























")
")








 Медицина
МедицинаПохожие презентации:


. Геморрагические диатезы")







Геморрагические диатезы
1. Геморрагические диатезы |
АЛГОРИТМЫ ДИАГНОСТИКИГЕМОРРАГИЧЕСКИХ СОСТОЯНИЙ
Бокарев И.Н., Смоленский В.С.,
Кабаева Е.В.
2. Геморрагические диатезы
К геморрагическим диатезам относят заболевания, в основекоторых лежат нарушения сосудистой стенки и различных
звеньев системы гемостаза, обусловливающие повышенную
кровоточивость или склонность к ее возникновению.
Геморрагические диатезы (ГД) – синдромы, характеризующиеся
избыточной кровоточивостью, обусловленной дефектом одного
или нескольких компонентов системы гемостаза.
В настоящем сообщении будет
рассмотрена диагностика лишь тех патологических состояний,
при которых ГД является ведущим признаком.
Причем внимание будет уделяться в
основном алгоритму диагностики этих состояний. Подробная
характеристика ГД из-за ограниченного объема статьи
опускается.
В обеспечении нормального гемостаза участвуют тромбоциты
(тромбоцитарный компонент), факторы свертывания крови
(плазменный компонент) и сосудистая стенка (сосудистый
компонент). Фибринолитическая система обеспечивает
растворение избыточных тромботических масс.
3. Эпидемиология
На земном шаре около 5 млн. человекстрадают первичными
геморрагическими проявлениями.
Учитывая, что вторичные геморрагии,
такие как ДВС-синдром в
предагональном состоянии, не всегда и
фиксируются, можно себе представить
широкую распространенность
геморрагических диатезов.
4. Этиология и патогенез
Патогенез наследственных геморрагическихсостояний определяется нарушением нормальных
гемостатических процессов: аномалиями
мегакариоцитов и тромбоцитов, дефицитом или
дефектом плазменных факторов свертывания крови,
неполноценностью мелких кровеносных сосудов.
Приобретенные геморрагические диатезы
обусловлены ДВС-синдромом, иммунными
поражениями сосудистой стенки и тромбоцитов,
токсикоинфекционными поражениями кровеносных
сосудов, заболеваниями печени, воздействиями
лекарственных средств.
5. Классификация
1. Геморрагические диатезы, обусловленные дефектом тромбоцитарногозвена
— недостаточность количества тромбоцитов
— функциональная неполноценность тромбоцитов
— сочетание количественной и качественной патологии тромбоцитов
2. Геморрагические диатезы, обусловленные дефектом прокоагулянтов
(гемофилии) — недостаточное их количество, необходимое для
формирования фибрина
— недостаточная функциональная активность отдельных прокоагулянтов
— наличие в крови ингибиторов отдельных прокоагулянтов
3. Геморрагические диатезы, обусловленные дефектом сосудистой стенки
— врожденные
— приобретенные
4. Геморрагические диатезы, обусловленные избыточным фибринолизом
— эндогенным (первичным и вторичным)
— экзогенным
5. Геморрагические диатезы, обусловленные сочетанием нарушений
различных компонентов системы гемостаза (болезнь Виллебранда,
ДВС-синдром и пр.)
6.
Данная классификация не включает все известныегеморрагические диатезы. Их более 300. Она
является схемой принципов классифицирования
геморрагических состояний, соблюдая которые
можно рубрифицировать не только любое из
известных геморрагических состояний, но и каждое
вновь обнаруженное.
Классификация тромбоцитопений предполагает их
подразделение в зависимости от основной причины,
их вызывающей. Этих причин несколько: нарушенное
воспроизводство, повышенное разрушение,
депонирование и разведение тромбоцитов. Причины
тромбоцитопений изложены ниже.
7.
1. Физические факторы— радиация
2. Химические факторы
— хлотиазид, цитостатики, уремия
3. Биологические факторы
— опухоли и т. д.
4. Уменьшение тромбоцитопоэза
— остеомиелофиброз
5. Врожденная гипоплазия мегакариоцитов
6. Авитаминоз (витамины В12, фолиевая кислота)
1. Иммунные
— лекарственная аллергическая тромбоцитопения
— посттрансфузионная аллергическая тромбоцитопения
— при коллагенозах
— при лимфолейкозах
— синдром Верльгофа
— изоиммунная неонатальная тромбоцитопения
— трансиммунная неонатальная тромбоцитопения
— вирусные инфекции
2. Неиммунные
— Болезнь Бернара — Сулье
— Синдром Вискотт — Олриджа
— Синдром Мэй — Хегглина
8. Тромбоцитопатия
Тромбоцитопатий — вторая группа геморрагических состояний,обусловленных неполноценностью тромбоцитарного компонента
гемостаза. Она объединяет заболевания, проявляющиеся
качественной неполноценностью тромбоцитов при сохранности их
количества. Она получила название тромбоцитопатий.
За последние годы в классификации тромбоцитопатий произошли
серьезные изменения. Суть их заключается в том, что многие
нозологические формы, характерной особенностью которых была
кровоточивость, оказались неоднородными.
Попытки увязать ту или иную особенность функциональных
нарушений тромбоцитов с поражением или особенностями
развития других органов или систем (синдром Херманского —
Прудлак, Чедияк — Хигаши и пр.) в этом плане также
демонстрируют определенный полиморфизм. Все это заставило
врачей концентрировать внимание на конкретной патологии
функции тромбоцитов, которая и легла в основу.
9. Различают следующие виды тромбоцитопатий:
1) тромбоцитопатия с нарушением адгезиитромбоцитов;
2) тромбоцитопатия с нарушением агрегации
тромбоцитов: а) к АДФ, б) к коллагену, в) к
ристомицину, г) тромбину, д) адреналину;
3) тромбоцитопатия с нарушением реакции
высвобождения;
4) тромбоцитопатия с дефектом «пула
накопления» высвобождающихся факторов;
5) тромбоцитопатия с дефектом ретракции;
6) тромбоцитопатия с сочетанием
вышеизложенных дефектов.
10.
Кроме констатации тромбоцитарныхдефектов, необходимо дополнять
диагностику заболевания обязательным
указанием количественной стороны
тромбоцитарного звена (гипотромбоцитоз,
гипертромбоцитоз, нормальное количество
тромбоцитов), а также констатацией
сопутствующей патологии.
Обобщены заболевания, в основе которых
лежит дефицит определенных плазменных
факторов свертывания крови (может быть их
все правильнее называть гемофилии).
11.
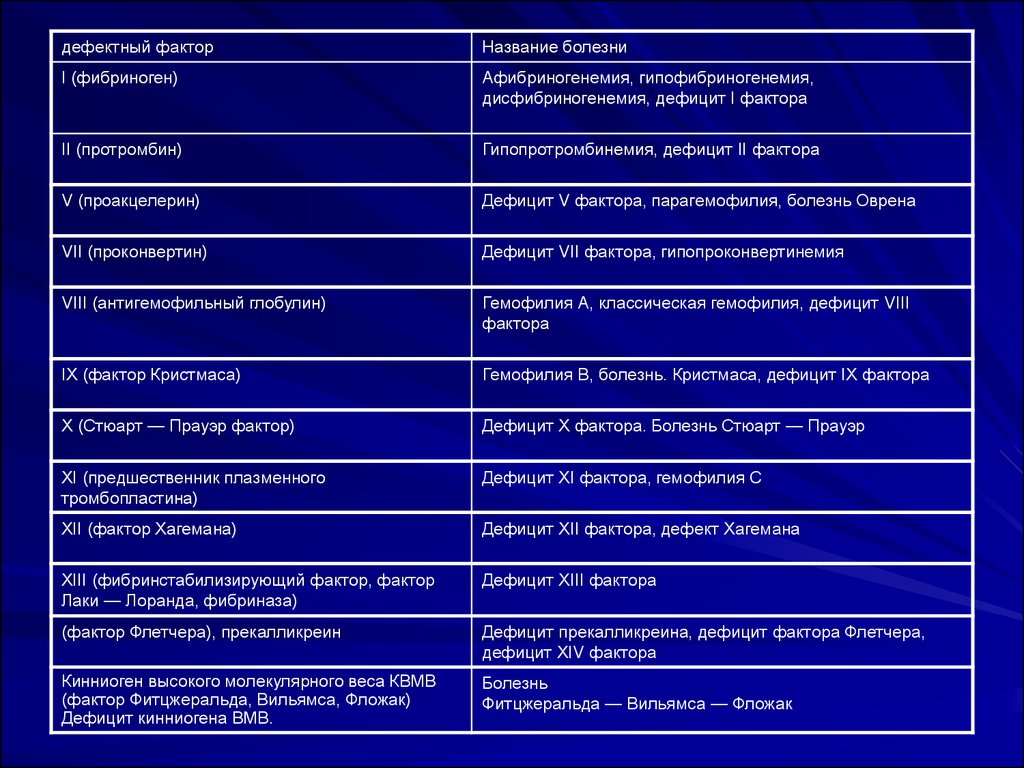
дефектный факторНазвание болезни
I (фибриноген)
Афибриногенемия, гипофибриногенемия,
дисфибриногенемия, дефицит I фактора
II (протромбин)
Гипопротромбинемия, дефицит II фактора
V (проакцелерин)
Дефицит V фактора, парагемофилия, болезнь Оврена
VII (проконвертин)
Дефицит VII фактора, гипопроконвертинемия
VIII (антигемофильный глобулин)
Гемофилия А, классическая гемофилия, дефицит VIII
фактора
IX (фактор Кристмаса)
Гемофилия В, болезнь. Кристмаса, дефицит IX фактора
X (Стюарт — Прауэр фактор)
Дефицит X фактора. Болезнь Стюарт — Прауэр
XI (предшественник плазменного
тромбопластина)
Дефицит XI фактора, гемофилия С
XII (фактор Хагемана)
Дефицит XII фактора, дефект Хагемана
XIII (фибринстабилизирующий фактор, фактор
Лаки — Лоранда, фибриназа)
Дефицит XIII фактора
(фактор Флетчера), прекалликреин
Дефицит прекалликреина, дефицит фактора Флетчера,
дефицит XIV фактора
Кинниоген высокого молекулярного веса КВМВ
(фактор Фитцжеральда, Вильямса, Фложак)
Дефицит кинниогена ВМВ.
Болезнь
Фитцжеральда — Вильямса — Фложак
12.
Классификация сосудистых заболеваний,протекающих с геморрагическими
проявлениями, предполагает их
подразделение в зависимости от
локализации поражения морфологических
структур сосуда.
Различают заболевания с поражением самого
эндотелия и заболевания с поражением
субэндотелия.
13.
Поражения эндотелия подразделяются на врожденные иприобретенные. Представителем врожденных
повреждений эндотелия является наследственная
геморрагическая телеангиэктазия (болезнь Рандю —
Ослера).
Среди приобретенных поражений эндотелия различают
заболевания воспалительного и иммунного характера,
повреждения, обусловленные механическими
факторами. Воспалительные и иммунные
приобретенные геморрагические состояния — это
болезнь Шенлейн — Геноха, узелковый артериит,
аллергический гранулематоз, васкулиты при
инфекционных заболеваниях и воздействиях лекарств.
В эту же подгруппу объединяются хронические
воспалительные инфильтраты, такие как гранулематоз
Вегенера, височный артериит, артериит Такаясу. Среди
механических повреждений эндотелия различают
ортостатическую пурпуру и саркому Капоши.
14.
Геморрагические заболевания, обусловленныенарушениями субэндотелиальных структур,
также подразделяются на врожденные и
приобретенные. Среди врожденных выделяют
синдром Эйлерс — Данлоса, эластическую
псевдоксантому, синдром Марфана, а также
болезнь несовершенного остеогенеза.
В приобретенные дефекты субэндотелия
объединяют геморрагические состояния при
амилоидозе, сенильной пурпуре,
кортикостероидной пурпуре, простой пурпуре и
геморрагические состояния при сахарном
диабете.
15. Таблица 1. Дифференциально-диагностическое значение кровоточивости
Вид кровоточивостиВероятность
ГД
Другие наиболее частые причины повышенной
кровоточивости
Спонтанные геморрагии
Носовые кровотечения
±
Местный дефект (ринит, дефект сосудов сплетения
Киссельбаха) или артериальная гипертензия
Десневые кровотечения
±
Пародонтоз
Меноррагии
±
Полипы, эрозии, опухоли гениталий
Гематурия
±
Местное повреждение урологического тракта
(камни, опухоли, полипы)
Желудочно-кишечные
кровотечения
±
Язвенные поражения слизистой, опухоли
желудочно-кишечного тракта
Кровохарканье
±
Тромбоэмболия легочной артерии, рак легких или
туберкулез
Примечание. ± – ГД мало вероятен; ++ – ГД вероятен.
16. Таблица 1. Дифференциально-диагностическое значение кровоточивости
Вид кровоточивостиВероятность
ГД
Другие наиболее частые причины повышенной
кровоточивости
Реакция на травму
Петехии, экхимозы
++
Глубокие подкожные гематомы
("синяки")
++
Гемартрозы
++
Длительные или обильные
кровотечения:из порезов
++
при удалении зубов
++
при тонзиллэктомии
++
во время или после операции
++
Пупочные кровотечения (при
рождении)
++
Повышенная кровоточивость в ответ на травму
свидетельствует о наличии у больного ГД, а
степень кровоточивости и гемостатические
средства, необходимые для ее устранения,
указывают на выраженность ГД
Примечание. ± – ГД мало вероятен; ++ – ГД вероятен.
17. КЛИНИЧЕСКИЕ МЕТОДЫ ДИАГНОСТИКИ
КЛИНИЧЕСКИЕ МЕТОДЫ ДИАГНОСТИКИВажную диагностическую информацию можно получить в ходе
осмотра и расспроса больного. В табл. 1 перечислены
возможные геморрагические прявления и их
дифференциально-диагностическое значение.
Тип кровоточивости в большинстве случаев зависит от вида
нарушения в системе гемостаза (табл. 2).
На основании данных клинического обследования и анамнеза
устанавливают тяжесть ГД, тип кровоточивости, время
появления жалоб, характер ГД (врожденный или
приобретенный), тип наследования.
Поиск причины и нозологический диагноз ГД облегчаются в тех
случаях, когда геморрагии в сочетании с другими симптомами
образуют синдром, характерный для определенных
нозологических форм (табл. 3), или когда геморрагии возникают
на фоне заболеваний или состояний, способных вызывать то
или иное нарушение в системе гемостаза (табл. 4).
18. Таблица 2. Зависимость характера кровоточивости от вида нарушения в системе гемостаза
Вид геморрагийХарактер кровотечений
тромбоцитарно-сосудистый
дефект
дефект плазменного компонента
Кровотечения в результате
поверхностных повреждений
Частые, профузные и
длительные
Редкие, не очень выраженные
Спонтанные кровоподтеки и
гематомы
Небольшие и поверхностные,
часто множественные
Обширные и глубокие, обычно
изолированные
Кожная и слизистая пурпура
Очень часто
Возникает в редких случаях
Кровоизлияния в суставы
Очень редки
Часты
Кровотечения вследствие
глубоких повреждений, удаления
зубов и т.п.
Обычно начинаются сразу.
Часто прекращаются под
влиянием местных средств
Возникают с запозданием, почти
не прекращаются под влиянием
местных гемостатических
средств
Наиболее частые проявления
Пурпура и экхимозы, эпистаксис,
меноррагии, желудочнокишечные кровотечения
Глубокие кровоизлияния (могут
быть без видимых причин или
после травм), особенно
суставные и мышечные,
длительные отсроченные
кровотечения после
повреждений
19. ЛАБОРАТОРНЫЕ МЕТОДЫ ДИАГНОСТИКИ
ЛАБОРАТОРНЫЕ МЕТОДЫ ДИАГНОСТИКИДанные лабораторных исследований имеют решающее значение для
постановки диагноза. Нужно помнить следующее: изменения
лабораторных тестов часто обнаруживаются только в
момент геморрагического эпизода; нормальные лабораторные
показатели у больных, имевших в анамнезе повышенную
кровоточивость, не свидетельствуют об отсутствии у них ГД (в таких
случаях рекомендуются повторные, часто многократные обследования);
часть лабораторных тестов, применяемых для исследования системы
гемостаза, недостаточно чувствительны (например, определение
времени свертывания крови).
Даже результаты такого теста, как определение активированного
частичного тромбопластинового времени (АЧТВ), изменяются у больного
гемофилией лишь при снижении недостающего фактора до уровня
менее 10% от нормы.
Нужно отметить, что и признаки кровоточивости появляются обычно
тогда, когда содержание какого-либо фактора становится ниже этого
критического уровня.
При некоторых видах ГД (аутоэритроцитарной сенсибилизации,
повышенной чувствительности к собственной ДНК, гемоглобину и др.) не
удается выявить нарушений системы гемостаза даже с помощью
современных методов.
20. Таблица 3. Диагностическая значимость геморрагий при их сочетании с другими симптомами
Клинические симптомы, отмечаемые наряду с ГДНаиболее вероятный диагноз
Распространенные геморрагии кожи и слизистых
Лихорадка
Сепсис, острый промиелоцитарный лейкоз
Выраженные кожные геморрагии, вплоть до некроза
кожи
Лихорадка
Артериальная гипертензия
Молниеносная пурпура
Распространенные геморрагии кожи и слизистых
Тромботическая тромбоцитопеническая пурпура
(синдром Мошковича)
Лихорадка
Неврологические нарушения (преходящие)
Умеренные кожные геморрагии
Гемолитическая анемия
Острая почечная недостаточность
Гемолитико-уремический синдром (синдром Гассера)
Кожная пурпура (полиморфная, симметричная)
Болезнь Шенлейна – Геноха
Артрит крупных суставов
Лихорадка
Кожные и слизистые геморрагии
Гемолитическая анемия
Синдром Фишера – Эванса
Умеренные кожные и слизистые геморрагии
Тромбоцитемия
Феномен Рейно, преходящие приступы ишемии мозга и
рецидивирующий тромбоз
21.
22. К ДИАГНОСТИКЕ ДВС - СИНДРОМА
Под диссеминированным внутрисосудистым свертываниемкрови (ДВС-синдром) понимают нарушение кровообращения в
микроциркуляторном русле из-за диффузного отложения в нем
фибрина и тромбоцитарных агрегатов. ДВС-синдром не является
самостоятельным заболеванием, но он осложняет течение многих
болезней.
Существует около ста клинических ситуаций, при которых
развивается ДВС-синдром. Это прежде всего опухоли (37%),
инфекционные заболевания (36%), лейкозы (14%),
шоковые состояния, особенно инфекционный шок (8,7%).
Кровотечения при ДВС-синдроме возникают вследствие одного
или нескольких изменений свойств крови, таких как потребление
факторов свертывания, тромбоцитопения, нарушение функции
тромбоцитов, активация реактивного фибринолиза и действие
продуктов деградации фибрина (ПДФ).
В сложных случаях диагностике помогает определение уровня
ПДФ и Д-димера, которые при ДВС-синдроме резко повышены.
23. Таблица 4. Наиболее частые причины геморрагий при отдельных патологических состояниях
Вид патологииНаиболее вероятные причины кровоточивости
Опухоли
ДВС-синдром, тромбоцитопения (метастатическое поражение костного мозга - КМ),
прорастание сосудов
Инфекционные заболевания
ДВС-синдром, тромбоцитопения (угнетение КМ; аутоиммунное поражение
тромбоцитов)
Острый лейкоз
ДВС-синдром, тромбоцитопения (поражение КМ)
Шоковое состояние
ДВС-синдром
Состояние после экстракорпорального кровообращения и оксигенации
Тромбоцитопения (отложение тромбоцитов на диализных мембранах)
Побочная реакция на прием лекарства
Васкулиты (гиперчувствительность), тромбоцитопения (угнетение КМ, повышенное
разрушение тромбоцитов иммунными механизмами), тромбоцитопатия
Хронический алкоголизм
Тромбоцитопения
Заболевания печени, сопровождающиеся печеночно-клеточной
недостаточностью
Снижение синтеза факторов свертывания крови в гепатоцитах, тромбоцитопения
(при гиперспленизме)
Обтурационная желтуха
Снижение синтеза факторов протромбинового комплекса (II, VII, IX, X) из-за
дефицита витамина К
Хронический миелопролиферативный синдром (болезнь Вакеза,
хронический миелолейкоз)
Тромбоцитемия
Миеломная болезнь
Сосудистые нарушения, тромбоцитопатия, тромбоцитопения
Макроглобулинемия Вальденстрема
Сосудистые нарушения, тромбоцитопатия, тромбоцитопения
Криоглобулинемия
Сосудистые нарушения, тромбоцитопатия, тромбоцитопения
Амилоидоз
Сосудистые нарушения, тромбоцитопатия, тромбоцитопения
Гипотиреоз
Тромбоцитопения (гипоплазия КМ)
Уремия
Тромбоцитопения (гипоплазия КМ), тромбоцитопатия
Гемотрансфузии
Тромбоцитопения в результате иммунной аллергической реакции, при разведении
большим количеством "старой" крови, не содержащей тромбоцитов, ДВС-синдром
Коллагенозы (системная красная волчанка, ревматоидный артрит,
дерматомиозит и др.)
Тромбоцитопения (повышенное разрушение в результате иммунных механизмов),
ингибиторная гемофилия(антитела к какому-либо фактору свертывания), васкулит
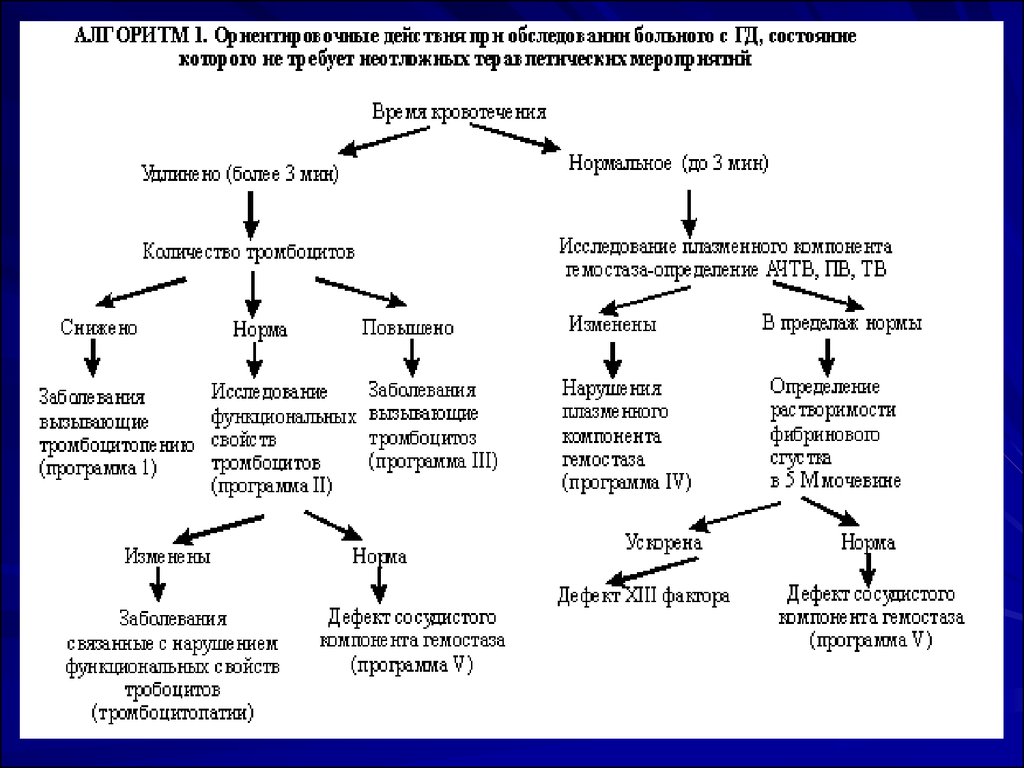
24. ПРОГРАММА 2. Обследование больных с ГД, состояние которых не требует неотложного вмешательства.
Если клинико-анамнестические данные больного с ГД не позволяютопределить направление, в котором следует искать причину повышенной
кровоточивости, целесообразно начать исследование с определения времени
кровотечения (ВК), как показано в Алгоритме 1.
Первоочередное изучение тромбоцитарного компонента гемостаза логично
еще и потому, что 80% всех случаев повышенной кровоточивости связано с
патологией тромбоцитов, в 18 - 20% случаев причина кровотечения –
нарушения плазменного компонента гемостаза и лишь в 1 - 2% – дефект
сосудистой стенки.
Аутоиммунные процессы обусловливают медикаментозную аллергическую
тромбоцитопению; посттрансфузионную аллергическую тромбоцитопению;
тромбоцитопению при заболеваниях соединительной ткани (системная красная
волчанка и др.), гемолитической аутоиммунной анемии и гипертиреозе,
хроническом лимфолейкозе; идеопатическую тромбоцитопеническую пурпуру
(болезнь Верльгофа).
Последний диагноз выставляется только после исключения всех
вышеперечисленных заболеваний.
Ускоренное разрушение (потребление) тромбоцитов на периферии
вследствие неиммунных процессов может возникать при ДВС-синдроме,
алкоголизме, гиперспленизме, массивных переливаниях "старой" крови, после
экстракорпорального кровообращения.
Вышеуказанные факторы и заболевания, приводящие к тромбоцитопении,
исключаются (или подтверждаются) на основании соответствующих
анамнестических и клинических данных (см. Алгоритм 2).
25.
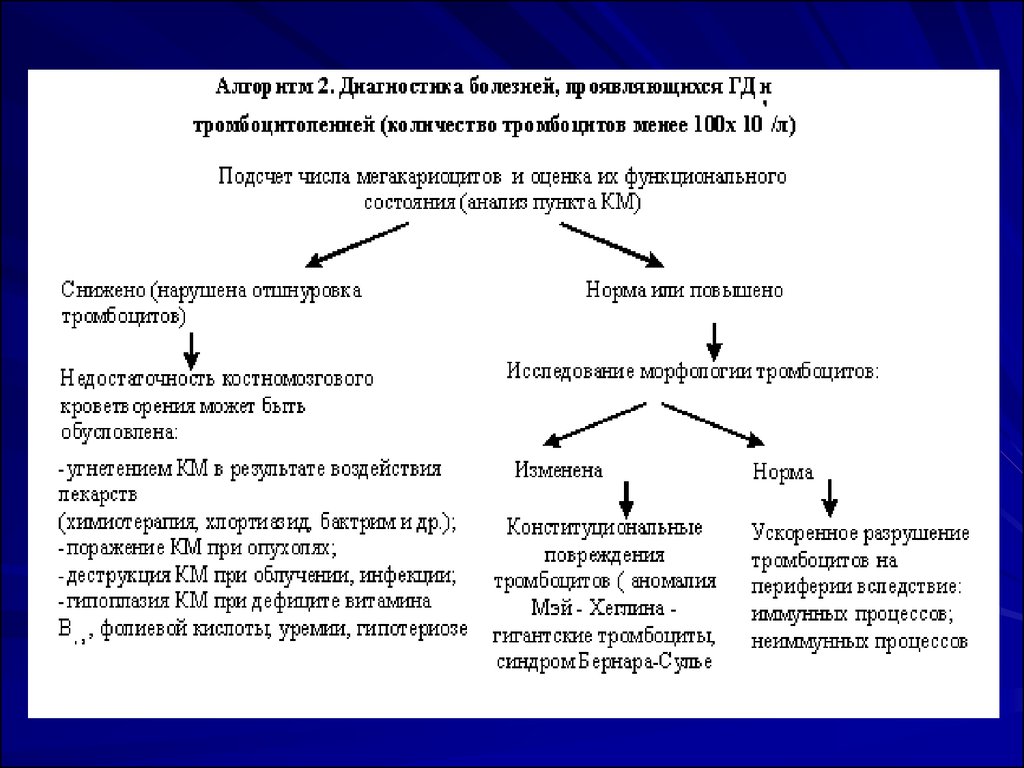
26. ПРОГРАММА 3. Диагностика болезней, проявляющихся ГД, удлинением времени кровотечения при нормальном количестве тромбоцитов
ПРОГРАММА 3. Диагностика болезней, проявляющихся ГД, удлинениемвремени кровотечения при нормальном количестве тромбоцитов
Такое сочетание лабораторных показателей характерно как для
тромбоцитопатий, так и для сосудистых нарушений. Для исключения
дефекта тромбоцитов необходимо исследовать их функциональные
свойства, что доступно лишь специализированным лабораториям.
Общепринятым считается определение следующих показателей:
адгезия тромбоцитов (прилипание их к стеклу, коллагену); агрегация
(прилипание тромбоцитов друг к другу), индуцированная АДФ,
адреналином, коллагеном, тромбином, ристоцетином; реакция
высвобождения (III фактора, АДФ, b-тромбоглобулина и т.д.); ретракция
кровяного сгустка. Результаты этих исследований позволяют
диагностировать тромбоцитопатии, нозологическая принадлежность
которых обусловлена характерным нарушением тех или иных
функциональных свойств тромбоцитов или их сочетанием.
Изменения функциональных свойств тромбоцитов могут наблюдаться
при уремии, макроглобулинемии Вальденстрема, миеломной болезни,
заболеваниях печени и другой патологии, а также под действием ряда
лекарств (ацетилсалициловой кислоты, тиклопедина, сульфинпиразона,
дипиридамола, нестероидных противовоспалительных препаратов,
декстрана и др.).
Эти факторы вызывают не всегда однозначные изменения
функциональной активности тромбоцитов и, кроме того, могут выявлять
их ранее не манифестировавшие дефекты.
27. Таблица 5. Плазменные нарушения гемостаза (гемофилии)
Дефектный факторНазвание болезни
Синонимы заболевания
I (фибриноген)
Афибриногенемия,
гипофибриногенемия,
дисфибриногенемия
Дефицит I фактора
II (протромбин)
Гипопротромбинемия
Дефицит II фактора
V (проакцелирин)
Дефицит V)
Парагемофилия, болезнь Оврена
VII (проконвертин)
Дефицит VII фактора
Гипопроконвертинемия
VIII (антигемофилический глобулин)
Гемофилия А
Классическая гемофилия, дефицит VIII
фактора
Болезнь Виллебранда
Ангиогемофилия
IX (фактор Кристмаса)
Гемофилия В
Болезнь Кристмаса, дефицит IX фактора
Х (фактор Стюарта – Прауэр)
Дефицит Х фактора
Болезнь Стюарта – Прауэра
XI (предшественник плазменного
тромбопластина)
Дефицит XI фактора
Гемофилия С
XII* (фактор Хагемана)
Дефицит XII фактора
Симптом Хагемана
XIII (фибринстабилизирующий фактор,
фактор Лаки-Лорана, фибриназа)
Дефицит XIII фактора
–
XIV**(фактор Флетчера,
прекалликреин)
Дефицит прекалликреина
Дефицит фактора Флетчера, дефицит XIV
фактора
XV** (кининоген высокой
молекулярной массы – КВММ, фактор
Фитцжеральда, Вильямса, Фложак)
Дефицит кининогена ВММ
Болезнь Фитцжеральда, Вильямса, Фложак
* Дефицит XII, XIV и XV факторов свертывания крови не проявляется геморрагиями, хотя лабораторное обследование обнаруживает у этих больных нарушение
контактной активации (удлинение АЧТВ).
28. ПРОГРАММА 4. Диагностика болезней, проявляющихся ГД и увеличением количества тромбоцитов (500–600 х 109/л)
Повышение количества тромбоцитов может быть обусловлено следующимипричинами.
• Реактивный тромбоцитоз при опухолях с метастазами, хронических
инфекционных заболеваниях, спленэктомии (может достигать 10 х 12 л), обширном
повреждении тканей (переломы ног, большие операции, роды).
Отсутствие у больного вышеуказанных факторов, провоцирующих вторичное
повышение количества тромбоцитов, позволяет исключить реактивный
тромбоцитоз.
• Первичное миелопролиферативное заболевание – тромбоцитемия.
Тромбоцитемия представляет собой лишь одну из форм миелопролиферативного
синдрома, который проявляется также истинной полицитемией (болезнь Вакеза) и
хроническим миелолейкозом. Кроме того,
первичная геморрагическая тромбоцитемия в ходе своего развития со временем
может перейти в болезнь Вакеза или хронический миелолейкоз.
При обоих типах повышения количества тромбоцитов последние формируются
быстро и часто бывают неполноценными в функциональном отношении. Это
выражается двумя свойствами, которые могут существовать одновременно:
1) спонтанной агрегацией тромбоцитов, клинически проявляющейся феноменом
Рейно, преходящими приступами ишемии мозга, тромбозами селезеночной ,
воротной вены, вен нижних конечностей, пещеристого тела (приапизм), коронарных
сосудов сердца;
2) слабым ответом на действие физиологических индукторов с повышенной
наклонностью к геморрагиям слизистых оболочек, что проявляется носовыми
кровотечениями, кровавой рвотой, меленой, гематурией, кровохарканьем,
меноррагией.
29.
30. ПРОГРАММА5. Диагностика болезней, проявляющихся ГД, нормальным ВК и изменениями тестов плазменного гемостаза
ПРОГРАММА5.Диагностика болезней, проявляющихся ГД, нормальным ВК
и изменениями тестов плазменного гемостаза
Такое сочетание лабораторных показателей
характерно для гемофилий, т.е. ГД, вызванных
несостоятельностью того или иного белка
(прокоагулянта). В табл. 5 указаны возможные
нарушения плазменного компонента гемостаза и их
закрепившиеся названия. Тип кровоточивости,
характерный для всей группы гемофилий, уже
обсуждался ранее. Нужно лишь отметить, что
тяжесть геморрагического синдрома, как правило,
связана со степенью дефекта фактора свертывания.
Точный диагноз заболевания (указание конкретного
пораженного фактора или группы факторов)
устанавливается на основании лабораторных данных
(анализ результатов определения АЧТВ, ПВ, ТВ,
производства коррекционных проб и использования
дефицитных плазм).
31. Острая идиопатическая тромбоцитопеническая пурпура.
32.
Обширные гематомы у пациента сгемофилией
Острый гемартроз
коленного сустава у
больного
гемофилией.
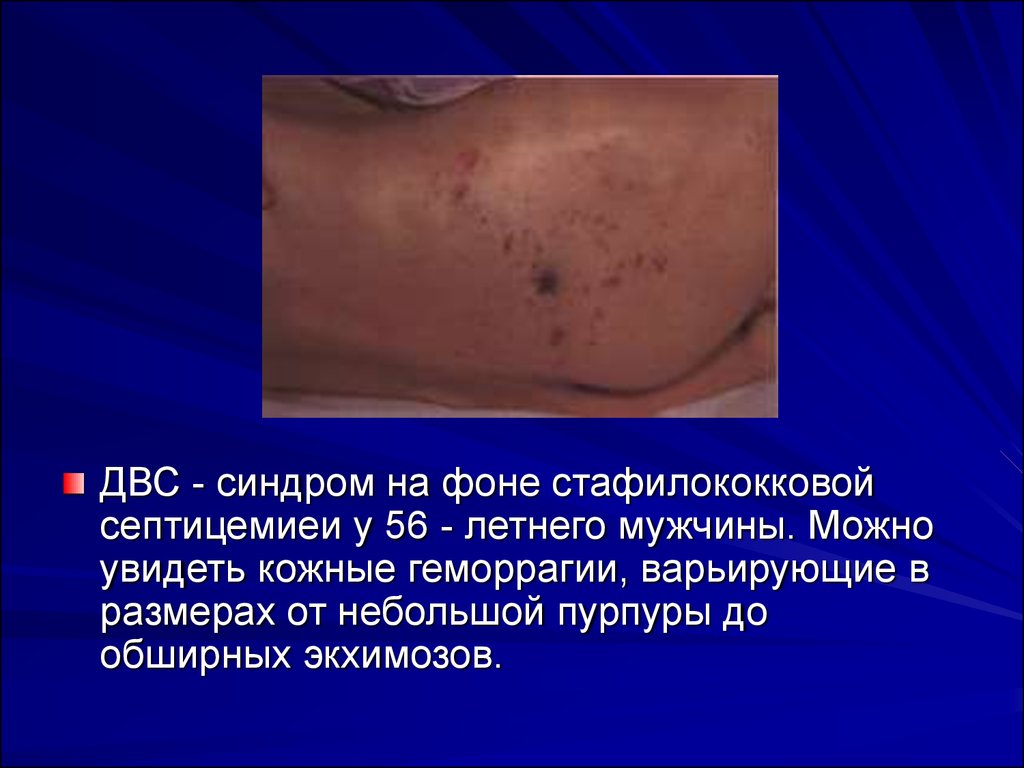
33.
ДВС - синдром на фоне стафилококковойсептицемиеи у 56 - летнего мужчины. Можно
увидеть кожные геморрагии, варьирующие в
размерах от небольшой пурпуры до
обширных экхимозов.
34.
Нарушения в плазменном компоненте гемостаза могутбыть не только врожденными, но и приобретенными.
Наиболее часто снижение уровня факторов
свертывания наблюдается при дисфункции
печеночных клеток, так как все факторы свертывания,
за исключением VIII, синтезируются гепатоцитом. В
первую очередь снижается уровень витамин Кзависимых факторов (II, VII, IX и X).
Аналогичная ситуация возникает при приеме
оральных антикоагулянтов – антивитаминов К.
Против белков свертывания могут образовываться
антитела (чаще против VIII фактора). Это
наблюдается при аутоиммунных заболеваниях, в
послеродовом периоде и при гиперчувствительности к
лекарствам (антибиотики, нитрофураны,
сульфаниламиды и др.).
35.
Диагностика избыточного фибринолиза как причины ГДпроводится в рамках этой же программы. Сам факт повышенного
фибринолиза устанавливается только лабораторным путем:
выявление удлинения ТВ, ускорения лизиса эуглобулинового
сгустка и повышения уровня ПДФ.
В качестве причины рассматриваются передозировка
тромболитических препаратов и ДВС-синдром. Первое
исключается на основании анамнестических данных, тактика
диагностики второго обсуждалась в соответствующем разделе.
Здесь же нужно обратить внимание на то, что при кровотечениях,
возникающих у больных после операций на предстательной
железе, небных миндалинах, при гиперменорее, язвенном
поражении желудочно-кишечного тракта, а также
посттравматических кровоизлияниях в среды глаза (гифемах)
следует предполагать наличие избыточного фибринолиза. Повидимому, он обусловлен локальным избытком плазмина, так как
с помощью вышеуказанных лабораторных методов в венозной
крови его определить не удается. Тем не менее назначение
ингибиторов фибринолиза для лечения этих кровотечений дает
хороший эффект.
36. ПРОГРАММА 6. Диагностика болезней, проявляющихся ГД при удлиненном или нормальном ВК и неизмененных тестах плазменного гемостаза
Как уже упоминалось ранее, нарушения тромбоцитарного сосудистого иплазменного компонентов гемостаза можно разграничить уже на основании типа
кровоточивости (см. табл. 2). Кроме того, патогномоничные симптомы некоторых
заболеваний сосудов настолько ярки, что не требуют предварительного
исследования тромбоцитарного компонента гемостаза.
Причины и механизмы нарушения сосудистой стенки разнообразны, но все они
приводят в конечном счете к неспособности взаимодействия тромбоцитов со
стенкой сосуда и к кровоточивости. Клинический диагноз основывается на
характере кожных и слизистых геморрагий в сочетании с особенностями
конкретной нозологической формы. Нозологический диагноз подтверждается на
основании морфологического изучения сосудов. С клинических позиций все
заболевания сосудистой стенки удобнее разделить на врожденные и
приобретенные.
К первым относятся: болезнь Рандю - Ослера - Вебера (наследственная
геморрагическая телеангиэктазия); синдром Элерса - Данлоса (генерализованная
фибродисплазия эластических волокон; сосудистые опухоли (гемангиомы).
Вторую группу представляют: васкулиты (болезнь Шенлейна – Геноха и др.);
сенильная пурпура; геморрагическая саркома Капоши; узловатая эритема;
болезнь Шамберга; болезнь Майокки (кольцевидная пурпура); пигментный
дерматит (Гужеро – Блюма); ползучая ангиома Хатчинсона.
Нужно учитывать возможность редких случаев скорбута (дефицита витамина
С), который наблюдается у одиноких старых людей с измененной психикой,
питающихся исключительно консервами в течение многих месяцев, а также о
возможности симуляции ГД, в частности приемом повышенных доз
антикоагулянтов или механически вызываемыми экхимозами, гематурией,
десневыми кровотечениями.
37. Примерная формулировка диагноза:
1. Иммунная тромбоцитопеническая пурпура,протекающая с геморрагиями на коже и на видимых
слизистых оболочках, десневыми, носовыми,
кишечными кровотечениями.
2. Гемофилия А (классическая гемофилия),
обусловленная дефицитом VIII фактора с
кровоизлияниями в мышцы и суставы, носовыми,
десневыми, кишечными, маточными кровотечениями.
3. Синдром диссеминированного внутрисосудистого
свертывания с кожными петехиями, кровоточивостью
слизистых оболочек, гематурией, кровохарканьем.
38. ЗАКЛЮЧЕНИЕ
В заключение нам хотелось бы еще раз подчеркнуть, что не во всех ситуацияхдиагностический поиск проходит через все предложенные программы алгоритма.
Тщательно собранный анамнез и клиническое обследование в ряде случаев
позволяют сразу же поставить правильный предположительный диагноз.
Основываясь на принципе выделения ведущего компонента в нарушении
гемостаза, ГД можно разделить на 5 групп.
1. ГД, обусловленные дефектом тромбоцитарного звена, возникающие в
результате:
– недостаточного количества тромбоцитов;
– функциональной неполноценности тромбоцитов;
– сочетания количественной и качественной патологии тромбоцитов.
2. ГД, обусловленные дефектом прокоагулянтов (гемофилии), возникающие в
результате:
– недостаточного количества одного или нескольких факторов, участвующих в
формировании фибрина;
– недостаточной активности вышеуказанных факторов;
– наличия ингибиторов отдельных прокоагулянтов.
3. ГД, обусловленные нарушениями сосудистой стенки.
4. ГД, обусловленные избыточным фибринолизом, который может быть
эндогенным (первичным и вторичным) и экзогенным.
5. ГД, обусловленные сочетанием нарушений различных компонентов системы
гемостаза.
К лабораторным исследованиям прибегают лишь для его подтверждения или
уточнения.