








 Кариотипы 47,ХХ,18+ или 47,ХУ,18+ Впервые описал J. Edwards в 1960 г. Среди новорожденных")




 - дефекты")

. При гемоглобинопатиях происходят нарушения пептидных цепей гемоглобина. В")


















 Медицина
МедицинаПохожие презентации:










Наследственные заболевания человека
1.
НАСЛЕДСТВЕННЫЕЗАБОЛЕВАНИЯ
ЧЕЛОВЕКА
Доцент кафедры биологии медицинской Демиденко Л.А.
2. Генетика человека
В 1929 г. советский генетик,невропатолог С.Н.Давиденков
организовал первую в мире
медико-генетическую
консультацию. Он первым в
мире поставил вопрос о
необходимости составления
каталога генов человека,
сформулировал понятие о
генетической гетерогенности
наследственных болезней
человека.
3. Наследственные заболевания – это заболевания, связанные с нарушениями генетического аппарата клетки. В настоящее время известно
более 2,5тысяч генетически обусловленных
болезней, при которых в первую очередь
страдает нервная система. Эти
заболевания протекают длительно,
многие неизлечимы и могут поражать все
органы и системы организма человека:
мозг, мышцы, печень, сердечнососудистую систему и т. д.
4. КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ ЧЕЛОВЕКА
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИГЕННЫЕ
МОНОГЕННЫЕ
АДОМИНИРУЮЩИЕ
ПОЛИГЕННЫЕ
ХРОМОСОМНЫЕ
МУЛЬТИ
ФАКТОРИАЛЬ НЫЕ
ИЗМЕНЕНИЕ
ЧИСЛА
ХРОМОСОМ
МОНОСОМИЯ
А - РЕЦЕССИВНЫЕ
ТРИСОМИЯ
Х - СЦЕПЛЕННЫЕ
ХРОМОСОМНЫЕ
ПЕРЕСТРОЙКИ
У - СЦЕПЛЕННЫЕ
5. ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомныезаболевания
связаны с аномалиями
числа или структуры
хромосом.
В основе этих заболеваний
лежат геномные мутации и
хромосомные аберрации.
Характерные признаки
хромосомных болезней:
маленький рост и вес при
рождении; черепно-лицевые
дисморфии; умственная
отсталость; многосистемные
поражения.
Только 3-5% наследуются.
6.
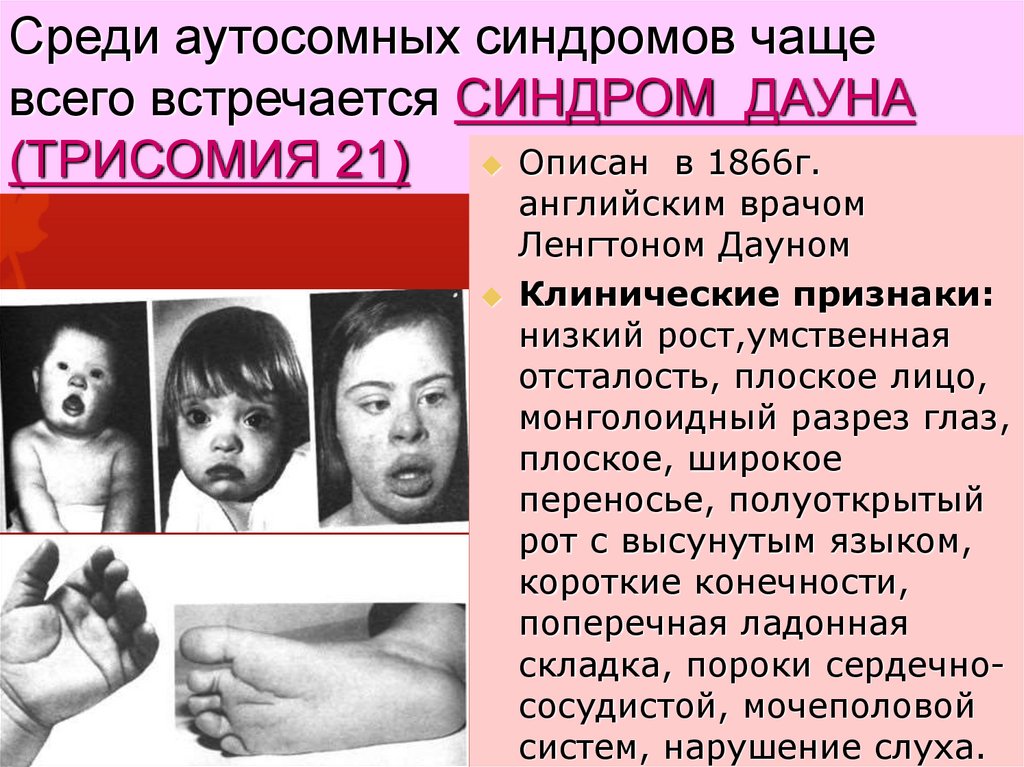
Среди аутосомных синдромов чащевсего встречается СИНДРОМ ДАУНА
(ТРИСОМИЯ 21) Описан в 1866г.
английским врачом
Ленгтоном Дауном
Клинические признаки:
низкий рост,умственная
отсталость, плоское лицо,
монголоидный разрез глаз,
плоское, широкое
переносье, полуоткрытый
рот с высунутым языком,
короткие конечности,
поперечная ладонная
складка, пороки сердечнососудистой, мочеполовой
систем, нарушение слуха.
7.
Одинаковочасто встречается как у
мальчиков, так и у девочек. Частота их
рождения (1:700-1:800 среди новорожденных)
связана с возрастом матери (после 35 лет риск
появления ребенка с синдромом Дауна
8. В основе этого заболевания лежит нерасхождение по 21-й паре аутосом во время мейоза. При этом у детей возникает кариотипы 47,
ХХ, 21+ или 47,ХУ, 21+.Возможна транслокационная природа
синдрома Дауна. В этом случае наблюдается
транслокация 21-й хромосомы на 21-ю, либо
на 15-ю.
Для прогноза потомства важно выяснить
причину заболевания (транслокация или
трисомия), что возможно путем исследования
кариотипа больного и родителей с
использованием дифференциальной окраски
хромосом.
9.
СИНДРОМ ПАТАУ(ТРИСОМИЯ 13)
Описан в 1960 г. К. Патау.
Кариотипы больных 47,ХХ,13+
или 47,ХУ,13+.
Частота синдрома Патау 1:7000
– 1:8000 среди новорожденных.
Продолжительность жизни всего
несколько месяцев; 95% умирают
до 1 года.
10. Клинические признаки: микроцефалия, расщепление мягкого и твердого неба, незаращение губы, недоразвитие или отсутствие глаз
(микрофтальмия или анофтальмия),полидактилия и синдактилия, пороки
внутренних органов: сердца, почек,
пищеварительной системы.
11. Синдром Эдвардса (трисомия 18) Кариотипы 47,ХХ,18+ или 47,ХУ,18+ Впервые описал J. Edwards в 1960 г. Среди новорожденных
синдром Эдвардсавстречается с частотой около 1:7000.
Продолжительность жизни 1,5 – 2 месяца.
Дети с этой аномалией чаще рождаются у
матерей, возраст которых старше 35 лет.
Клинические признаки:
Череп необычной формы:узкий лоб и широкий,
выступающий затылок, очень низко
расположенные, деформированные уши,
постоянный признак – недоразвитие нижней
челюсти. Пальцы рук широкие и короткие,
характерная аномалия кисти – поперечная
ладонная складка; тяжелые пороки сердца.
12.
СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА(МОНОСОМИЯ - Х) Кариотип больных 45,ХО
Впервые этот синдром был описан в 1925 г. эндокринологом
Н.А. Шерешевским, а затем изучался Г. Тернером в 1938 г.
Клинические признаки:
низкий рост (130-145 см.),
крыловидные кожные
складки на шее (шея
сфинкса), волосы на шее
растут очень низко,
бесплодие, врожденные
пороки сердца, гипоплазия
ногтей, снижение остроты
зрения и слуха, поперечная
ладонная складка, часто
отставание в умственном
развитии .
13.
СИНДРОМ КЛАЙНФЕЛЬТЕРА(47, ХХУ; 48,ХХУУ; 48,ХХХУ)
Описан в 1942 г.
Наблюдается у лиц с мужским фенотипом.
Частота встречаемости 2:1000
новорожденных мальчиков
Клинические признаки:
высокий рост, длинные конечности,
астенический тип телосложения: узкие
плечи, широкий таз, жироотложение по
женскому типу, характерная особенность –
недоразвитие семенников, отсутствие
сперматогенеза, наблюдается умственная
отсталость, выраженная в разной степени.
14. В результате хромосомных аберраций возникают заболевания, связанные с нарушениями структуры хромосом. Синдром 4р-
(Вольфа-Хиршхорна) –делеция в 4-ой паре аутосом.
Кариотипы 46,ХХ, 4р- или 46,ХУ,4р-.
Синдром описан авторами в 1965 г.
Синдром Вольфа-Хиршхорна
характеризуется следующими
признаками: микроцефалия, широкий и
клювовидный нос, асимметричный череп,
низко расположенные уши, задержка
психомоторного развития.
15.
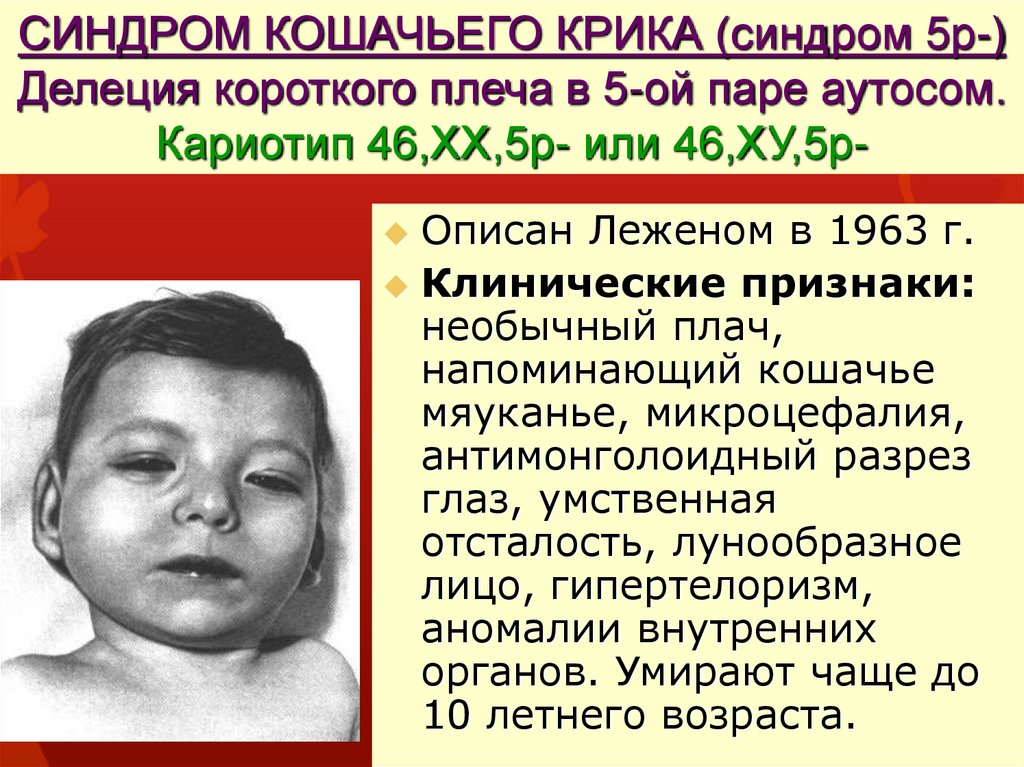
СИНДРОМ КОШАЧЬЕГО КРИКА (синдром 5р-)Делеция короткого плеча в 5-ой паре аутосом.
Кариотип 46,ХХ,5р- или 46,ХУ,5рОписан Леженом в 1963 г.
Клинические признаки:
необычный плач,
напоминающий кошачье
мяуканье, микроцефалия,
антимонголоидный разрез
глаз, умственная
отсталость, лунообразное
лицо, гипертелоризм,
аномалии внутренних
органов. Умирают чаще до
10 летнего возраста.
16. ГЕННЫЕ ЗАБОЛЕВАНИЯ возникают в результате точковых мутаций, затрагивающих структуру самого гена. При этом меняется молекулярное
строение ДНК. Этосвязано с нарушениями синтеза
белка в клетке, что ведет к
развитию различных молекулярных
заболеваний, которые наследуются
аутосомно-доминантно, аутосомнорецессивно, сцепленно с полом.
17. Генные болезни в настоящее время подразделяются на: - наследственные заболевания ферментных систем (энзимопатии) - дефекты
белков крови(гемоглобинопатии)
- дефекты структурных белков
(коллагеновые болезни)
- заболевания с невыясненными
первичными биохимическими дефектами
18. Среди энзимопатий различают следующие формы: 1. Болезни нарушения обмена аминокислот (фенилкетонурия – нарушение обмена
фенилаланина;альбинизм – дефекты синтеза
пигмента меланина; тирозиноз –
дефекты обмена тирозина).
2. Болезни нарушения обмена
углеводов (галактоземия – дефекты
метаболизма молочного сахара –
лактозы; мукополисахаридозы –
дефекты расщепления
полисахаридов)
19. 3. Болезни нарушения обмена липидов (липидозы). При гемоглобинопатиях происходят нарушения пептидных цепей гемоглобина. В
результате могутразвиваться серповидноклеточная
анемия, метгемоглобинемия, талассемия.
В основе возникновения
коллагеновых болезней лежат
генетические дефекты биосинтеза и
распада коллагена (структурного
компонента соединительной ткани).
Примером является болезнь Марфана.
20. Аутосомно-доминантный тип наследования
• 1. Болезнь встречается в каждом поколенииродословной.
• 2. Соотношение больных мальчиков и девочек
равное.
• 3. Болезнь у гомозигот протекает тяжелее, чем у
гетерозигот.
• 4. Вероятность рождения больного ребенка, если
болен один из родителей, равна 50%.
• 5. Возможны случаи, когда болезнь носит стертый
характер (неполная пенетрантность гена).
21.
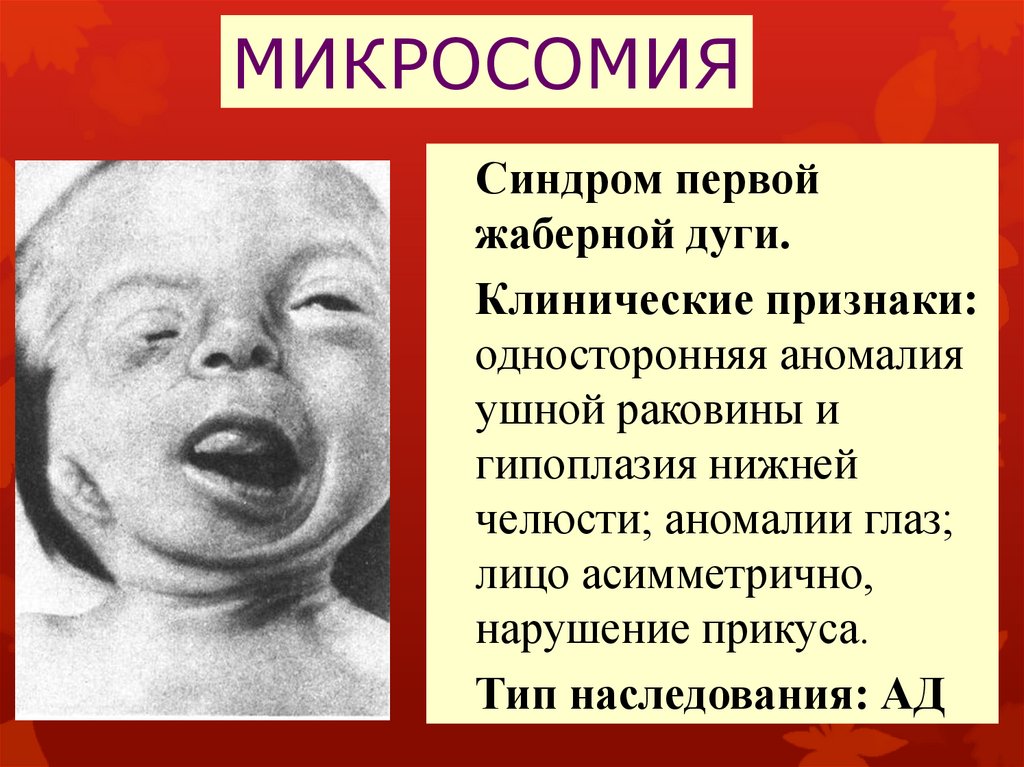
МИКРОСОМИЯСиндром первой
жаберной дуги.
Клинические признаки:
односторонняя аномалия
ушной раковины и
гипоплазия нижней
челюсти; аномалии глаз;
лицо асимметрично,
нарушение прикуса.
Тип наследования: АД
22.
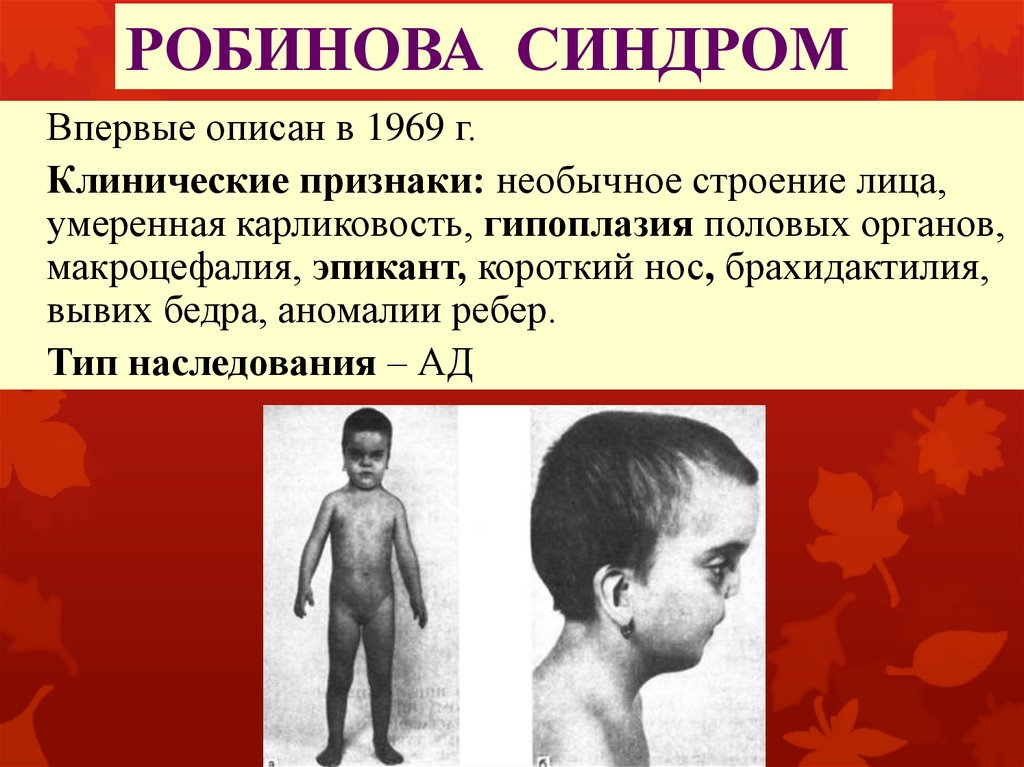
РОБИНОВА СИНДРОМВпервые описан в 1969 г.
Клинические признаки: необычное строение лица,
умеренная карликовость, гипоплазия половых органов,
макроцефалия, эпикант, короткий нос, брахидактилия,
вывих бедра, аномалии ребер.
Тип наследования – АД
23.
ВИЛЛЬЯМСА СИНДРОМВпервые описан в 1961 г.
Клинические признаки:
Необычное лицо, низкий рост, короткий нос,
полные щеки, маленькая нижняя челюсть,
умственная отсталость.
Тип наследования – АД
24.
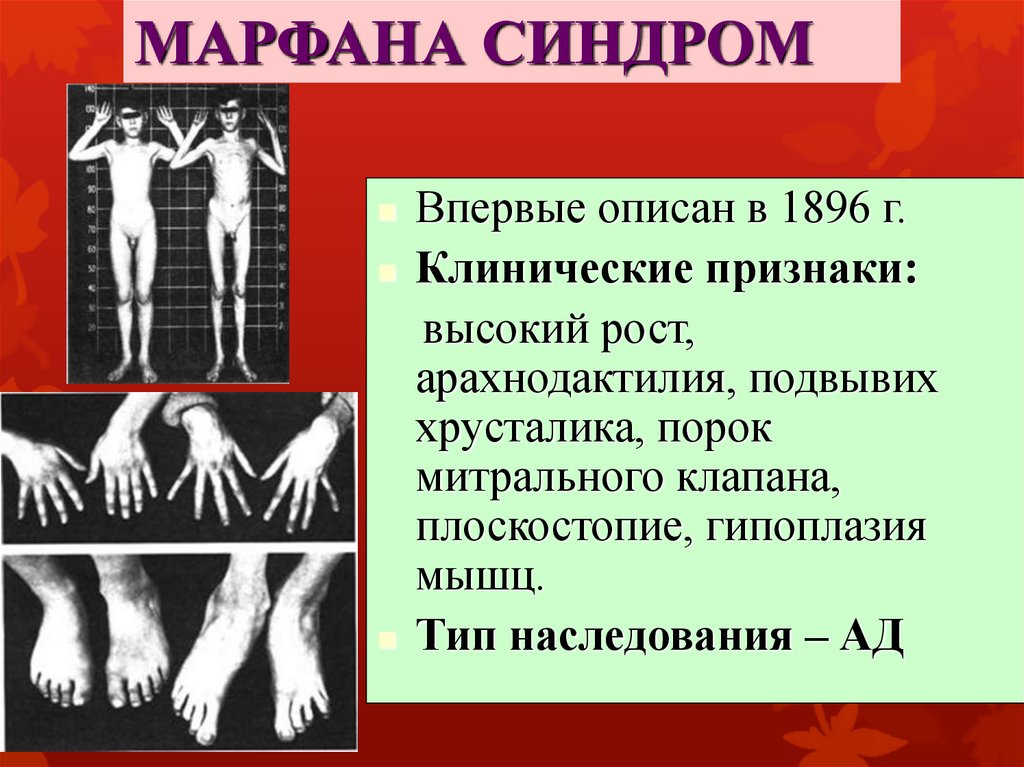
МАРФАНА СИНДРОМВпервые описан в 1896 г.
Клинические признаки:
высокий рост,
арахнодактилия, подвывих
хрусталика, порок
митрального клапана,
плоскостопие, гипоплазия
мышц.
Тип наследования – АД
25.
ПОЛИДАКТИЛИЯКлинические признаки:
существует два варианта:
тип А, при котором
дополнительный палец
функционален, и тип В,
когда дополнительный
палец недоразвит и
представляет собой
кожный вырост.
Тип наследования: АД
26.
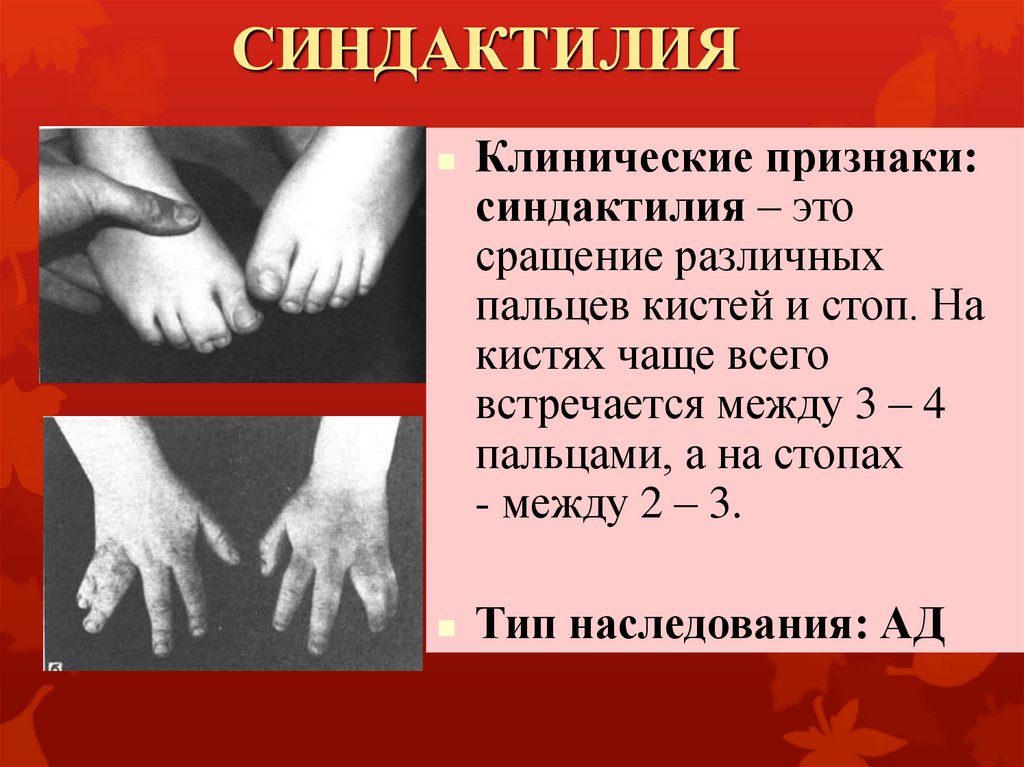
СИНДАКТИЛИЯКлинические признаки:
синдактилия – это
сращение различных
пальцев кистей и стоп. На
кистях чаще всего
встречается между 3 – 4
пальцами, а на стопах
- между 2 – 3.
Тип наследования: АД
27.
ОСТЕОГЕНЕЗКлинические признаки:
повышенная ломкость
трубчатых костей, ребер и
ключиц при минимальной
травме, деформации
конечностей, голубые склеры
глаз, «янтарные зубы»,
треугольное лицо, «рыбьи
позвонки».
Рентгенологически
выявляется истончение
костей.
Тип наследования: АД
28.
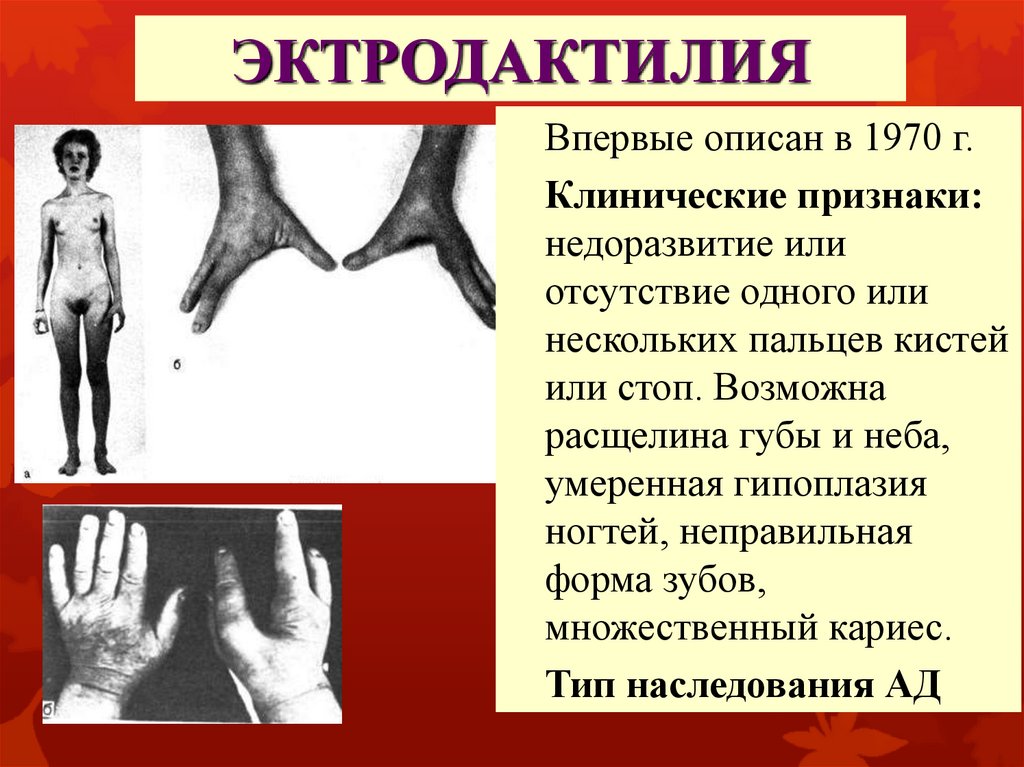
ЭКТРОДАКТИЛИЯВпервые описан в 1970 г.
Клинические признаки:
недоразвитие или
отсутствие одного или
нескольких пальцев кистей
или стоп. Возможна
расщелина губы и неба,
умеренная гипоплазия
ногтей, неправильная
форма зубов,
множественный кариес.
Тип наследования АД
29.
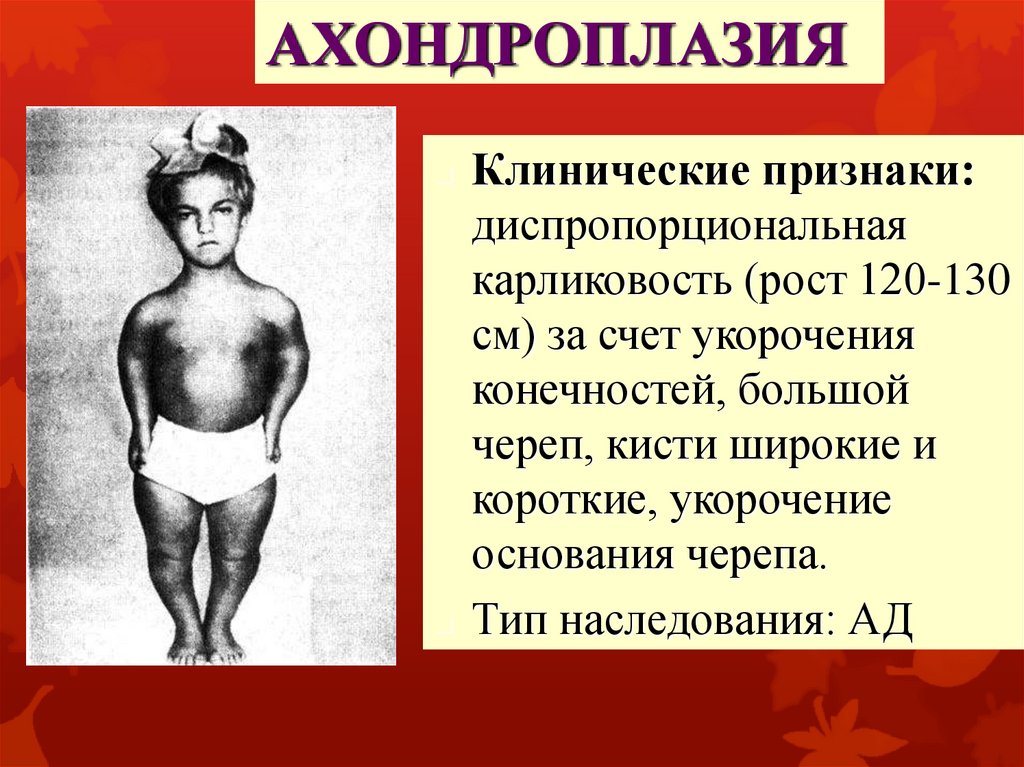
АХОНДРОПЛАЗИЯКлинические признаки:
диспропорциональная
карликовость (рост 120-130
см) за счет укорочения
конечностей, большой
череп, кисти широкие и
короткие, укорочение
основания черепа.
Тип наследования: АД
30.
ВИТИЛИГОКлинические признаки:
частичная депигментация
кожи; поражение обычно
симметричное на руках,
лице, шее. Больные очень
чувствительны к УФлучам (получают
солнечные ожоги),
повышен риск рака кожи.
Тип наследования: АД
31.
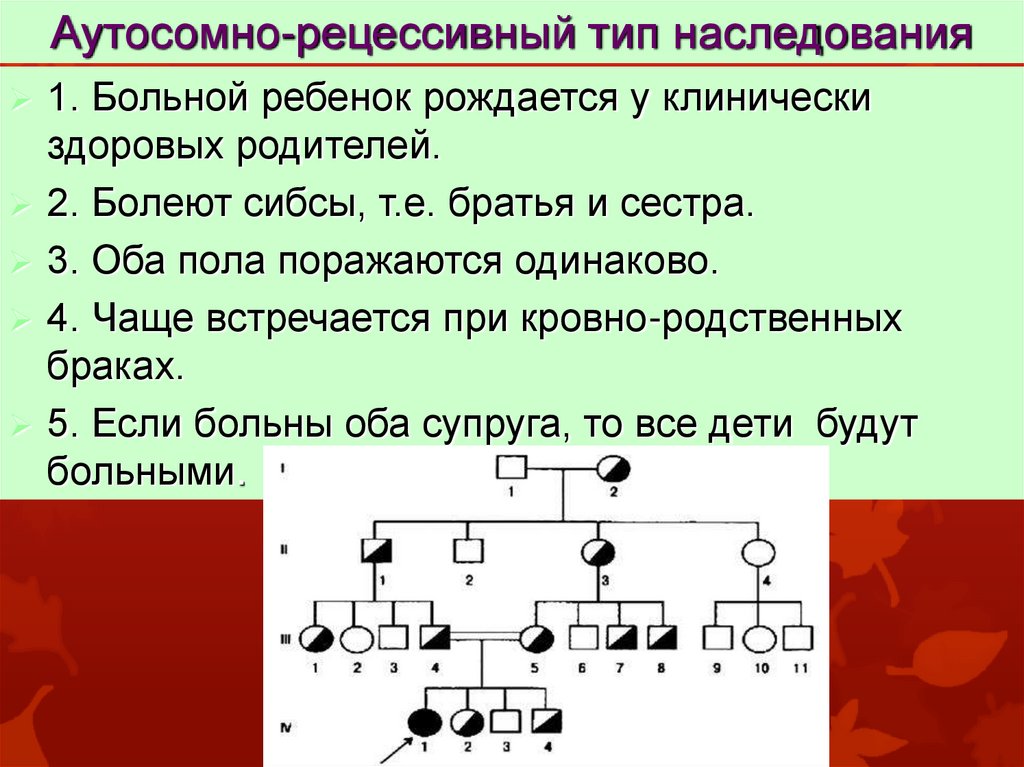
Аутосомно-рецессивный тип наследования1. Больной ребенок рождается у клинически
здоровых родителей.
2. Болеют сибсы, т.е. братья и сестра.
3. Оба пола поражаются одинаково.
4. Чаще встречается при кровно-родственных
браках.
5. Если больны оба супруга, то все дети будут
больными.
32.
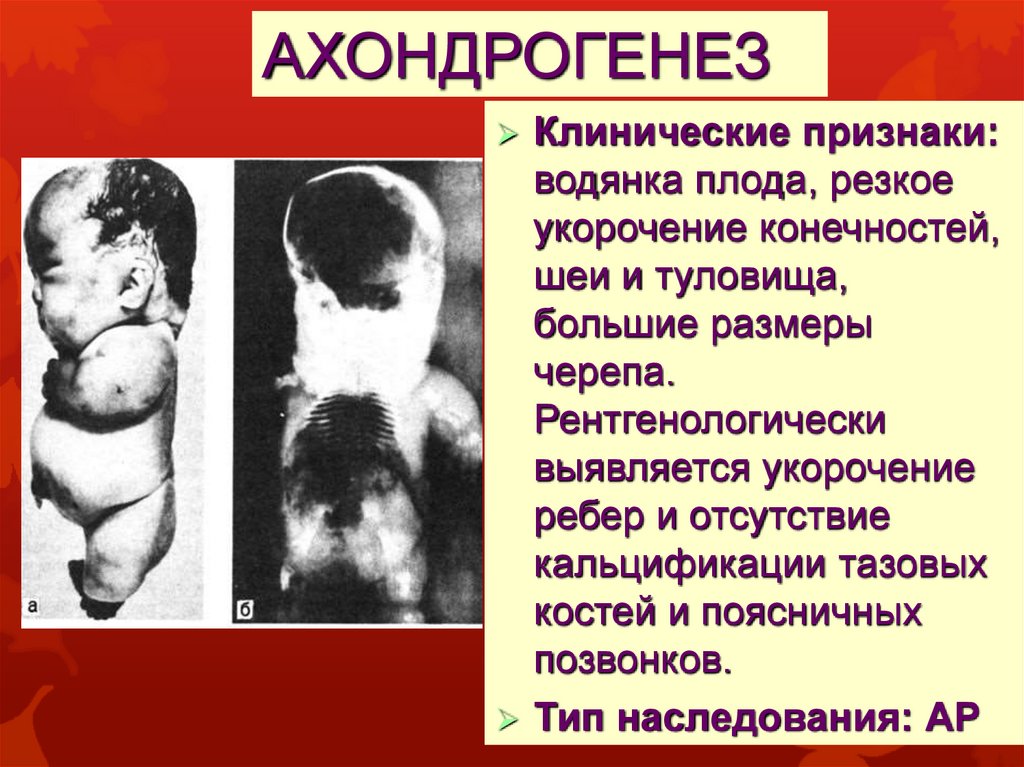
АХОНДРОГЕНЕЗКлинические признаки:
водянка плода, резкое
укорочение конечностей,
шеи и туловища,
большие размеры
черепа.
Рентгенологически
выявляется укорочение
ребер и отсутствие
кальцификации тазовых
костей и поясничных
позвонков.
Тип наследования: АР
33.
ЧЕРЕП В ФОРМЕ ТРИЛИСТНИКАКлинические признаки:
характерная форма черепа
(возникает вследствие
внутриутробного
зарастания швов) и лица,
высокий лоб, птоз,
клювовидный нос,
антимонголоидный разрез
глаз. Часто встречается в
сочетании с другими
аномалиями.
Тип наследования: АР
34.
НУНАН СИНДРОМВпервые описан в 1928 г.
Клинические признаки: гипертелоризм, эпикант,
низко посаженные уши, нарушение прикуса,
антимонголоидный разрез глаз, крипторхизм,
аномалии грудной клетки, низкий рост, пороки
сердца, умственная отсталость.
Тип наследования: АР
35.
КОККЕЙНА СИНДРОМВпервые описан в 1946 г.
Клинические признаки:
низкорослость,
старообразное лицо,
микроцефалия,
умственная отсталость,
дегенерация сетчатки,
деформации суставов,
килевидная грудная
клетка, тремор,
анорексия, крипторхизм.
Тип наследования: АР
36.
МУКОПОЛИСАХАРИДОЗСиндром Моркио описан в
1929 г.
Клинические признаки:
отставание в росте,
деформация позвоночника и
грудины, деформация
коленных суставов, короткая
шея и гипертрофия нижней
части лица, большой живот.
Смерть чаще от сердечной
патологии до 20 лет.
Тип наследования: АР
37.
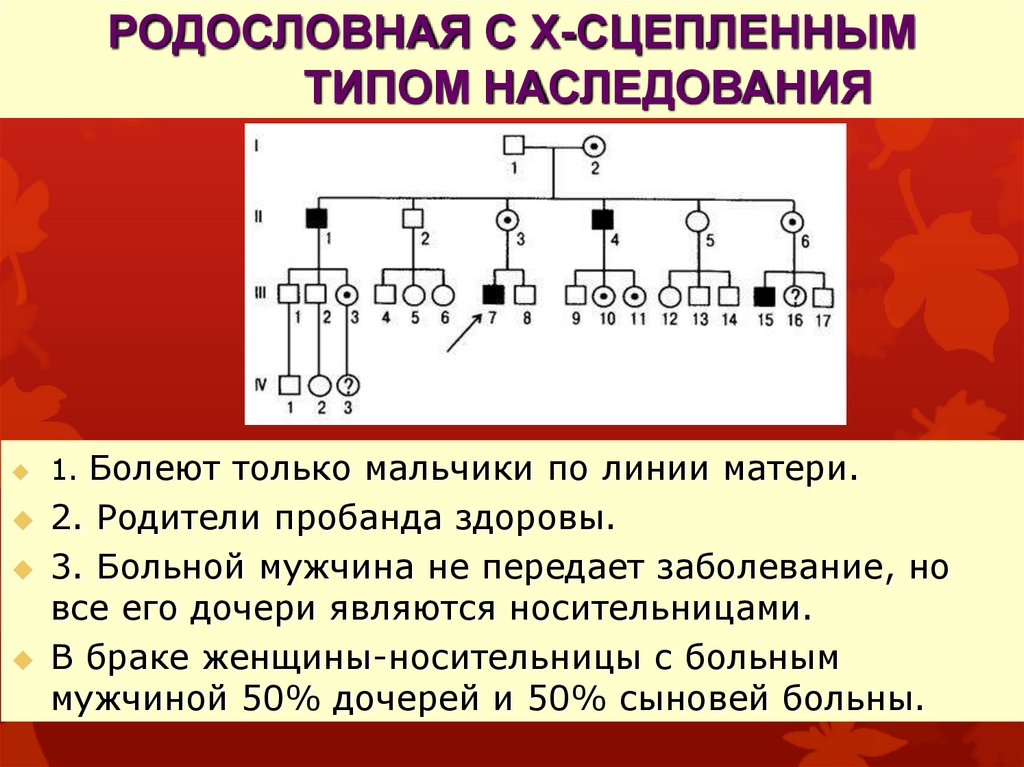
РОДОСЛОВНАЯ С Х-СЦЕПЛЕННЫМТИПОМ НАСЛЕДОВАНИЯ
1. Болеют только мальчики по линии матери.
2. Родители пробанда здоровы.
3. Больной мужчина не передает заболевание, но
все его дочери являются носительницами.
В браке женщины-носительницы с больным
мужчиной 50% дочерей и 50% сыновей больны.
38.
ГИДРОЦЕФАЛИЯКлинические признаки:
увеличение объема головы,
расширение желудочков мозга;
истончение и расхождение
костей черепа, диспропорция
мозговой и лицевой частей
черепа, косоглазие, умственная
отсталость и задержка
развития, расстройства
движений и координации,
атрофия белого вещества мозга.
Тип наследования: Хрецессив.
39.
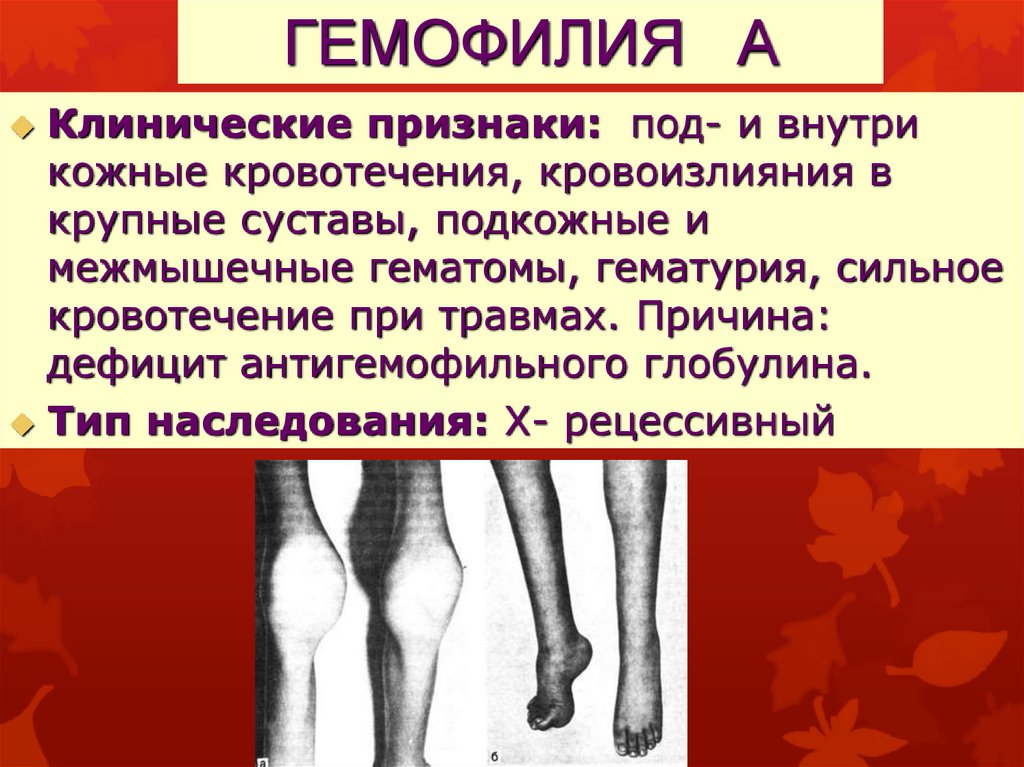
ГЕМОФИЛИЯ АКлинические признаки: под- и внутри
кожные кровотечения, кровоизлияния в
крупные суставы, подкожные и
межмышечные гематомы, гематурия, сильное
кровотечение при травмах. Причина:
дефицит антигемофильного глобулина.
Тип наследования: Х- рецессивный
40.
СИНДРОМ ЭЛЕРСА-ДАНЛООписан в 1657 г.
Клинические
признаки:
гиперрастяжимость
соединительной ткани
(нарушение синтеза
коллагена); кожа тонкая
как бумага; перегибание
пальцевых суставов на
90о, а локтевого и
коленного суставов на
10°; пороки внутренних
органов. Существует 8
типов.
Тип наследования: Хрецессив., АД, АР
41. МУЛЬТИФАКТОРИАЛЬНЫЕ ЗАБОЛЕВАНИЯ Для возникновения этих заболеваний требуется наследственная предрасположенность и определенные
условия внешней среды. На их долюприходится 92-93% всех хронических
неинфекционных заболеваний.
К мультифакториальным заболеваниям
относятся атеросклероз, варикоз,
гипертоническая болезнь, сахарный
диабет и др.
Наследование сложное, законам
Менделя не подчиняется.
42.
МЕДИКО-ГЕНЕТИЧЕСКОЕКОНСУЛЬТИРОВАНИЕ
ПРЕДУПРЕЖДЕНИЕ РОДСТВЕННЫХ
БРАКОВ.
ВЫЯВЛЕНИЕ ГЕТЕРОЗИГОТНЫХ
НОСИТЕЛЕЙ МУТАНТНОГО ГЕНА.
ДОРОДОВАЯ ДИАГНОСТИКА
НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
Главная его задача это прогнозирование
вероятности появления детей
с той или иной наследственной аномалией.
43.
Рекомендации, даваемые в медикогенетических консультациях направлены на то,чтобы консультируемые могли их учитывать и
добровольно принимать решение.
Врачи не рекомендуют браки между
близкими родственниками и между носителями
наследственных болезней.
Медицинское консультирование может выявить
наличие наследственных болезней, развитие
которых зависит от неблагоприятных
воздействий среды.
Своевременное проведение профилактических
мероприятий может предотвратить их
проявление.
44.
ТЕРМИНОЛОГИЧЕСКИЙ СЛОВАРЬ• Акроцефалия - высокий «башенный» череп.
• Алопеция – стойкое или временное выпадение
волос.
• Аменорея – отсутствие менструального цикла.
• Аплазия – полное отсутствие органа или части
его.
• Атрезия – отсутствие канала или естеств.
отверстий.
• Арахнодактилия – необычно длинные и тонкие
пальцы.
• Брахидактилия – укорочение пальцев.
• Витилиго – очаговая депигментация кожи.
• Гипертелоризм –широко расставленные глаза.
45.
ТЕРМИНОЛОГИЧЕСКИЙ СЛОВАРЬ
Прогерия – преждевременное старение
организма.
Птеригиум – крыловидные складки кожи.
Птоз – опущение внутренних органов или
века.
Синдактилия – сращение соседних
пальцев.
Страбизм – косоглазие.
Телеканти – латеральное смещение
внутренних углов глаз.
Тремор - дрожание конечностей, головы и
даже всего тела.
Энофтальм - глубокопосаженные глаза.
46.
• Гипертрихоз – избыточный рост волос.• Гипоплазия – недоразвитие органа.
• Гипогонадизм – недоразвитие половых
желез.
• Крипторхизм –отсутствие одного или обоих
яичек.
• Макроцефалия – чрезмерно большая голова.
• Микрогения –малые размеры нижней
челюсти.
• Микроцефалия – малые размеры
головного мозга.
• Полидактилия – увеличение количества
пальцев.
• Прогения – чрезмерное развитие нижней
челюсти.
47.
• Экзофтальм – смещение глазного яблокавперед, сопровождающееся расширением
глазной щели.
• Эпикант – вертикальная кожная складка у
внутреннего угла глаза.
• Анорексия - уменьшения аппетита.
• Гематома - полость, заполненная кровь.
• Гематурия - кровь в моче.
• Нистагм - непроизвольные ритмичные
судорожные движения глазных яблок.