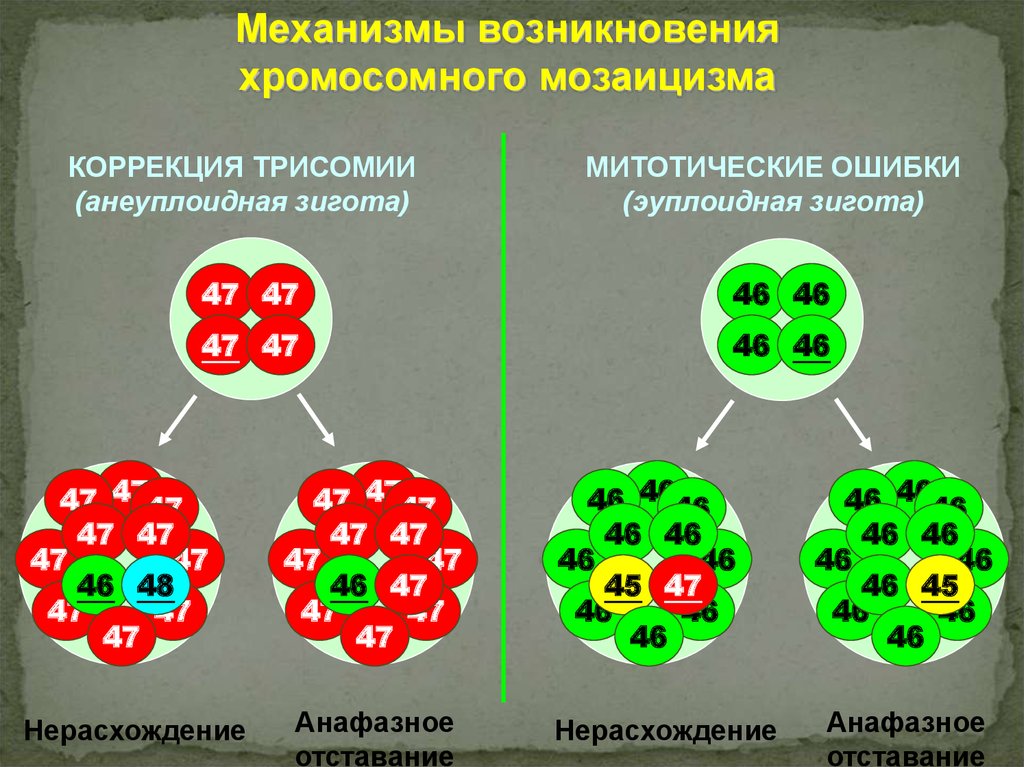
Механизмы возникновенияхромосомного мозаицизма
КОРРЕКЦИЯ ТРИСОМИИ
(анеуплоидная зигота)
МИТОТИЧЕСКИЕ ОШИБКИ
(эуплоидная зигота)
47 47
46 46
47 47
46 46
47 4747
47 47
47
47
46 48
47
47
47
Нерасхождение
47 4747
47 47
47
47
46 47
47
47
47
Анафазное
отставание
46 4646
46 46
46
46
45 47
46
46
46
Нерасхождение
46 4646
46 46
46
46
46 45
46
46
46
Анафазное
отставание
33.
Аутосомные – в аутосомах (неполовыххромосомах)
Сцепленные с Х-хромосомой
Сцепленные с Y-хромосомой
34.
Моносомии, ХОПолисомии , ХХХ, синдром Дауна
Анеуплоидии,
Полиплоидии (1п, 3п, 4п).
Геномные – изменение числа хромосом
. Полиплоидии (изменение числа
хромосом в кариотипе кратное
гаплоидному набору(1п, 3п, 4п)) и
гетероплоиди
35.
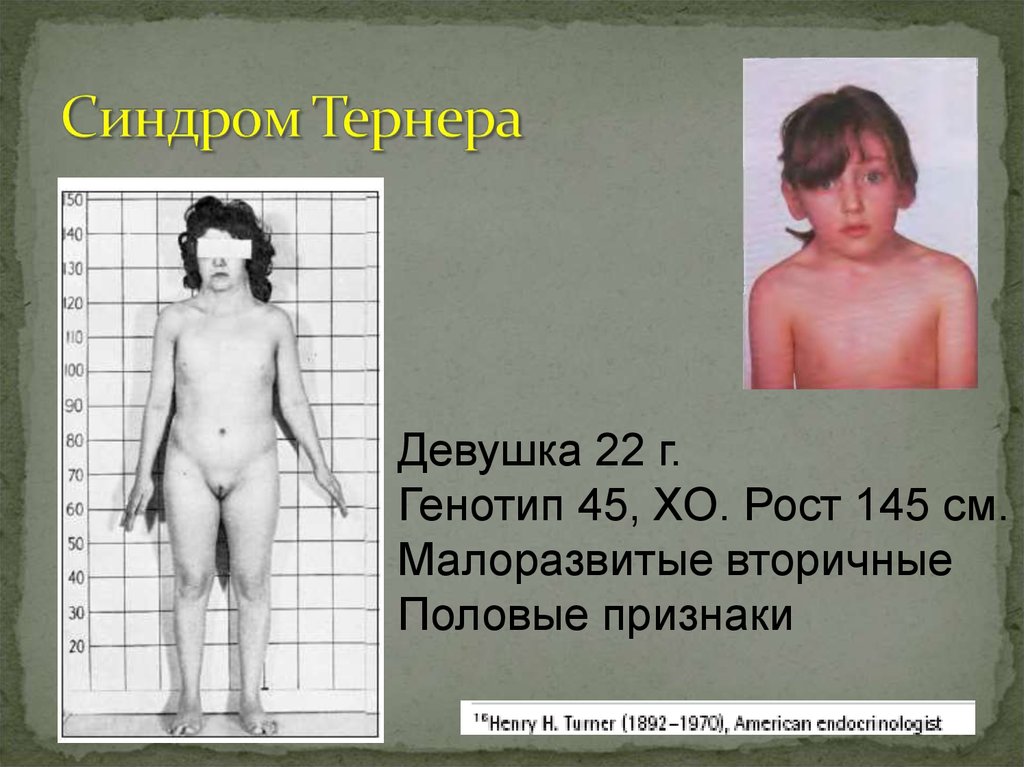
Генотип 47, ХХ / ХУ, 21+36.
Девушка 22 г.Генотип 45, ХО. Рост 145 см.
Малоразвитые вторичные
Половые признаки
37.
Синдром Кляйнефелтера (ХХУ, ХХХУ, ХХХХУ) 2-2,5случая на 1000.
Трисомия Х (ХХХ) 1:1000.
Синдром Шерешевского- Тернера (ХО) 1:3000.
38.
39.
ЯдерныеЦитоплазматические (немногочисленные, но
тяжелые митохондриальные болезни)
Митохондрии имеют
свою кольцевую ДНК
40.
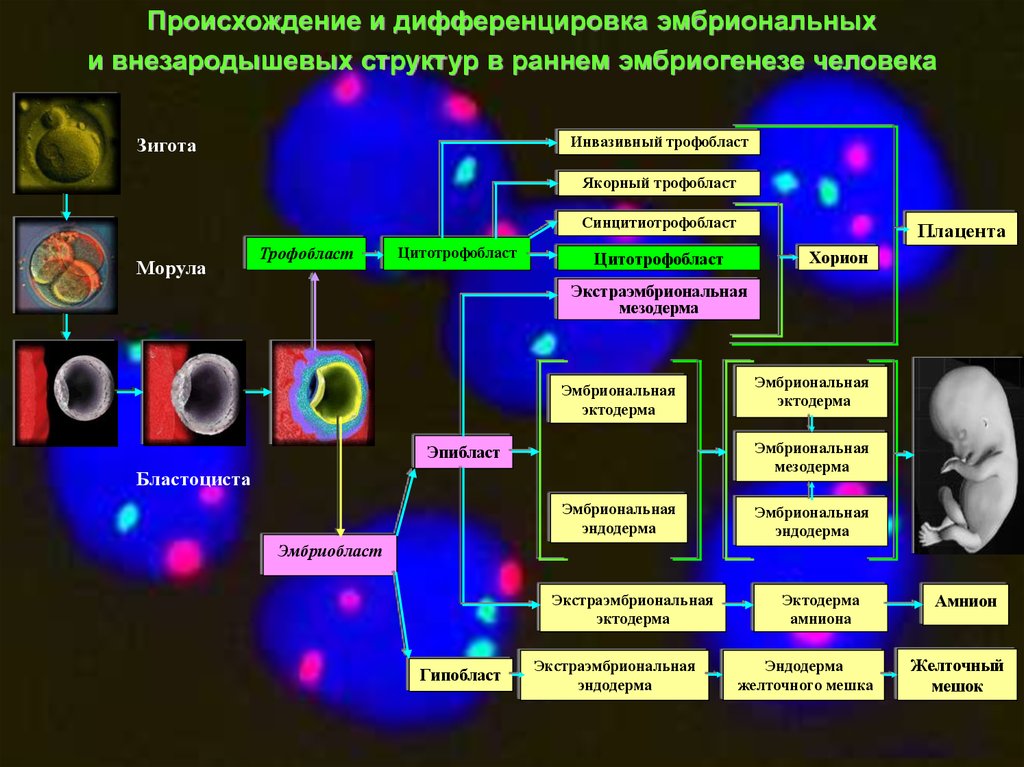
Происхождение и дифференцировка эмбриональныхи внезародышевых структур в раннем эмбриогенезе человека
Инвазивный трофобласт
Зигота
Якорный трофобласт
Синцитиотрофобласт
Морула
Трофобласт
Цитотрофобласт
Плацента
Хорион
Цитотрофобласт
Экстраэмбриональная
мезодерма
Эмбриональная
эктодерма
Эмбриональная
эктодерма
Эмбриональная
мезодерма
Эпибласт
Бластоциста
Эмбриональная
эндодерма
Эмбриональная
эндодерма
Эмбриобласт
Экстраэмбриональная
эктодерма
Гипобласт
Экстраэмбриональная
эндодерма
Эктодерма
амниона
Эндодерма
желточного мешка
Амнион
Желточный
мешок
41.
ГАМЕТОПАТИИФАКТОР
Заболевание или
ДЕЙСТВУЮЩИЙ НА патологическое
ПОЛОВЫЕ КЛЕТКИ
состояние
Бластопатии
1-15
после Сросшие
оплодотворения
Циклопия
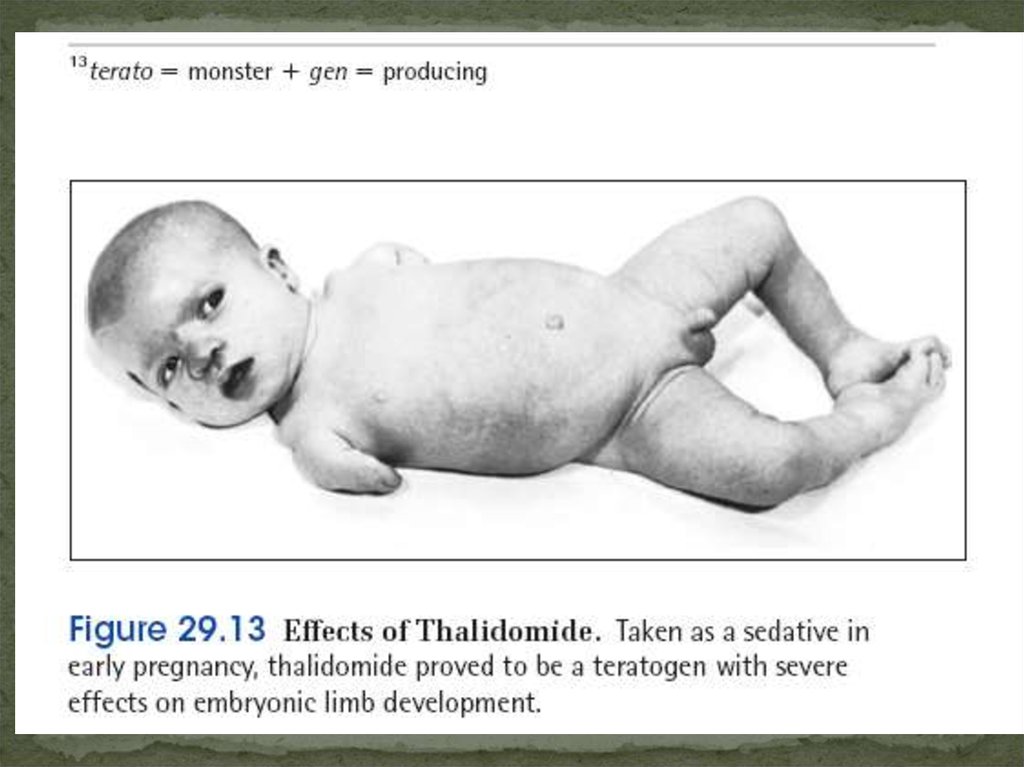
Эмбриопатии
С 16 дней до 8 недель Талитомидные,
берем
диабетические,
алкогольные,
медикаментозные,
вирус краснухи.
Фетопатии
От 9 недель берем до Крипторхизм, открытый
рождения
боталлов
проток
гипоплазия органа
близнецы
42.
Формы наследственной патологииЧастота на 1000
1. МОНОГЕННЫЕ
4.5 – 15.0
Аутосомно-доминантные
2.0 – 9.5
Аутосомно-рецессивные
2.0 – 3.5
Х-сцепленные
0.5 – 15.0
2. ХРОМОСОМНЫЕ
5.0 – 7.0
3. МУЛЬТИФАКТОРИАЛЬНЫЕ
7.0 – 10.0
4. ВРОЖДЕННЫЕ ПОРОКИ РАЗВИТИЯ 19.0 – 22.0
43.
Уродства – дефекты морфогенеза, наиболеетяжелые проявления ВПР
Дисплазии – деформации, морфологические
врожденные изменения, выходящие за рамки
общепринятых норм
Малые аномалии развития – стигмы
дизэмбриогенеза: синдактилия, ямочки на
щеках, искривление мизинца, дефекты, не
требующие медицинского вмешательства
Клинически значимые пороки развития –
врожденные аномалии требующие
медицинского вмешательства
44.
ЛС, алкоголь, марихуана, героин, кокаин, гипертемия,вещества, загрязняющие окружающую среду,
инфекционные заболевания:
Вирус краснухи
1 мес – 50% ВПР
2 мес – 22%
3-4 мес – 6-10%
ЦМВ
1-2% случаев всех беременностей
ВПГ II типа
< чем в 0,02% случаев, ВПР очень редко в
I триместре- смерть плода.
Токсоплазма
30% всех инфицированных беременных
45.
46.
47.
• Диагностика наследственных болезней• Анализ их распространенности в разных
популяциях и этнических группах
Медико-генетическое консультирование
Профилактика наследственных болезней
(пренатальная диагностика)
Изучение молекулярно-генетических основ
возникновения и формирования наследственных
заболеваний
Выявление факторов риска мультифакториальных
заболеваний
48.
Клинико- синдромологическийморфологические – например, УЗИ
биохимические – дефицит плазменного
фактора VIII
Клинико- генеалогический
составление родословной
близнецовый
цитогенетическое - кариотипирование
Молекулярная диагностика
последовательность расположения
нуклеотидов в генах или их фрагментах
49.
Генеалогическое исследованиеМетод предложен в 1965 году английским ученым Ф. Гальтоном.
Суть метода - построение родословной и ее анализ. Установление
наследственного
характера
заболевания
и
типа
наследования,
прогнозирование рождения больных детей в семьях с наследственной
патологией.
Анализ включает следующие этапы:
1. Установить, является ли заболевание единичным в семье или
имеется несколько случаев (семейный характер)? Если признак
встречается несколько раз среди поколений, то можно предположить, что он
имеет наследственную природу;
2.
Определить
тип
наследования
признака.
Составление
родословной начинают с пробанда. Ведется краткая запись данных о
каждом члене рода с указанием его родства по отношению к пробанду.
Генеалогические данные следует получать не менее чем от трех
поколений родственников по восходящей и боковой линиям, о всех членах
семьи с указанием внуков. Вносят сведения о выкидышах, абортах,
мертворожденных детях, бесплодных браках и др., включая рано умерших.
Обязательно следует указывать возраст проявления патологии. Чем больше
поколений прослежено в родословной, тем она полнее и соответственно
тем выше шансы на получение достоверных результатов.
50.
ОСНОВНЫЕ ТЕРМИНЫПробанд – больной или носитель
определенного признака, наследование
которого необходимо изучить, то есть
лицо, по отношению к которому
строится родословная.
Сибсы - дети одной родительской пары
(братья и сестры).
51.
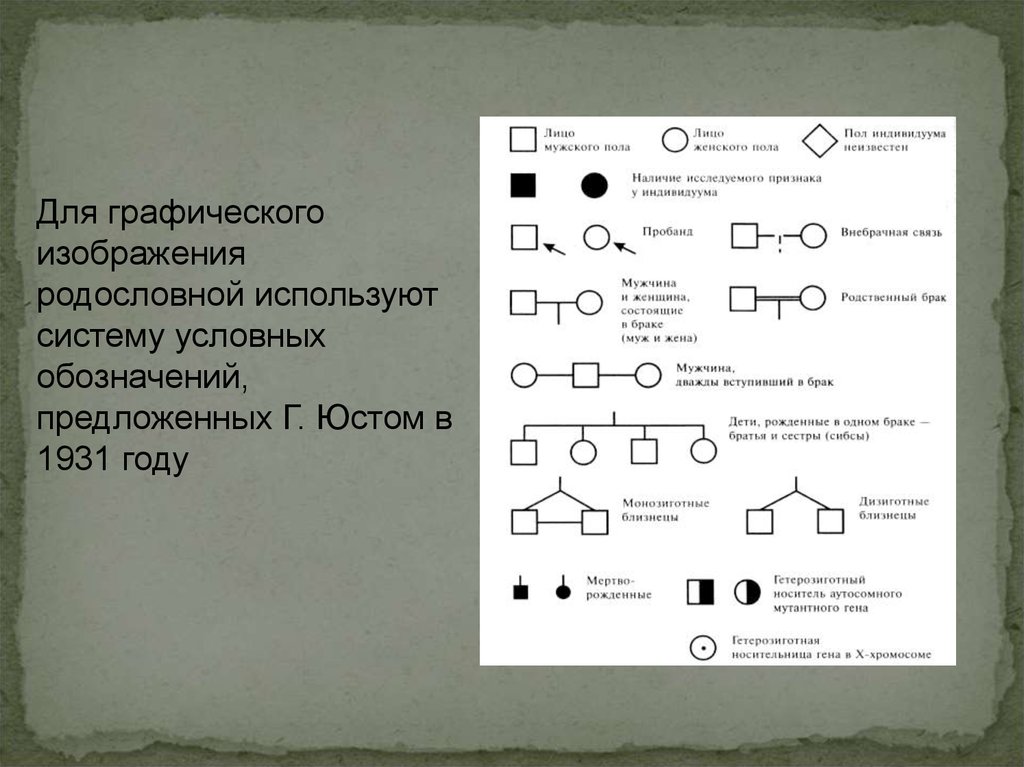
Для графическогоизображения
родословной используют
систему условных
обозначений,
предложенных Г. Юстом в
1931 году
52.
53.
1) болезнь родителей - наличие2) заболевание у женщин и мужчин
3) заболеваемость в поколениях (вертикальный
характер распределения заболевания)
4) вероятность рождения больного ребенка
5) заболеваемость среди сибсов
(горизонтальный характер распределения
заболевания)
54.
Аутосомно-доминантный тип наследования1. Патологический признак встречается в каждом поколении родословной.
2. Признак проявляется у гомозиготных и гетерозиготных носителей гена. Если
сибсов в данном поколении много, то соотношение больных и здоровых сибсов
приближается к 3:1.
3. Оба пола поражаются в равных пропорциях. Отец и мать одинаково передают
мутантный ген дочерям и сыновьям; возможна передача болезни от отца к сыну.
4. Проявления болезней может развиваться не сразу после рождения.
5. Не всегда пенетрантность патологических проявлений 100 % (полная). Это
связано с тем, что в поколениях могут встречаться гетерозиготы, но без выраженных
признаков болезни. О гетерозиготности свидетельствует появление болезни у части
их детей.
6. Различная выраженность клинических проявлений. В зависимости от
экспрессивности и пенетрантности мутантного гена клинические проявления
болезни могут варьировать. Доминантный ген может обладать разной степенью
экспрессивности или степенью выраженности (тяжестью заболевания), что
затрудняет установление типа наследования. При высокой экспрессивности гена
развивается тяжелая, часто с летальным исходом, форма заболевания, а при
низкой – человек внешне может быть здоров. Как правило, многие аутосомнодоминантные заболевания в гомозиготном состоянии протекают в более тяжелой
форме, чем в гетерозиготном.
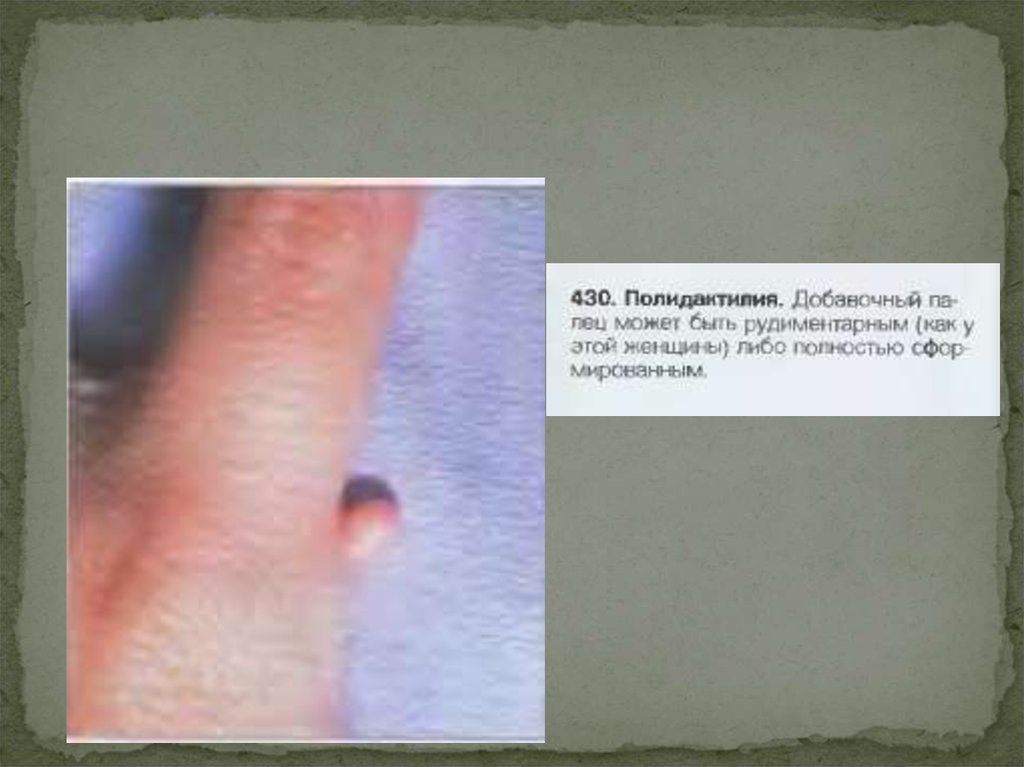
По аутосомно-доминантному типу наследуется полидактилия (шестипалость),
брахидактилия (короткопалость), ахондроплазия (карликовость), синдром
Марфана и др.
55.
Аутосомно-рецессивный тип наследования1. Заболевание встречается в родословной редко и не во всех поколениях.
2. Риск рождения больного ребенка составляет 25 %.
3. Вероятность заболевания у девочек и мальчиков одинакова.
4. Признак может проявиться у детей, родители которых были здоровы, так
как являлись гетерозиготными носителями мутантного гена.
5. Мутантный ген проявляет свое действие только в гомозиготном
состоянии, в гетерозиготном состоянии он может существовать во многих
поколениях, не проявляясь фенотипически.
6. Частота возникновения наследственных рецессивно-аутосомных
болезней находится в прямой зависимости от степени распространенности
мутантного гена среди населения: повышается в изолированных
популяциях и в семьях с высоким процентом близкородственных браков.
7. Возможна различная степень экспрессивности и пенетрантности
признака.
К аутосомно-рецессивным заболеваниям относятся: фенилкетонурия,
галактоземия, альбинизм и др. Заболевания, как правило,
диагностируются в раннем возрасте.
56.
Х-сцепленный доминантный типнаследования
1. Фенотипическое проявление заболевания будут
иметь как гомозиготы, так и гетерозиготы.
2. У здоровых отцов сыновья и их дети будут
здоровыми, так как от отца им может быть передана
только Y-хромосома.
3. Дочери гомо- и гетерозиготы будут фенотипически
больными.
4. В литературе описаны родословные при некоторых
болезнях с этим типом передачи, у которых нет сибсов
мужского пола, так как сильная степень поражения
вызывает их гибель еще внутриутробно.
57.
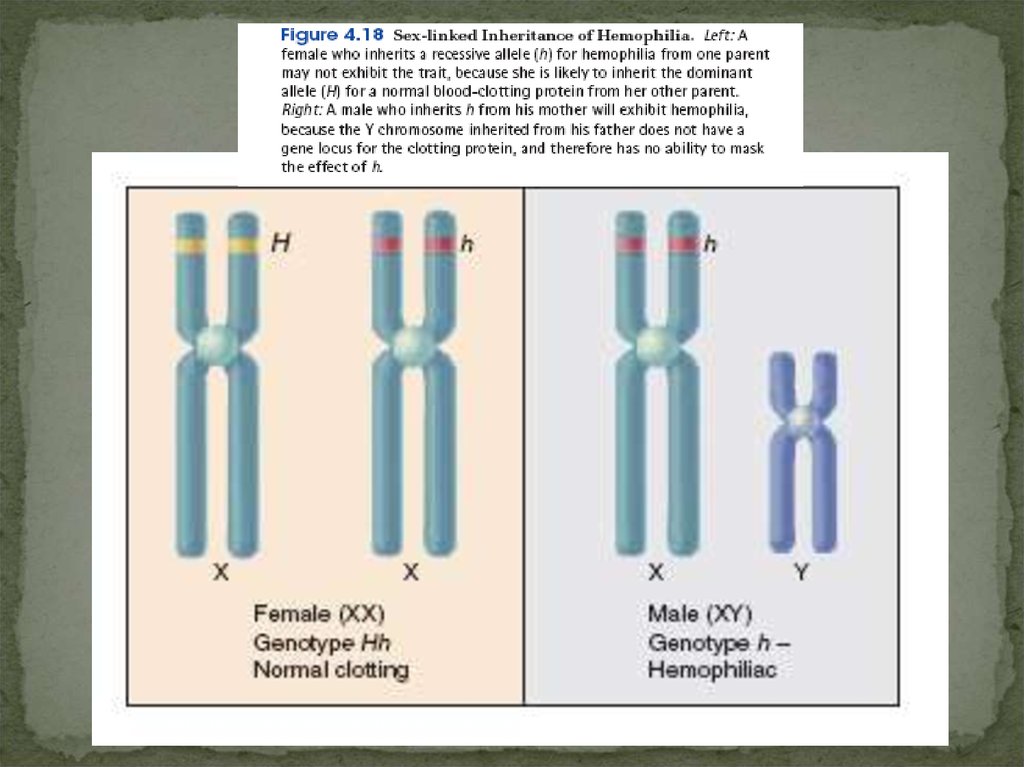
Х-сцепленный рецессивный типнаследования
1. Сыновья будут больными, так как имеют только одну Xхромосому (гемизиготность).
2тистическое соотношение здоровых и больных мужчин составляет
1:1.
Гемофилия,
миопатия
Дюшена,
некоторые
формы
дальтонизма.
Женщины могут болеть такими заболеваниями как гемофилия,
дальтонизм. Такие случаи возможны, но они исключительно
редки, так как являются результатом встречи гетерозиготной
женщины и больного мужчины.
58.
Y-сцепленный тип наследования1. Передается только сыновьям (голандрическое наследование).
С Y-хромосомой наследуются такие признаки, как гипертрихоз (наличие
волос по краю ушных раковин), кожные перепонки между
пальцами, развитие семенников, интенсивность роста тела,
конечностей и зубов.
59.
Цитогенетическое исследованиеОсуществляется диагностика хромосомных болезней с помощью
цитогенетического анализа.
Материалом для цитогенетических исследований служат клетки
человека, практически любых тканей и органов, на любой стадии
клеточного цикла, в митозе и мейозе, лимфоциты периферической
крови, клетки костного мозга, фибробласты, клетки опухолей и
эмбриональных тканей и др.
Показания к проведению исследования:
1. Возраст женщины на момент родов 35 лет и более.
2. Подозрение на хромосомные болезни.
3. Множественные врожденные пороки развития у уже
родившихся детей.
4. Несколько случаев спонтанных абортов, мертворождений,
врожденных пороков развития у детей.
5. Существенная задержка умственного и физического развития у
ребенка.
6. Нарушение репродуктивной функции.
60.
Различают прямые и непрямые методы исследования хромосом.1. Прямые методы применяются при исследовании тканей,
обладающих высокой митотической активностью (костный мозг,
клетки лимфатических узлов, ткани эмбриона на ранних стадиях
развития и хорион/плацента на любом сроке беременности) и при
исследовании мейотических хромосом.
2. Непрямые методы включают получение препаратов хромосом из
любой ткани после стимулирования пролиферации клеток в
культуральных условиях в течение времени — от нескольких часов
(кратковременные культуры) до нескольких лет (перевиваемые
культуры).
В клинической цитогенетике широко применяются методы анализа
буккального эпителия и лимфоцитов периферической крови.
61.
трисомия по хромосоме 662.
БЛИЗНЕЦОВЫЙ МЕТОДПредложил Ф. Гальтон в 1875 году для разграничения роли
наследственности и среды в развитии различных признаков у человека.
Суть метода - сравнение проявления признака в разных группах близнецов.
Учитывают частоту встречаемости признаков и различия их генотипов.
Различают две группы близнецов – монозиготные и дизиготные.
Монозиготные близнецы развиваются из одной оплодотворенной
яйцеклетки в результате нарушения ее дробления (расхождение
зародышевых клеток обычно на стадии двух бластомеров). Они однополые,
генетически идентичны, так как имеют 100 % общих генов.
Дизиготные близнецы появляются в результате одновременного
оплодотворения
двух
отличных
яйцеклеток
двумя
разными
сперматозоидами. Дизиготные близнецы имеют в среднем около 50 % общих
генов и могут быть как однополыми, так и разнополыми.
Результатом сравнения этих двух групп близнецов является расчет
показателей
соответствия
(конкордантности)
и
несоответствия
(дискордантности), а также вычисление частоты возникновения признака в
каждой группе близнецов.
63.
Конкордантность (С) – показатель идентичности пары близнецов поопределенному
признаку;
соответствует
доле
(%)
сходных
(конкордантных) по изучаемому признаку пар среди обследованных пар
близнецов для каждой группы:
СMZ = (n MZ /NMZ) х100 %, СDZ = (n DZ /NDZ) х100 %,
где MZ – монозиготная группа близнецов, DZ – дизиготная группа
близнецов, n – число пар соответствующей группы, у которых признак
отмечен у обоих близнецов, N – общее число обследованных близнецовых
пар этой же группы.
Дискордантность (D) – показатель, который рассчитывается для
дизиготных близнецов как доля дискордантных (т. е. признак выявлен
только у одного близнеца из пары) по изучаемому признаку среди всех
обследованных дизиготных пар близнецов:
DDZ = 100 % – СDZ
где DZ – дизиготная группа близнецов, СDZ – показатель идентичности
пары близнецов по определенному признаку.
64.
КОНКОРДАНТНОСТЬ НЕКОТОРЫХПРИЗНАКОВ ЧЕЛОВЕКА У БЛИЗНЕЦОВ
Признаки
Группы крови
Форма бровей
Цвет глаз
Цвет волос
Папиллярные линии кистей рук
Косолапость
Грыжа спинного мозга
Синдром Дауна
Рахит
Корь
Скарлатина
Дифтерит
Рак
Эпилепсия
Слабоумие
Шизофрения
Маниакально-депрессивный
психоз
Конкордантность,%
Монозиготные близнецы
Нормальное развитие
100
100
99,5
97
92
Заболевания
23
77
89
88
95
84
50
16
67
91
80
Дизиготные близнецы
77
19
64
51
28
23
40
2
33
7
22
87
47
38
14
3
53
13
65.
ПОПУЛЯЦИОННО-СТАТИСТИЧЕСКИЙ МЕТОДИзучают наследственные признаки в больших группах населения в одном или нескольких
поколениях. С использованием статистической обработки получаемых данных.
Методом можно рассчитать частоту встречаемости в популяции аллелей гена и
соответствующих генотипов по этим аллелям, выяснить распространение в ней различных
наследственных признаков, в том числе заболеваний.
Изучить мутационный процесс, роль наследственности и среды в формировании
фенотипического полиморфизма человека по нормальным признакам, а также в
возникновении болезней, особенно с наследственной предрасположенностью.
Основой для выяснения генетической структуры популяции является закон генетического
paвновесия Харди-Вайнберга. Отражает закономерность, в соответствии с которой при
определенных условиях соотношение аллелей генов и генотипов в генофонде популяции
сохраняется неизменным в ряду поколений.
Математическим выражением закона Харди-Вайнберга служит формула:
(pA + ga)2 = 1,
где р и g – частоты встречаемости аллелей А и а соответствующего гена. Анализ частот
встречаемости разных признаков в популяции в случае их соответствия закону ХардиВайнберга позволяет утверждать, что признаки обусловлены разными аллелями одного гена.
66.
БИОХИМИЧЕСКОЕ ИССЛЕДОВАНИЕОсновано на том, что при мутациях ферментов и транспортных белков, их
можно выявлять по накоплению патологических метаболитов.
Субстраты ферментных реакций или транспортируемые через мембрану
вещества
(аминокислоты, сахара, глюкозаминоглюканы, кетокислоты)
накапливаются в моче и сыворотке крови.
Например, при недостатке фенилаланингидроксилазы в крови и тканях
накапливается фенилаланин и продукты его переаминирования –
фенилпировиноградная и фенилуксусная кислота. Обе кислоты
токсически воздействует на мозг ребенка.
67.
Показаниями к биохимическимисследованиям служат:
1. Неясные и затяжные формы желтух у детей в период новорожденности и первого года
жизни.
2. Хроническое расстройство пищеварения неинфекционной природы (диарея, рвота,
гепатомегалия, трудности с кормлением).
3. Аномалии развития скелета не рахитической природы.
4. Нарушение речи, органов зрения, слуха.
5. Дефекты поведения, двигательная расторможенность, мышечная гипотония.
6. Гиперрефлексия, судорожный синдром.
7. Умственная отсталость.
8. Необычные волосы, ногти, лицо.
9. Стойкие изменения в моче – протеинурия, гематурия, лейкоцитурия, фосфатурия,
глюклзурия.
Данные методы применяют при подозрении на наследственные болезни обмена веществ. Они
могут быть многоэтапными (скрининговые) или сразу строго направленными на определенную
патологию.
В практике здравоохранения Российской Федерации используется двухэтапная программа:
1. Просеивающая программа (массовый и селективный скрининг).
2. Методы, подтверждающие диагноз.
В нашей стране осуществляется диагностика фенилкетонурии, врожденного гипотиреоза,
адреногенитального синдрома, врожденных аномалий развития нервной трубки и болезни
Дауна.
68.
МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКОЕ ИССЛЕДОВАНИЕПозволяет диагностировать наследственные болезни на уровне
генетических дефектов.
Прямая диагностика мутаций включает несколько методов:
- определение нуклеотидной последовательности (секвенирование)
в генах или их фрагментах;
- выявление нарушения места рестрикции; аллельспецифическую
гибридизацию с синтетическими олигонуклеотидными зондами;
- химическое и ферментативное расщепление ДНК в местах
неправильного сшивания оснований;
- регистрацию
изменения
электрофоретической
подвижности
мутантных молекул ДНК;
- трансляцию белкового продукта in vitro,
- метод нуклеотидной последовательности
69.
Спектр наследственных болезней, выявляемых с помощью методов ДНКдиагностики в Российской Федерации (по данным П. В. Новикова и О. В. Евграфова)Наследственные заболевания
Гематологический научный центр Минздрава РФ (г. Москва)
Болезнь Виллебранда
Гемофилия А
Гемофилия В
Семейная гиперхолестеринемия
-талассемия
Медико-генетический центр РАМН (г. Москва)
Агаммаглобулинемия
Атаксия Фридрейха
Атаксия-телеангиэктазия
Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация)
Ломкость Х-хромосомы
Миодистрофия Дюшенна-Беккера
Муковисцидоз
Невральная амиотрофия
Недостаточность 21-гидроксилазы (гиперплазия коры
надпочечников)
Спинальная амиотрофия
Спинально-бульбарная амиотрофия
Фенилкетонурия
Хорея Гентингтона
Чистая дисгенезия гонад, XX-мужчины и некоторые другие
нарушения полового развития
Х-сцепленная невральная амиотрофия
Локализация генов
12pter-pl2
Xq28
Хр27.1-27.2
19р13.2-р13.1
11р15.5
Xq21.3-q22
9ql3-q21.1
11q23.1
13q14.3-q21.1
Xq27.3
Хр21.2
7q31.2
1q22, 17р11.2
6р21.3
5ql3
Xq11-ql2
12q24.1
4pter-pl6.3
Yр
Xql3.1
70.
Центр охраны здоровья матери и ребенка (г. Москва)Недостаточность 21-гидроксилазы (гиперплазия коры
6р21.3
надпочечников)
Научно-исследовательский институт неврологии РАМН (г. Москва)
Хорея Гентингтона
4pter-pl6.3
Институт экспериментальной медицины РАМН (г. С.-Петербург)
Муковисцидоз
7q31.2
Недостаточность 1-антитрипсина
14q31-q32.3
Семейная гиперхолестеринемия
19р13.2-р13.1
Институт акушерства и гинекологии им. Д.О. Отта РАМН (г. С.-Петербург)
Болезнь Виллебранда
12pter-pl2
Болезнь Леша-Нихена
Xq26-q27.2
Болезнь Хантера
Xq28
Гемофилия А
Xq28
Гемофилия В
Хр27.1-27.2
Ломкость Х-хромосомы
Xq27.3
Миодистрофия Дюшенна-Беккера
Хр21.2
Миотоническая дистрофия
19ql3.2-ql3.3
Муковисцидоз
7q31.2
Спинально-бульбарная амиотрофия
Xq11-ql2
Фенилкетонурия
12q24.1
Хорея Гентингтона
4pter-pl6.3
Петербургская медицинская академия (г. С.-Петербург)
Недостаточность ацил-СоА-дегидрогеназы
lp31
Фенилкетонурия
12q24.1
-талассемия
11р15.5
Институт медицинской генетики РАМН (г. Томск)
Миодистрофия Дюшенна-Беккера
Хр21.2
71.
ИММУНОГЕНЕТИЧЕСКОЕ ИССЛЕДОВАНИЕМетоды позволяют выявлять наследственные (первичные) иммунодефициты, оценить
совместимость матери и плода, определить прогноз при болезнях с наследственным
предрасположением. Показаниями к проведению могут служить следующие так называемые
«настораживающие признаки»:
1. Частые заболевания отитом (не менее 6-8 раз в течение одного года).
2. Несколько подтвржденных серьезных синуситов (не менее 4-6 раз в течение одного года).
3. Более двух подтвержденных пневмоний в течение одного года.
4. Повторные глубокие абсцессы кожи или внутренних органов.
5. Потребность в длительной терапии антибиотиками для купирования инфекции (до 2 мес. и
более).
6. Потребность во внтуривенном введении антибиотика для купирования инфекции.
7. Не менее двух глубоких инфекций, таких как менингит, остеомиелит, сепсис.
8. Отставание грудного ребенка в росте и массе.
9. Наличие в семье первичных иммунодефицитов, факты ранних смертей от тяжелых
инфекций или наличие одного из перечисленных симптомов.
10. Персистирующая молочница или грибковое поражение кожи в возрасте старше 1 года.
Изучение иммунного статуса при иммунодефицитах должно включать изучение количества и
функциональной активности основных компонентов иммунной системы (фагоцитарная
система, система комплемента, Т- и В-системы иммунитета). Методы, применяемые для
оценки функционирования этих систем, условно разделены на тесты 1-го и 2-го уровней.
Тесты 1-го уровня являются ориентировочными и направлены на выявление грубых дефектов
в иммунной системе. Тесты 2-го уровня являются функциональными и направлены на
обнаружение конкретной «поломки» в иммунной системе
72.
Диагностика наследственных болезней бывает первичной и вторичной,суть которой не допустить рождения больного ребенка. Направления
первичной диагностики включают:
1) планирование семьи: проспективное (до рождения ребенка) и
ретроспективное консультирование (после рождения больного ребенка);
2) пренатальная диагностика с элиминацией патологических эмбрионов и
плодов.
Вторичная диагностика предусматривает коррекцию болезни после
рождения (нормокопирование) - формирование здорового фенотипа при
патологическом генотипе.
73.
Показаниями к пренатальной диагностикеявляются:
1. Возраст женщины старше 35-37 лет, мужчины старше 45 лет.
2. Рождение ребенка с хромосомной или генной болезнью.
3. Наличие в семье точно установленного наследственного заболевания.
4. Носительство матерью гена Х-сцепленного рецессивного заболевания.
5. Наличие в анамнезе у беременных спонтанных абортов в ранние сроки
беременности, мертворождений, рождение детей с пороками развития и
хромосомной патологией.
6. Беременные из зоны повышенного радиационного фона.
7. Наличием кровно-родственных браков.
8. Наличие сходных случаев заболевания в семье и среди отдаленных
кровных родственников.
9. Рецидивирующее, хроническое, длительно неподдающееся лечению
течение заболевания.
10. Наличие редко встречающихся специфических симптомов или их
сочетание.
11. Множественное поражение органов и систем органов.
12. Наличие более 5 врожденных морфогенетических вариантов,
называемых также микроаномалиями развития.
13. Врожденный характер заболевания.
74.
В настоящее время применяют непрямые и прямые методыпренатальной диагностики. При непрямых методах исследуют
беременную женщину (например, исследование сыворотки крови на
альфа-фетопротеин).
При прямых методах исследуют плод.
Методы пренатальной диагностики бывают инвазивные и
неинвазивные.
К инвазивным относятся - преимплантационная диагностика,
которая осуществляется на 1-2 клетках, взытых на ранних стадиях
дробления (8-18 клеток) от оплодотворенной in vitro яйцеклетки;
хорионбиопсия
(взятие
эпителия
ворсинок
хориона);
плацентобиопсия; амниоцентез (получение амниотической жидкости
и клеток плода); кордоцентез (забор крови из пуповины); биопсия
кожи, печени, мышц плода; везикоцентез (прокол стенки мочевого
пузыря плода для получения его мочи); фетоскопия (осмотр плода
фиброоптическим эндоскопом, введенным в амниотическую полость
через брюшную стенку матки, метод позволяет осмотреть плод,
пуповину, плаценту, произвести биопсию).
К неинвазивным относятся – ультразвуковое исследование (УЗИ),
скрининговое исследование сыворотки крови беременной женщины
(определение
альфа-фетопротеина
(АФП),
хорионического
гонадотропина,
неконъюгированного
эстриола,
75.
Программа пренатального тестирования беременныхПримечание: * концентрация вещества зависит от срока беременности в 6 нед. 1,5 мкг/мл.; 12-14 нед. - 30 мкг/мл, 16-18 нед. - 31-44мкг/мл; 20 нед. - 10 мкг/мл.