")














")




 Медицина
МедицинаПохожие презентации:










Особенности разработки методики теста «Растворение» (ключевые параметры; процесс разработки)
1. Особенности разработки методики теста «Растворение» (ключевые параметры; процесс разработки)
Выполнила:ординатор группы ФО-3301
Воронович Алина
2.
Тест «Растворение» используется для определения количествалекарственного вещества, которое в условиях, указанных в
частной фармакопейной статье, за определенный
промежуток времени должно высвобождаться в среду
растворения из твердой дозированной лекарственной
формы.
3. Ключевые параметры
тип аппарата;среда растворения;
время отбора проб;
скорость вращения мешалки /скорость потока среды растворения;
аналитический метод количественного определения ДВ, высвободившихся в среду растворения;
количество ДВ, которое должно высвободиться в среду растворения за нормируемое время,
выраженное в процентах от заявленного содержания.
4. Прибор для растворения
• Выбор прибора должен быть основан на знании состава, физикохимических свойств, технологии производства ЛП и поведенияЛФ в in vitro системе.
• Считается, что ЛМ больше подходит для оценки
таблетированных ЛФ, аппарат ВК – для капсул и ЛФ, которые
всплывают или медленно распадаются.
• Способом решения проблемы растворения плохо растворимых
ФС является использование аппарата «Проточная ячейка».
• Рекомендуемая рабочая скорость вращения для ВК – 100 об/мин,
для ЛМ – 50 об/мин, не рекомендуется использовать скорость
вращения более 150 об/мин.
5. Выбор аппарата для конкретной ЛФ
6. Выбор аппарата для конкретной ЛФ
7. Среды растворения
• Выбор среды растворения является критическим при разработке теста. Состав среды растворения подбирают длякаждого конкретного ЛП, принимая во внимание природу ФС, ее минимальную ионизацию и участок ЖКТ, в
котором должно проходить растворение и всасывание ФС.
В качестве среды чаще всего используется вода, искусственный желудочный сок или растворы кислоты
хлористоводородной (НСI) разных концентраций, буферные растворы с диапазоном рН от 4,1 до 8,0 (в единичных
случаях – 8,5 и выше).
• Использование воды в качестве среды растворения в общем случае не рекомендуется, так как у воды отсутствует
буферная емкость, а рН, поверхностное натяжение, электропроводность, содержание СО2 зависят от источника
воды и могут изменяться в процессе исследования под влиянием свойств самой ФС, абсорбции и реабсорбции
диоксида углерода воздуха.
• Для ЛП с ФС I и III классов рекомендуется использовать обычные среды без применения ПАВ, допустимо
использование воды .
• Если процесс растворения проходит медленнее, чем опорожнение желудка, рекомендуется определять профиль
растворения ЛП в нескольких средах растворения с различными значениями рН.
• Растворимость плохо растворимых ФС может быть рН-зависимой либо рН-независимой. Слабые основания лучше
растворяются в средах с кислым значением рН, однако их растворимость понижается у пожилых пациентов и при
приеме антацидных ЛП. Напротив, слабые кислоты растворяются в нижних отделах ЖКТ.Поэтому они показывают
профили замедленного высвобождения, даже если ЛФ растворяется быстро в щелочной среде.
• Для рН-независимых соединений, малоионизируемых и липофильных ФС рекомендуется буферный раствор с
добавлением ПАВ. Оптимальным считается комбинация ПАВ + кислота, либо буферный раствор, или желчные
кислоты в виде смесей.
8. Среды растворения
• Допустимо, но нежелательно использование органических растворителей, например изопропанола;использование сред с рН более 7,4, т. к. в этом случае отсутствует физиологическая релевантность [88].
• В частных случаях возможно использование ферментов, солей, ПАВ, смешанных водно-органических
растворителей (например 30% пропанола). Однако валидность такого метода ставится под сомнение. В любом
случае добавление ПАВ, органических растворителей и т. п. должно быть обосновано на стадии разработки теста
«Растворение» [64]. Недопустимо использование органических растворителей, таких как пиридин, ПАВ в
концентрациях более 4%, сильнощелочных сред (рН 13). Максимально допустимое рН среды – 9,5.
• Подходы к разработке методики для ЛП с ФС IV класса практически не отличаются от ЛП с ФС II класса. При
подборе сред рекомендуется учитывать область абсорбции в ЖКТ.
• В случае изучения опасности преждевременного высвобождения ФС из ЛП при приеме алкоголя пациентами,
принимающими пролонгированные ЛП, допустимо использование среды с 40% содержанием этанола
• При несоответствии спецификациям ЛП в форме твердых или мягких желатиновых капсул или таблеток,
покрытых оболочкой, в состав которых входит желатин, в среду допустимо добавление пепсина или
панкреатина.
• Метод деаэрации должен быть валидирован и оговорен в каждом случае, так как может заметно влиять на
профиль растворения ФС..
Температура среды растворения обычно составляет 37±0,5 °С (при исследовании ЛС для внутреннего применения)
Объем среды растворения, как правило, должен быть в 20 раз больше такового для получения насыщенного раствора субстанции, содержащейся в
ЛП. Обычно таблетки или капсулы растворяют в 500 мл буферного раствора, но возможно увеличение объема до 900(1000) мл.
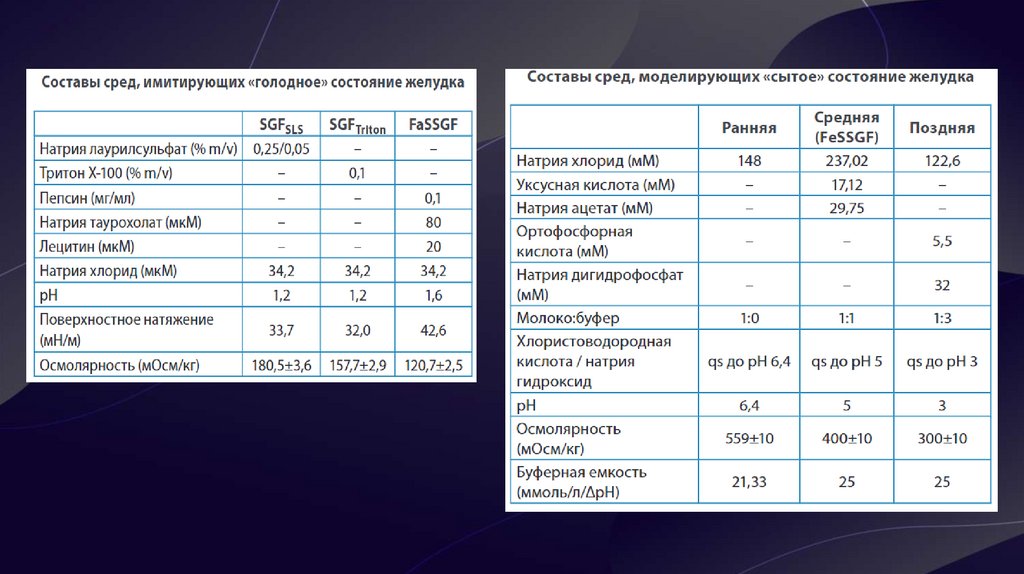
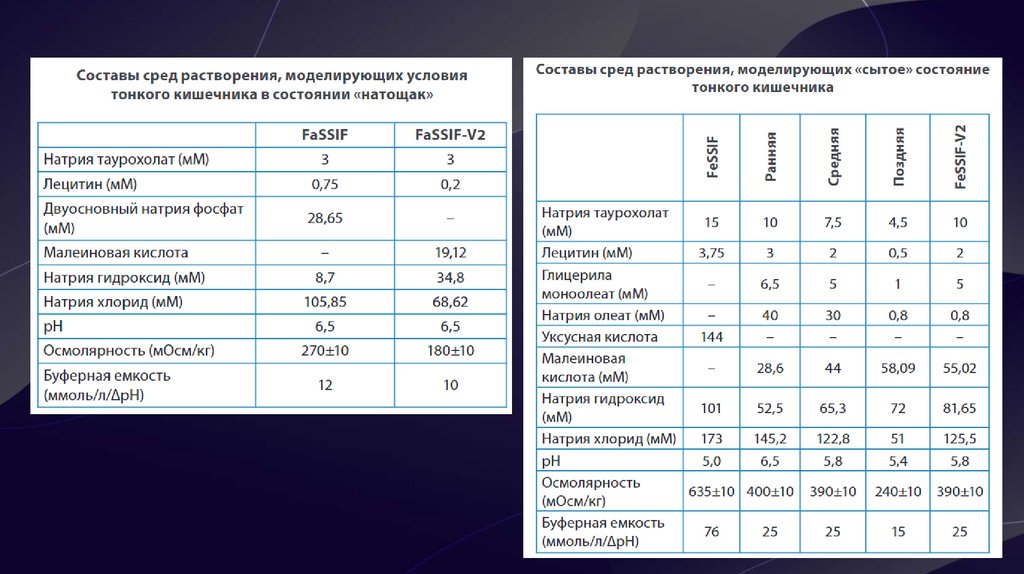
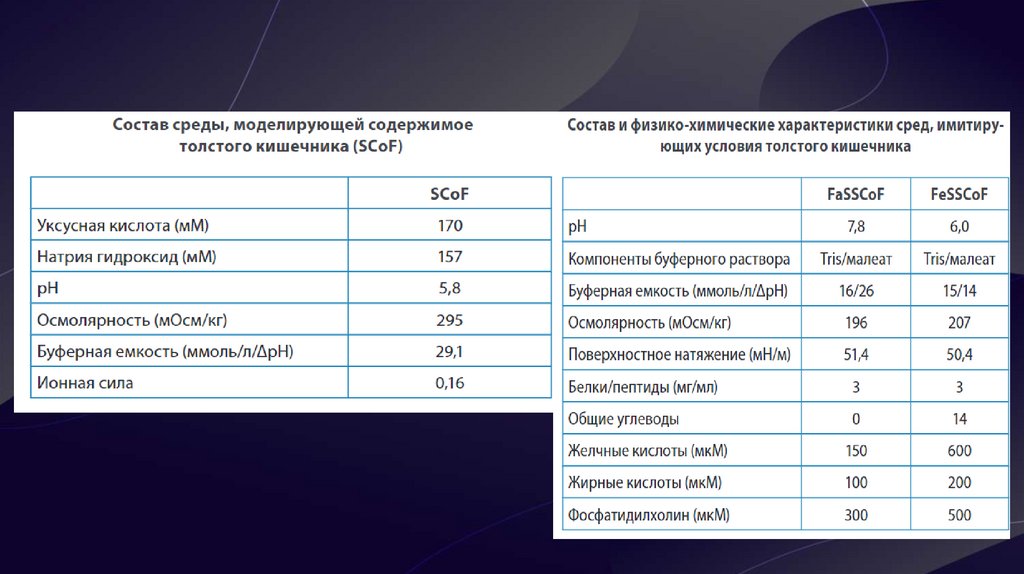
9. Биорелевантные среды
• Биорелевантные среды – это среды растворения,максимально приближенные к внутренним жидкостям
человеческого организма (кишечный, желудочный сок) по
химическому составу и по физико-химическим свойствам
(рН, осмолярность, буферная ёмкость, поверхностное
натяжение).
• C помощью биорелевантных сред возможно
смоделировать влияние приёма пищи на скорость и
полноту растворения препаратов. Более того, применение
данных сред для препаратов, относящихся ко 2 и 4 классам
БКС, наглядно продемонстрировало корреляцию профилей
растворения in vitro и фармакокинетических кривых in vivo,
а также позволило предсказать поведение липофильных,
малорастворимых веществ и абсорбцию ЛВ, относящихся к
3 классу БКС. Они подходят для для испытаний с высокой
дискриминаторной способностью, в процессе разработки
ЛП на 2 и 3 стадиях клинических испытаний.
• Однако такие среды не пригодны для фармакопейных
тестов, т. к. имеют высокую стоимость и могут вызвать
затруднения при количественном определении ФС.
10.
11.
12.
13. Время отбора проб
• Временная точка (или точки) отбора проб зависят от целейпроведения исследования или испытания. В случае
проведения теста «Растворение» (для целей контроля
качества) время отбора проб подбирают в зависимости от
вида ЛФ:
• 1 точка в диапазоне от 30 до 60 мин (45 мин согласно ОФС
«Растворение») для ЛФ немедленного высвобождения,
• 2 точки (по одной для кислотной и щелочной стадии) для ЛФ
с отложенным высвобождением,
• не менее 3 точек отбора проб для пролонгированных ЛФ
14. Фильтрация
• Перед определением количества ФС, перешедшей в средурастворения, отобранные пробы фильтруют.
• Фильтрующие материалы из нейлона/полиамида не
подходят для этих целей из-за высокой абсорбирующей
способности и влияния на результаты испытания.
• Фильтром выбора является гидрофильный фильтр из
политетрафторэтилена.
15. Методы количественного определения
высокоэффектУФ-
ивная
спектрофотоме
жидкостная
трия (СФМ)
хроматографи
я(ВЭЖХ)
газожидкостна
я
массспектромет
потенциометри
хроматография
рия (МС),
ческое
(ГЖХ)
И др.
титрование
• Рекомендуется подбирать аналитический метод в зависимости от цикла жизни ЛП.
• Методом выбора считается СФМ. Однако он недопустим при влиянии плацебо более 2%.
• В этом случае предпочтительнее метод ВЭЖХ. Он также используется при низкой концентрации
субстанции, двух и более активных компонентах в ЛП.
• Для низкодозовых ЛП возможно снижение объема среды либо применение ВЭЖХ-МС.
• ОФС допускает использование нескольких единиц данной ЛФ («Объединенный образец») на
каждый сосуд для растворения, однако при этом происходит увеличение вариабельности
результатов.
16. Нормы растворения
• При разработке методики теста «Растворение» необходимо установитьнорму количества высвободившегося в среду растворения ЛВ от
заявленного 63 содержания.
• В случае если в состав ЛС входят несколько действующих веществ, то для
каждого из них раздельно устанавливают нормы и время растворения. В
некоторых случаях, например, для малорастворимых ЛС 2 и 4 класса БКС,
таких как карбамазепин, FDA рекомендует устанавливать нормы для двух
временных точек отбора проб (например, 15 мин и 45 мин) .
• Для пролонгированных ЛС устанавливают нормы не менее чем по 3 точкам,
при этом первая точка нормируется как «не более…», вторая – «не более…
не менее…», третья – «не менее».
• Для кишечнорастворимых твердых дозированных ЛФ нормируется
количество вещества, высвободившегося во время буферной стадии; для
кислотной стадии норма высвобождения не должна превышать 10% [278].
17. Сравнение норм растворения оригинальных ЛП и дженериков
• Для инновационных ЛС разработка методик теста «Растворение»является наиболее сложным процессом, при этом дизайн исследований
и их объем определяются физико-химическими и
фармакокинетическими свойствами активного вещества. Выбор
методики испытания должен учитывать результаты экспериментов на
животных: полученные экспериментально основные
фармакокинетические параметры лекарственного вещества позволяют
обосновать условия проведения теста, прежде всего среду растворения.
Технология производства и состав ЛФ определяют выбор прибора и
гидродинамические параметры (скорость вращения аппарата)
испытания. Нормы теста «Растворение» — время растворения (время
отбора проб) и количество активного вещества, перешедшее в среду
растворения к определенному моменту времени, — устанавливают на
основании экспериментальных исследований в соответствии с
требованиями Общей фармакопейной статьи «Растворение для твердых
дозированных лекарственных форм».
• Оценка соотношения между полученными фармакокинетическими
параметрами и нормами теста «Растворение» позволяет определить
область применения методики: четкая корреляция между результатами
экспериментов in vitro и in vivo может сделать в последующем
необязательным проведение исследований биоэквивалентности в
случае изменения состава ЛП или процесса его производства . При
отсутствии корреляции тест может служить только методом контроля
качества препарата.
• При подборе условий теста кинетики растворения
для воспроизведенных ЛС ценным источником
информации может стать база данных FDA по тесту
«Растворение». Подходы к установлению норм
растворения для дженериков делятся на три
категории в зависимости от наличия фармакопейного
теста для ЛП и от характера теста «Растворение»,
используемого для препарата сравнения:
1. Тест «Растворение» ЛП имеется в USP. В этом
случае он и является тестом для контроля качества
дженерика.
2. Тест «Растворение» ЛП отсутствует в USP; доступен
тест препарата сравнения, внесенного в список ВОЗ.
В этом случае рекомендуется определять профили
растворения, используя метод, одобренный для
препарата сравнения.
• 3. Тест отсутствует в USP и недоступен тест препарата
сравнения. Необходимо проведение сравнительного
испытания растворения в разных условиях теста. Во
всех случаях получают профили растворения.
Параметры растворения устанавливаются на основе
данных биоэквивалентности.
18. Процесс разработки теста «Растворение»
• Разработка теста «Растворение» оригинального препаратаосуществляется в несколько этапов.
Заканчивают разработку
На первом – проводят
На следующем этапе
установлением (по
исследования на животных
определяют нормы
возможности) корреляции
и расчет
растворения. Чаще всего
между результатами,
фармакокинетических
используются такие
полученными in vitro и in
параметров. Основываясь
параметры, как время
vivo (IVIVC). Затем
на полученных данных и с
растворения и количество
формулируют условия
учетом свойств ФС,
ФС, перешедшее в среду
теста «Растворение». При
осуществляют научно
растворения к
отсутствии корреляции
обоснованный выбор
определенному моменту
тест может использоваться
условий растворения ФС.
времени.
только как метод контроля
качества ЛП
19. Методика испытания (ОФС)
• В сосуд аппарата для растворения помещают определенный объем среды растворения.Доводят температуру среды растворения до (37 ± 0,5) °С.
• При использовании аппарата «Вращающаяся корзинка», если нет других указаний в ФС или
НД, помещают по одной единице лекарственной формы в каждую из 6 сухих корзинок
аппарата. Опускают корзинки в среду растворения и включают мотор, вращающий
перемешивающее устройство.
• При использовании аппарата «Лопастная мешалка», если нет других указаний в ФС или НД, по
одной единице лекарственной формы помещают непосредственно в каждый из 6 сосудов со
средой растворения до начала вращения мешалки. Для предотвращения всплывания таблеток
и капсул на поверхность среды растворения комплектность аппарата должна предусматривать
соответствующее грузило в виде проволоки из инертного материала или стеклянной спирали,
удерживающее таблетки или капсулы на дне сосуда. Допускается использование других
альтернативных грузил. Необходимо соблюдать осторожность для того, чтобы избежать
оседания пузырьков воздуха на поверхности таблетки или капсулы.
• При использовании аппарата «Проточная ячейка» помещают 1 шарик диаметром 5,0 ± 0,5 мм
и затем стеклянные шарики подходящего размера, обычно 1,0 ± 0,1 мм (входят в комплект
аппарата), на дно конической части проточной ячейки для предотвращения прохождения
жидкости в трубку. Единицу лекарственной формы, если нет других указаний в
фармакопейной статье или нормативной документации, помещают в ячейку или
непосредственно в слой стеклянных шариков. Закрывают аппарат фильтрующей системой.
20.
• 1 группа: таблетки; таблетки, покрытые оболочкой; гранулы (времярастворения которых превышает 5 мин); гранулы, покрытые оболочкой;
капсулы;
• 2 группа: таблетки, покрытые кишечнорастворимой оболочкой;
кишечнорастворимые капсулы, гранулы и другие кишечнорастворимые
твердые дозированные лекарственные формы;
• 3 группа: таблетки, капсулы и гранулы с пролонгированным
высвобождением.
Для твердых дозированных лекарственных форм 2 группы может
использоваться одна из 2 альтернативных методик проведения испытания
«Растворение».
Для твердых дозированных лекарственных форм 3 группы аппарат, методика
испытания и аналитический метод определения содержания действующего
вещества в растворе должны быть описаны в фармакопейной статье или
нормативной документации.
21. Методика 1
• Испытание проводят в две стадии.• 1-я стадия (кислотная). По 750 мл хлористоводородной кислоты раствора 0,1 М, если нет других указаний в ФС
или НД, помещают в каждый из 6 сосудов для растворения. Доводят температуру среды растворения до (37 ±
0,5) °С. Помещают по 1 таблетке или по 1 капсуле, если нет других указаний в ФС или НД, в каждый из 6 сосудов
для растворения, включают мотор перемешивающего устройства. Через 2 ч, если нет других указаний в ФС или
НД, отбирают аликвоту и сразу же продолжают процесс растворения в щелочной среде, как описано ниже.
• Отобранную аликвотную часть раствора анализируют по методике, описанной в ФС или НД. Результаты
испытаний на 1-й стадии считаются удовлетворительными, если количество действующего вещества,
перешедшего в среду растворения, соответствует критериям раздела «Интерпретация результатов».
• 2-я стадия (щелочная). В каждый из 6 сосудов для растворения прибавляют по 250 мл натрия фосфата раствора
0,2 М (Na3PO4 × 12H2O), температура которого составляет (37 ± 0,5) °С (перемешивающее устройство аппарата
продолжает работать). Доводят рН среды растворения до 6,80 ± 0,05 с помощью хлористоводородной кислоты
раствора 2 М или натрия гидроксида раствора 2 М.
• Продолжают процесс растворения в течение 45 мин, если нет других указаний в ФС или НД. После отбора
пробы раствора проводят определение содержания действующего вещества в растворе по методике,
описанной в фармакопейной статье или нормативной документации. Результаты испытаний на 2-й стадии
считаются удовлетворительными, если количество действующего вещества, перешедшего в среду растворения,
соответствует критериям раздела «Интерпретация результатов»
• Примечание. Процедура добавления натрия фосфата раствора 0,2 М и доведения рН среды растворения до
заданного значения должна проводиться в течение не более 5 мин.
22. Методика 2
• Испытание проводят в две стадии.• 1-я стадия (кислотная). По 1000 мл хлористоводородной кислоты раствора 0,1 М, если нет других указаний в ФС или НД,
помещают в каждый из 6 сосудов для растворения. Доводят температуру среды растворения до (37 ± 0,5) °С. Помещают по 1
таблетке или по 1 капсуле, если нет других указаний в фармакопейной статье или нормативной документации, в каждый из 6
сосудов для растворения, включают мотор перемешивающего устройства. Через 2 ч, если нет других указаний в
фармакопейной статье или нормативной документации, отбирают аликвоту и сразу же продолжают процесс растворения в
щелочной среде, как описано ниже.
• Отобранную аликвотную часть раствора анализируют по методике, описанной в фармакопейной статье или нормативной
документации. Результаты испытаний на 1-й стадии считаются удовлетворительными, если количество действующего вещества,
перешедшего в среду растворения, соответствует критериям раздела «Интерпретация результатов»
• 2-я стадия (щелочная). Из каждого сосуда для растворения удаляют хлористоводородной кислоты раствор 0,1 М и помещают
по 1000 мл фосфатного буферного раствора рН 6,8 (2) с температурой (37 ± 0,5) °С. Допустимо переносить испытуемые единицы
твердой дозированной лекарственной формы из сосудов для растворения, содержащих хлористоводородной кислоты раствор
0,1 М, в сосуды для растворения, содержащие по 1000 мл фосфатного буферного раствора рН 6,8 (2) с температурой (37 ± 0,5)
°С.
• Процесс растворения продолжают в течение 45 мин, если нет других указаний в ФС или НД. Затем отбирают аликвоту и сразу
же анализируют по методике, описанной в фармакопейной статье или нормативной документации. Результаты испытания на 2й стадии считаются удовлетворительными, если количество действующего вещества, высвободившегося в среду растворения,
соответствует критериям раздела «Интерпретация результатов» (табл. 2).
• Примечание. Приготовление фосфатного буферного раствора рН 6,8 (2). Хлористоводородной кислоты раствор 0,1 М и натрия
фосфата раствор 0,2 М (Na3PO4 × 12H2O) смешивают в соотношении 3:1 и при необходимости доводят рН полученного
раствора до 6,80 ± 0,05 с помощью хлористоводородной кислоты раствора 2 М или натрия гидроксида раствора 2 М.