



 аминокислот")




")




































 - дефект")













 Медицина
Медицина Химия
ХимияПохожие презентации:

")




")


")
Специфические пути обмена отдельных аминокислот
1. ФГБОУ ВО «Кемеровский государственный медицинский университет» МЗ РФ Кафедра медицинской биохимии
специфические пути обмена отдельныхаминокислот
2. Обмен аминокислот: источники и пути использования
3. Общие пути катаболизма аминокислот в клетках
Дезаминирование (отщепление аминогруппы от АК)Трансаминирование (переаминирование- перенос
аминогруппы на α-кетокислоту)
Декарбоксилирование (отщепление СО2 )
Реакции по радикалам
4.
5. Судьба продуктов катаболизма (дезаминирования) аминокислот
6. ПУТИ ИСПОЛЬЗОВАНИЯ ДИКАРБОНОВЫХ АМИНОКИСЛОТ
7. Включение безазотистых остатков аминокислот в ЦТК
8.
9.
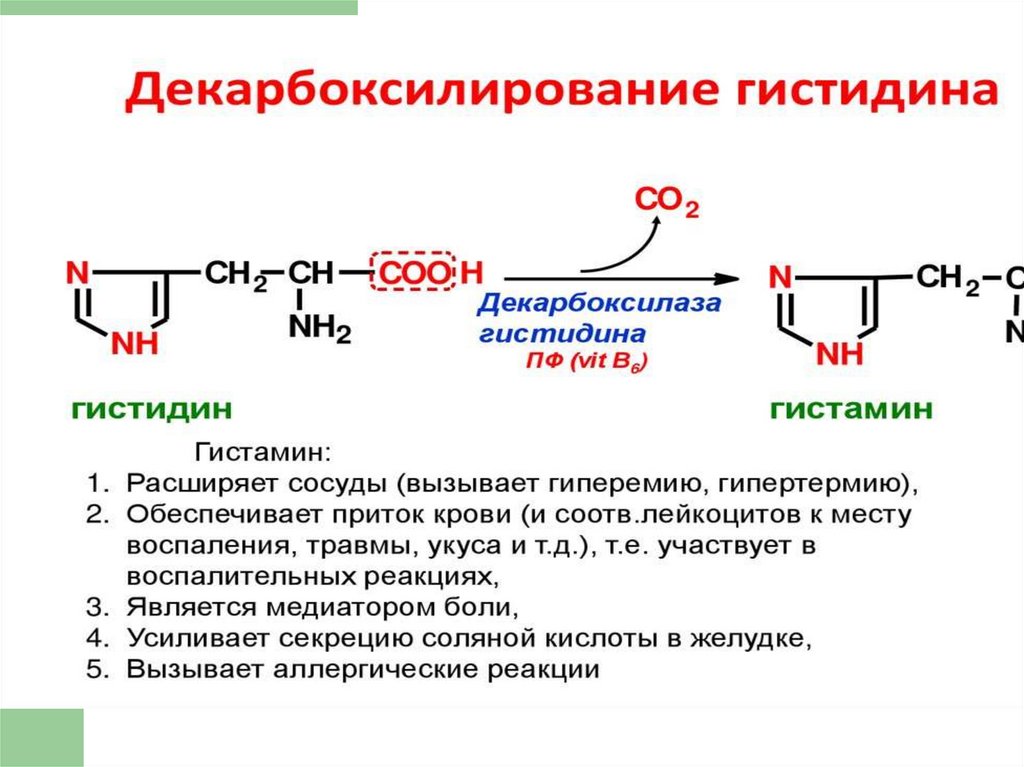
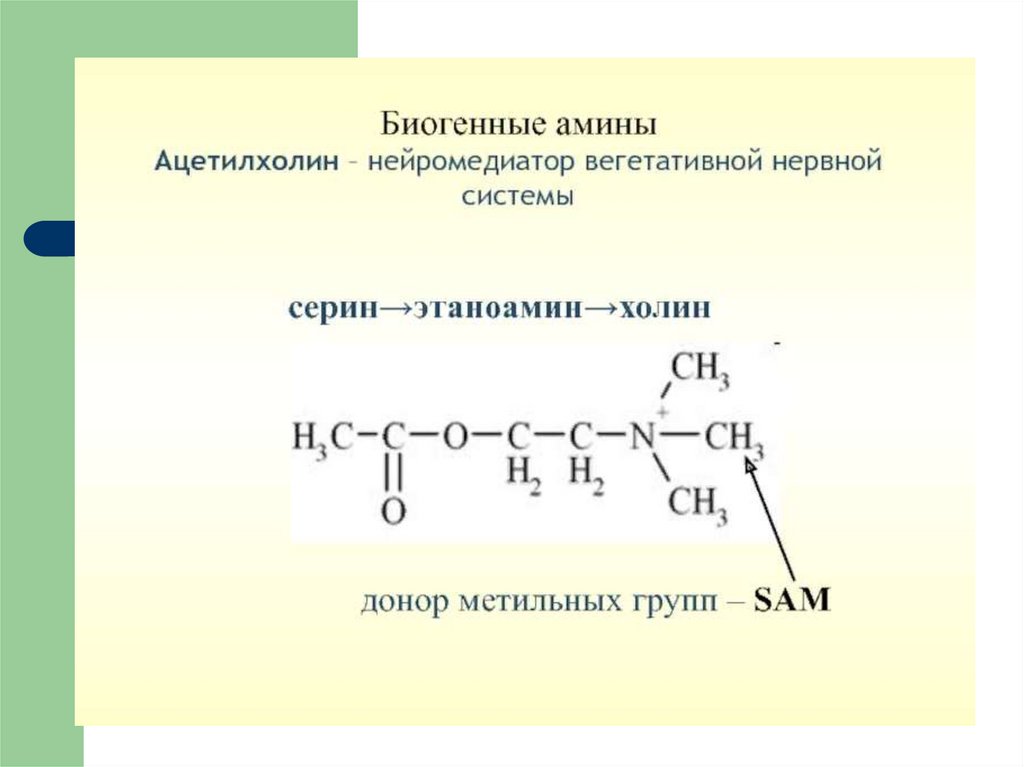
10. ЭФФЕКТЫ ГИСТАМИНА
11. Клинические проявления действия гистамина и антигистаминовых препаратов (блокаторов Н- рецепторов)
12.
13.
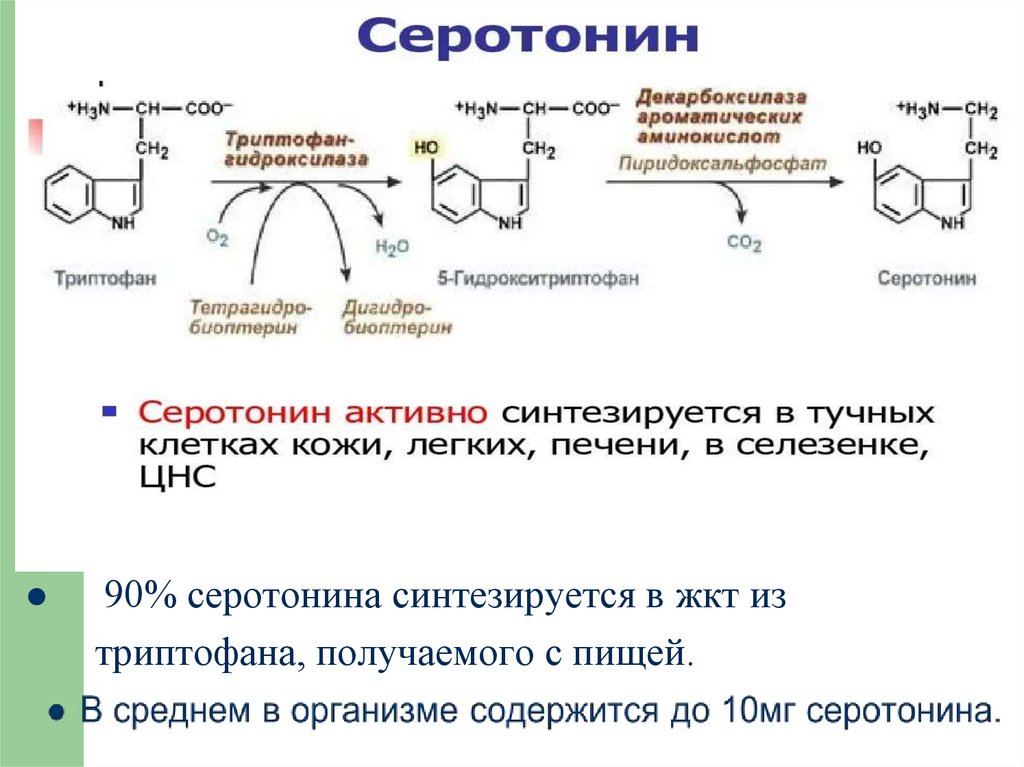
90% серотонина синтезируется в жкт изтриптофана, получаемого с пищей.
14.
15.
16.
17.
18. МЕЛАТОНИН- производное серотонина
19.
20.
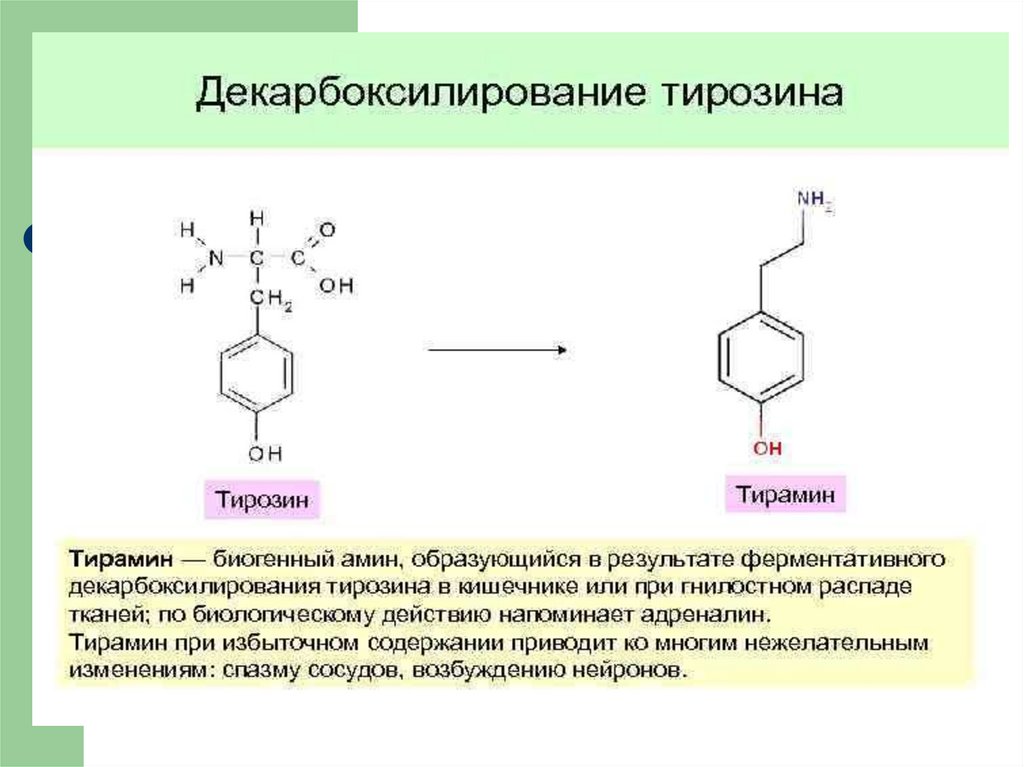
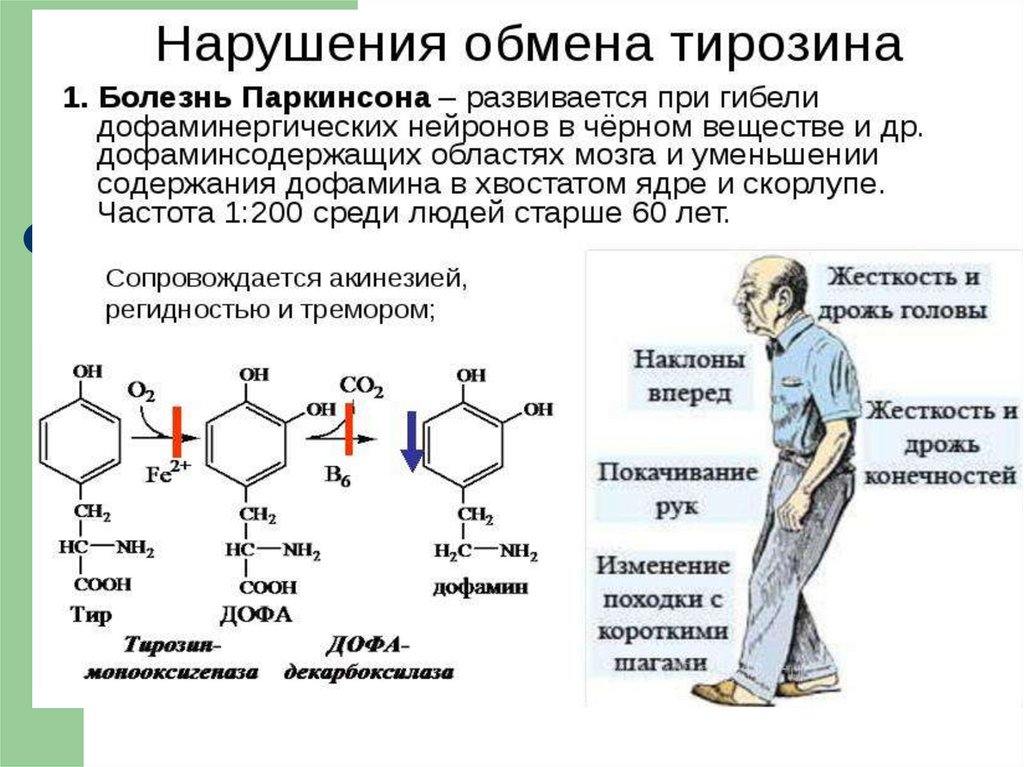
21. ОБМЕН ФЕНИЛАЛАНИНА и ТИРОЗИНА
22.
23.
24.
25.
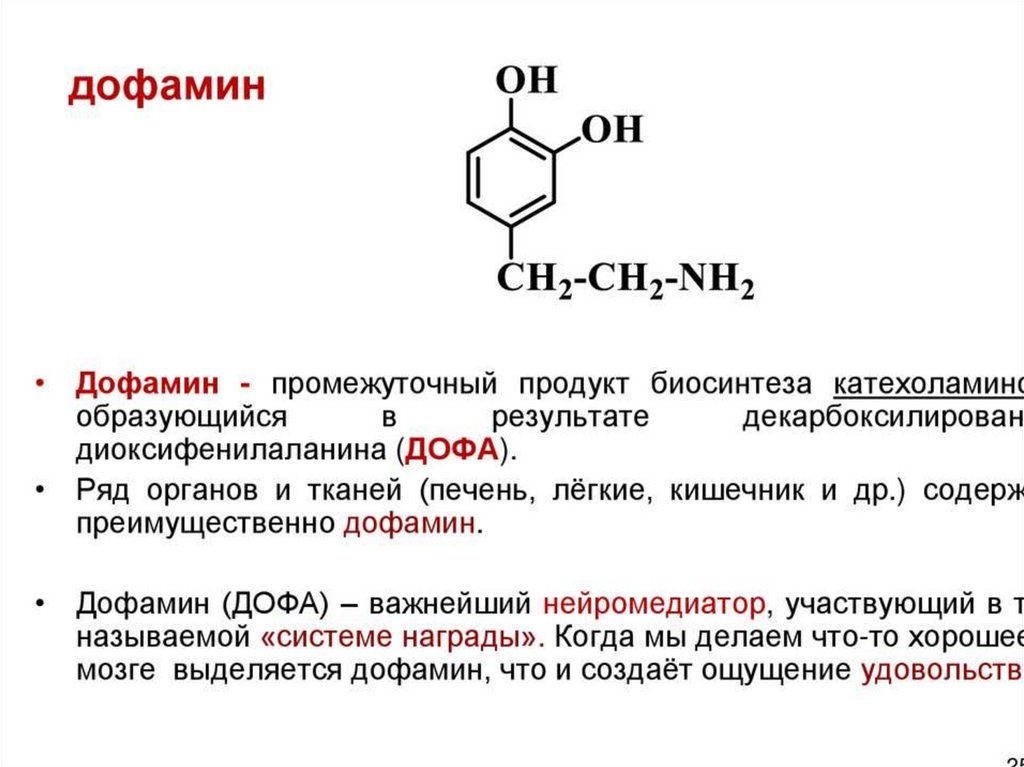
Роль дофамина в организмеКак гормон:
• повышает артериальное давление, частоту и силу
сердечных сокращений;
• расслабляет гладкую мускулатуру желудка и кишечника;
• увеличивает фильтрацию жидкости, кровоток в почках,
ускоряет выделение натрия с мочой.
Как нейромедиатор оказывает влияние на:
• формирование мотивации;
• чувство удовольствия, чувство удовлетворения;
• ощущение награды и желания,;
• эмоциональные реакции, сопровождающие двигательную
активность.
26.
27.
Нарушение обмена дофамина наблюдается пришизофрении. ( гиперсекреция в височной доле, или
недостаток в др структурах.)
Шизофрения – это серьезное расстройство психики,
при котором больной страдает галлюцинациями
(чаще всего слуховыми), у него искажается
интерпретация реальности, начинается бред (ложные
фиксированные убеждения), нарушения мышления,
поведения. Социальная активность человека зависит
от степени тяжести заболевания. Примерно 10 %
больных совершают самоубийство, около 80 %
испытывают депрессию хотя бы раз в жизни.
28.
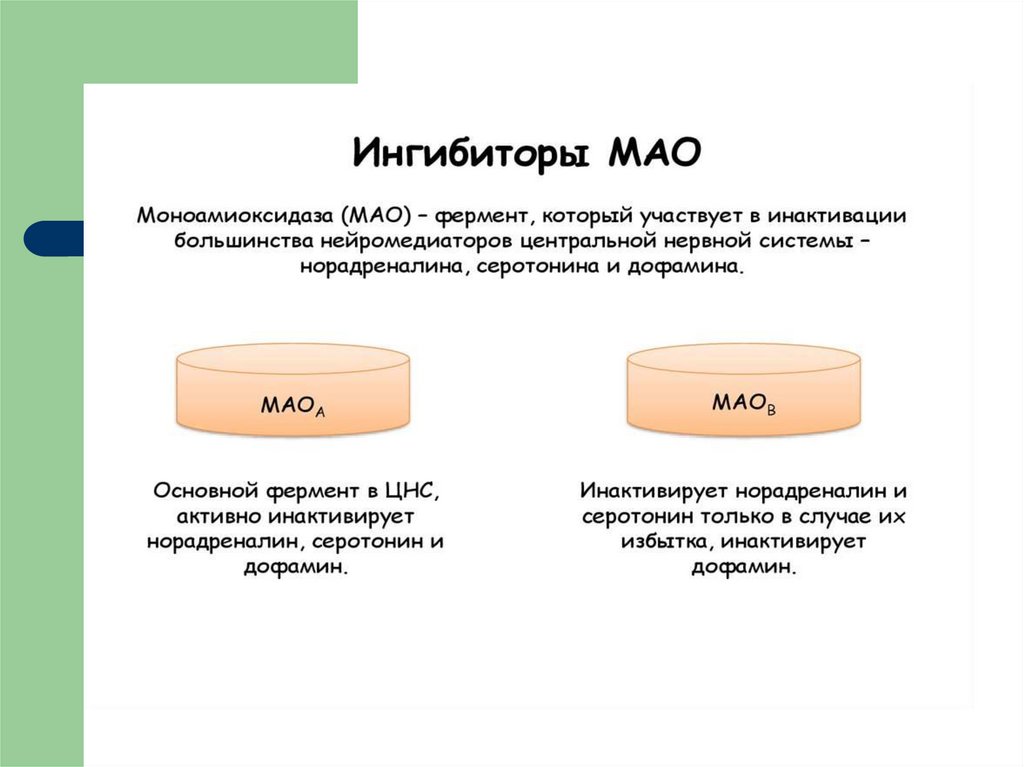
29. ИНАКТИВАЦИЯ БА
30. Ингибиторы моноаминоксидазы
ИМАО, MAOI — биологически активныевещества, способные ингибировать фермент
моноаминоксидазу, содержащийся в нервных
окончаниях, препятствуя разрушению этим
ферментом различных моноаминов (серотонина,
норадреналина, дофамина, фенилэтиламина,
триптаминов) и тем самым способствуя
повышению их концентрации в синаптической
щели.
31.
32.
33.
34.
35. Химизм реакций окисления фенилаланина в тирозин
36.
37. ФЕНИЛКЕТОНУРИЯ - тяжелое наследственное заболевание, которое характеризуется главным образом поражением нервной системы.
Фенилкетонурия (ФКУ) – развивается в результатеврожденного дефекта фермента, отвечающего в организме
человека за нормальный обмен фенилаланина.
В результате мутации гена, контролирующего синтез
фенилаланинмонооксигеназы (фенилаланингидроксилазы),
развивается метаболический блок, вследствие чего
основным путем преобразования фенилаланина
становится дезаминирование и синтез токсических
производных - фенилпировиноградной, фенилмолочной и
фенилуксусной кислот.
В крови и тканях значительно увеличивается содержание
фенилаланина (до 0,2 г/л и более при норме 0,01-0,02 г/л).
Существенную роль в патогенезе болезни играет
недостаточный синтез тирозина, который является
предшественником катехоламинов и меланина...
38. Отсутствие в печени фермента фенилаланингидроксилазы препятствует нормальному превращению фенилаланина пищи в тирозин
Поэтому фенилаланин используется лишь присинтезе белка, а избыток накапливается в
клетках печени и попадает в кровоток, где
количество фенилаланина является токсичным
для клеток мозга. Почки не справляются с его
реабсорбцией, в результате чего он выводится с
мочой в виде фенилПВК. Именно наличие этого
фенилкетона в моче дало основание назвать
соответствующее патологическое состояние
фенилкетонурией - ФКУ.
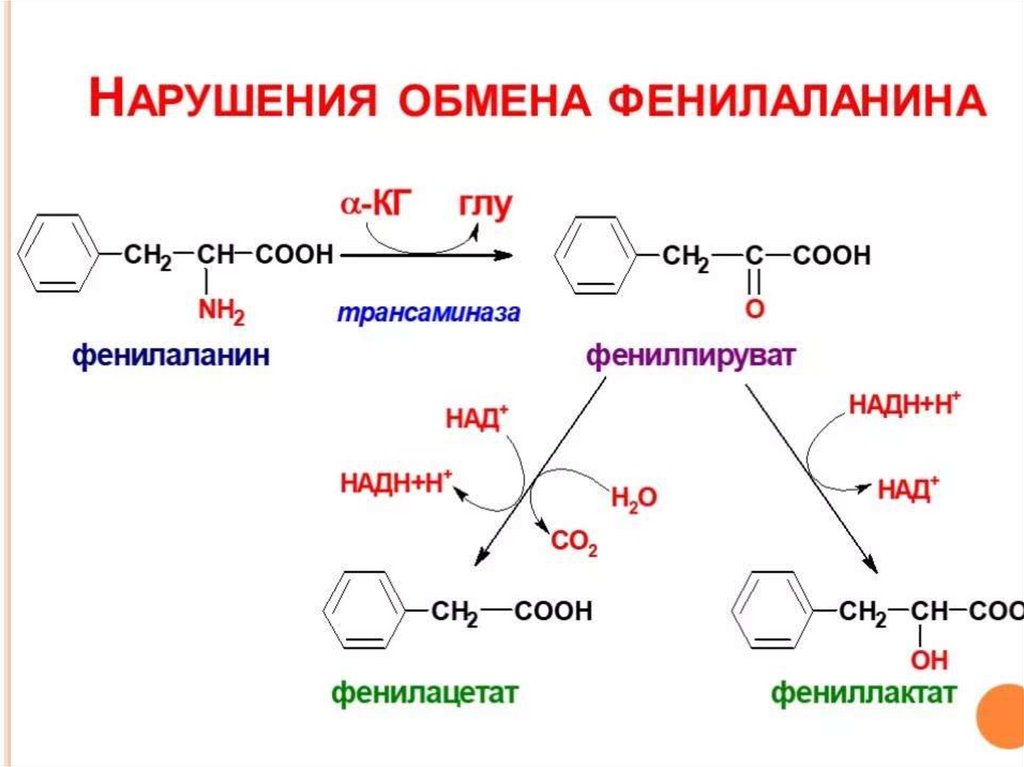
39. ОБМЕН ФЕНИЛАЛАНИНА при ФКУ
40. ПРОЯВЛЕНИЯ ФКУ
При заболевании нарушаются обменные процессы,особенно важные для развивающегося мозга ребенка.
В крови и других жидкостях организма накапливается в
большом количестве фенилаланин и повышено образуются
такие вещества как фенилпировиноградная,
фенилмолочная и фенилуксусная кислоты, которые
выделяются в повышенных количествах с мочой.
Следствием нарушенного обмена в мозге является
тяжелое психическое недоразвитие. Если не предпринято
своевременное лечение, то больные на всю жизнь
остаются глубокими инвалидами.
41. В патогенезе ФКУ имеют значение следующие механизмы:
Прямое токсическое действие на ЦНС фенилаланина и егопроизводных вызывает:
Нарушение в обмене белков, липо- и гликопротеидов;
Нарушение транспорта аминокислот;
Нарушение метаболизма гормонов;
Нарушение обмена моноаминовых нейромедиаторов
(катехоламинов и серотонина);
Нарушение функции печени - диспротеинемия,
генерализованная гипераминоацидемия, повышение
ДОФА, метаболический ацидоз, нарушение окислительной
и белоксинтезирующей функции клеточных органелл.
42. Варианты ФКУ
Фенилкетонурия 1.Классическая
фенилкетонурия
(ФКУ)
описана
А.Folling.,1934г.
Заболевание
наследуется
аутосомно-рецессивно
и
вызвано мутацией гена, локализующегося в длинном плече
12 хромосомы.
В основе болезни лежит дефицит фермента фенилаланин4-гидроксилазы,
обеспечивающего
превращение
фенилаланина в тирозин. Как следствие накопление в
тканях и жидкостях больного организма фенилаланина и
его производных: фенилпировиноградная, фенилмолочная,
фенилуксусная
кислоты,
фенилэтиламин,
фенилацетилглютамин и др.
Частота классической ФКУ среди новорожденных по
данным массового скрининга в среднем колеблется от
1:5000 до 1:10000 по разным регионам России
43. Варианты ФКУ
Фенилкетонурия 2.Впервые атипичная ФКУ описана I.Smith, 1974г.
Заболевание связано с дефицитом
дигидроптеридинредуктазы. Нарушается восстановление
активной формы тетрагидробиоптерина, участвующего в
качестве кофактора в гидроксилировании фенилаланина,
тирозина, и триптофана.
Частота заболевания составляет 1:100000 новорожденных.
Рано начатое лечение способствует нормализации
фенилаланина в крови, однако не предупреждает
появление клинической симптоматики, которая развивается
в начале второго полугодия жизни. Фенилкетонурию 2
называют диеторезистентной ФКУ.
44. Варианты ФКУ
Фенилкетонурия 3. Этот вариант болезни описал S.Kaufman в 1978 г. Заболевание связано с
недостаточностью 6-пируваттетрагидроптеринсинтетазы,
фермента участвующего в процессе синтеза
тетрагидробиоптерина. Развивающиеся при этом
расстройства сходны с нарушениями, наблюдаемыми при
ФКУ 2.
Частота болезни составляет 1:30000 новорожденных.
Фенилкетонурия 3 также диеторезистентна.
Другие варианты ФКУ: Эти формы ФКУ связаны с
нарушением альтернативных путей обмена фенилаланина.
Формируется метилминдальная ацидурия и
парагидроскифенилуксусная ацидурия.
45. Материнская фенилкетонурия.
Заболевание развивается у детей женщин,страдающих ФКУ и не получающих диету в
зрелом возрасте. Патогенез мало изучен,
предполагается, что он сходен с патогенезом
остальных форм ФКУ. Тяжесть поражения плода
коррелирует с уровнем фенилаланина в плазме
матери. Так как эмбрион особенно чувствителен к
тератогенным воздействиям, рекомендуется
начинать диету еще до наступления
беременности. В суточном рационе использовать
менее 15-20 мг/кг фенилаланина.
46. Клинические проявления ФКУ
При рождении больные фенилкетонурией не отличаются отдругих новорожденных. Манифестация ФКУ происходит
обычно в возрасте 2-6 месяцев.. Уже на втором месяце
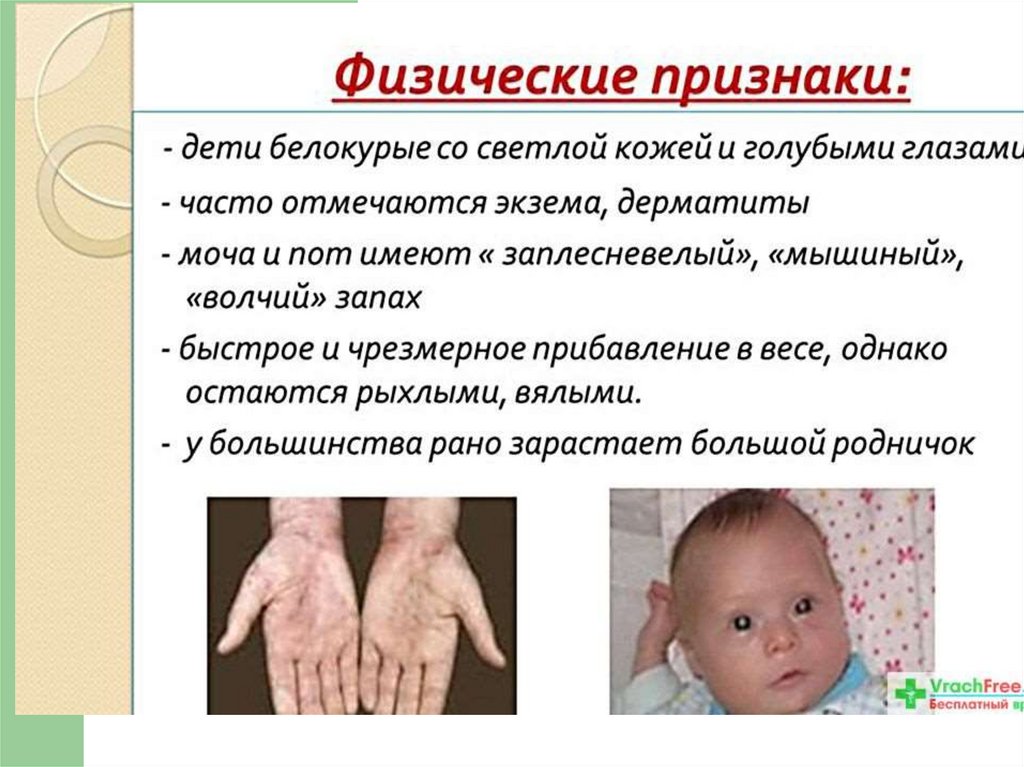
можно заметить некоторые физические признаки:
посветление волос, радужек глаз, что особенно заметно у
детей, родившихся с темными волосами. Многие дети
очень быстро и чрезмерно прибавляют в весе, однако
остаются рыхлыми, вялыми. У большинства из них рано
зарастает большой родничек. Чаще всего явные признаки
болезни обнаруживаются на 4-6 месяце жизни, когда дети
перестают реагировать радостью на обращение к ним,
перестают узнавать мать, не фиксируют взгляд и не
реагируют на яркие игрушки, не переворачиваются на
живот, не сидят.
47. Клинические проявления ФКУ
По мере прогрессирования болезни могут наблюдатьсяэпилептиформные приступы - развернутые судорожные и
бессудорожные типа кивков, поклонов, вздрагиваний,
кратковременных отключений сознания. Гипертония
отдельных групп мышц проявляется своеобразной "позой
портного" (поджатые ноги и согнутые руки). Могут
наблюдаться гиперкинезы, атаксия, тремор рук, иногда
парезы по центральному типу. Дети нередко белокурые со
светлой кожей и голубыми глазами, у них часто отмечаются
экзема, дерматиты. Обнаруживается склонность к
артериальной гипотензии.
48. Клинические проявления ФКУ
Проявлениями болезни служат: вялость ребенка,отсутствие интереса к окружающему; повышенная
раздражительность, беспокойство; срыгивание, рвота;
судорожные эквиваленты: сосательные автоматизмы,
атетозные движения; судорожный синдром;
заплесневелый, мышиный, волчий запах мочи и пота.
При отсутствии лечения формируется задержка статикомоторного и психоречевого развития, умственная
отсталость достигает, как правило, глубокой степени
(идиотия или имбецильность, глубокая психическая
инвалидность).
49.
50.
51. Клинико-лабораторная диагностика ФКУ
В течение многих лет соответствующим диагностическим тестомслужит реакция между фенилпировиноградной кислотой,
которая выделяется с мочой ребенка, и хлорным железом.
При положительной реакции появляется типичное зеленое
окрашивание. Кроме того, образуются и выводятся с мочой
другие аномальные метаболиты, такие как фенилмолочная и
фенилуксусная кислоты. Последнее соединение «пахнет
мышами», так что болезнь легко диагностировать по запаху;
именно так она и была впервые обнаружена.
ФКУ может быть диагностирована на основе обнаружения
следующих признаков:
стойкой гиперфенилаланинемии (более 240 ммоль/л);
вторичного дефицита тирозина;
экскреции фенилкетонов с мочой (проба Феллинга на экскрецию
фенилпировиноградной кислоты).
52.
53. Лечение фенилкетонурии и прогноз
Если ничего не предпринимать, фенилкетонурия приводит кразвивитию олигофрении.
.
Главным способом лечения является диетотерапия,
ограничивающая поступление в организм фенилаланина;
приступить к ней нужно немедленно после установления
диагноза. При ранней диагностике это гарантирует
нормальное нервно-психическое развитие ребенка.
Диетотерапия, как единственный эффективный метод
лечения ФКУ, должна применятся с первых месяцев жизни
ребенка, тогда поражение мозга не разовьется. Важно
ограничить количество потребляемого фенилаланина
таким образом, чтобы обеспечить его поступление в
организм в количествах, необходимых и достаточных для
роста и развития, но предотвратив его накопление в
жидкостях тела.
54.
55. Очень важно!
Кроме диетотерапии необходим постоянный медицинскийконтроль за умственным и физическим развитием ребенка.
Применение диетотерапии на позднем этапе не вернет ребенку
нормального интеллекта. Дети, у которых это заболевание не
диагностируют сразу при рождении, а выявляют по умственной
отсталости, не могут быть излечены.
По достижении 12-14 лет такие дети могут переходить на
нормальное питание и никаких признаков отравления
фенилаланином у них не будет.
Однако женщина, которая в детстве переболела ФКУ, должна
снова перейти на диету и употреблять только продукты с
пониженным содержанием фенилаланина перед зачатием, и
оставаться на этой диете во время беременности и кормления
грудью. Если она не сделает этого, то ее ребенок подвергается
риску замедленного физического и умственного развития, даже
если его отец не является носителем гена ФКУ.
56. ФКУ- проблема социальная
57.
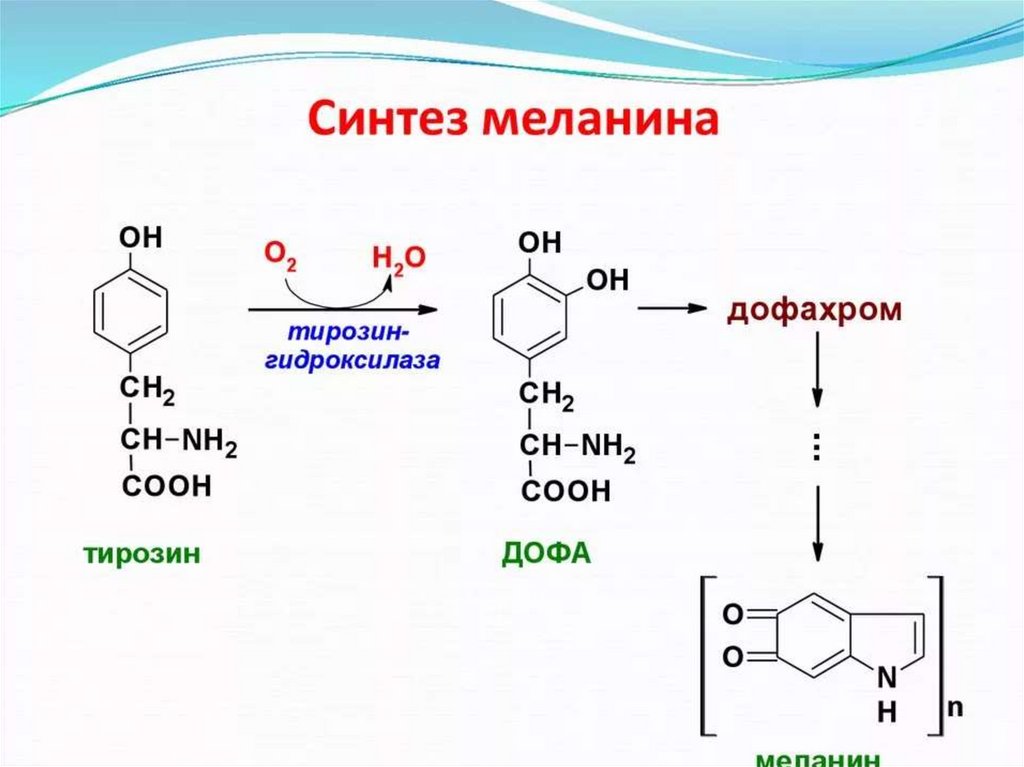
58. Альбинизм - наследственно обусловленное нарушение синтеза пигментов (меланинов: эумеланинов и феомеланинов) - дефект
тирозиназы.Цвет кожи, глаз волос зависит от
распределения меланоцитов и количества в них
меланинов
59.
60. Альбинизм
Альбиносы чащерождаются
в
семьях
чернокожих.
Известны
случаи
частичного
альбинизма. Люди с такой
болезнью
имеют
пятнистую кожу.
Также
снижена острота зрения и
светобоязнь.
Если
среди
представителей
европеоидной
расы альбинизм воспринимается
как
косметическая
особенность
внешности, то у представителей негроидной расы
он
приравнивается
к
тяжелым
врожденным
уродствам.
Частота заболевания
1:20 000.
61.
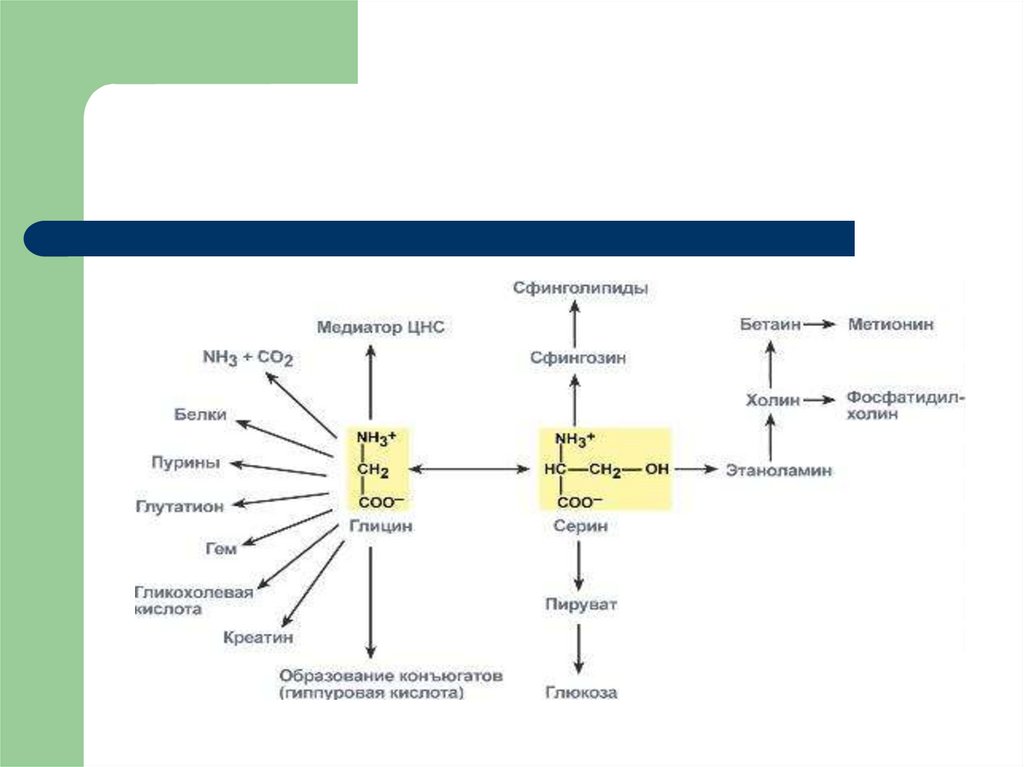
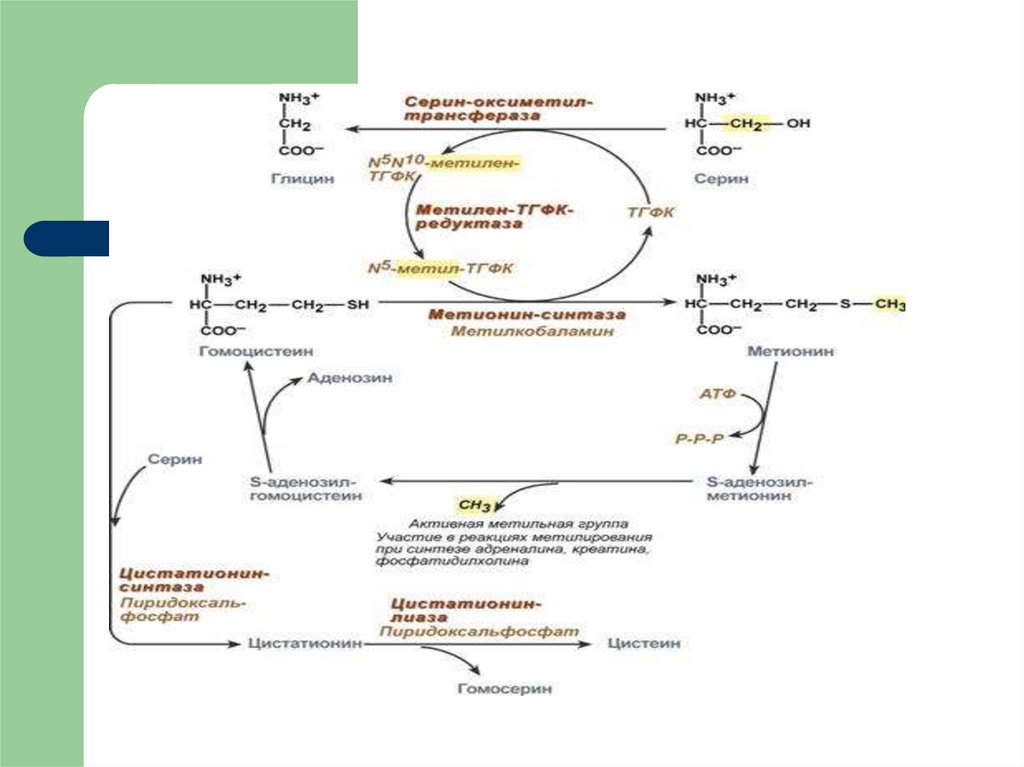
62. ОБМЕН СЕРИН И ГЛИЦИНА Серин и глицин превращаются друг в друга. Роль реакции состоит в образовании активной формы
тетрагидрофолиевой кислоты – N5,N10-метилен-ТГФК.63.
64.
Образованный в реакции распада серина до глицинаN5,N10-метилен-тетрагидрофолат (активная
форма витамина В9) при участии фермента метиленТГФК-редуктазы превращается в N5-метил-ТГФК. Его
метильный остаток участвует в метионинсинтазной реакции реметилирования гомоцистеина в
метионин. В результате перемещения метильной группы и
отщепления аденозина остается гомоцистеин
В печени, кроме метил-ТГФК, источником метильной
группы может быть вещество бетаин (триметилглицин).
65. Обмен метионина
Метионин присоединяет аденозильный остаток ипревращается в активную форму метионина
– S-аденозилметионин, участвующий во многих
реакциях метилирования, в частности, при
синтезе креатина, карнитина,
фосфатидил
холина, адреналина. В результате
перемещения метильной группы и отщепления
аденозина остается гомоцистеин
66. S-аденозилметионин
67.
68. Нарушение обмена метионина: дефект метиленфолатредуктазы или цистатионин-синтазы
Гомоцистеин, растворенный в плазме, провоцируетсвободнорадикальное окисление липидов в липопротеинах
крови и тем самым их задержку в крови, ускоряет агрегацию
тромбоцитов, вызывает повреждение эндотелия сосудов.
Гомоцистеинемия считается фактором риска и обнаруживается в
30% случаев атеросклероза, тромбозов, ишемической болезни
сердца. Она выявляется при болезни Альцгеймера, нарушениях
беременности – невынашивание, мертворождения.
Одновременно назначается диета со сниженным содержанием
метионина, что достигается специальным подбором продуктов,
бедных этой аминокислотой.
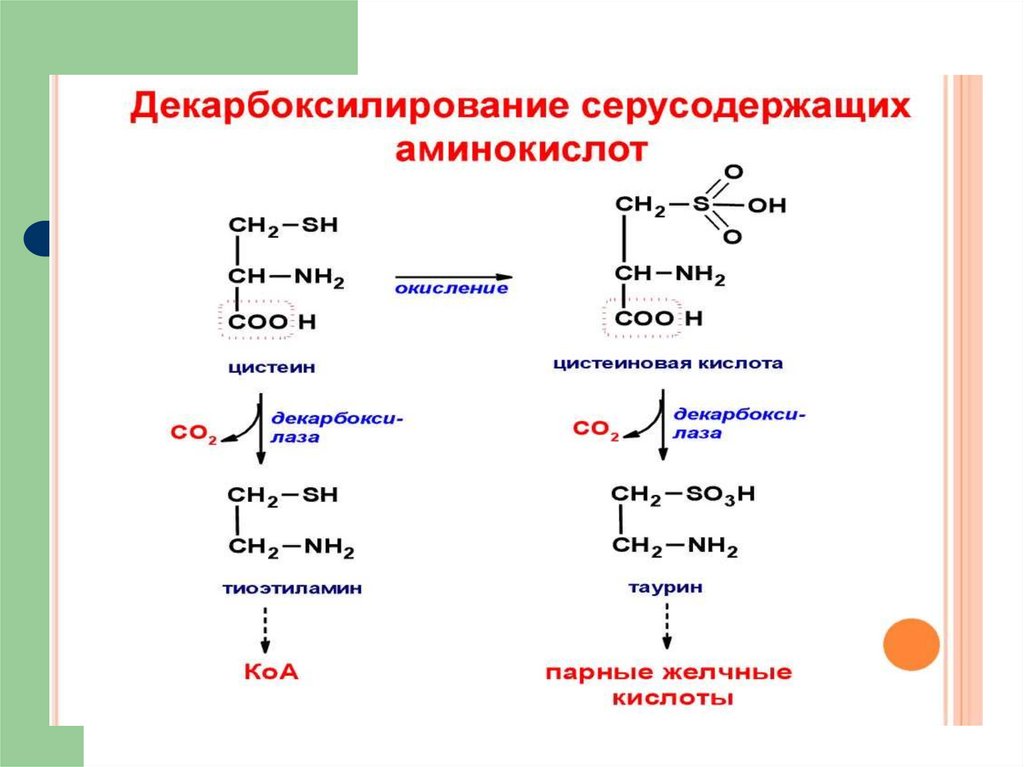
69. метаболизм цистеина
Цистеин является чрезвычайно важнойаминокислотой в связи с тем, что это
единственный источник органической серы для
клеток организма. В результате реакций
метаболизма эта сера переходит в состав других
серусодержащих веществ –
фосфоаденозинфосфосерная
кислота (ФАФС), коэнзим А, глутатион,
сульфированные производные углеводов
(хондроитинсульфат, кератансульфат, дермат
ансульфат) или выводится почками в виде
сульфатов.
70. Пути использования цистеина
71. Синтез таурина
72. функции таурина
является обязательным компонентомактивных форм желчных кислот
играет роль внутриклеточного
антиоксиданта,
играет роль тормозного нейромедиатора
73. Нарушение обмена цистеина: дефект цистатионин-синтазы, гиповитаминоз фолиевой кислоты, В12 и В6
ГОМОЦИСТИНУРИЯПри дефекте цистатионин-синтазы применяется лечение
витамином В6 в дозе 250-500 мг/день. При дефекте метилентетрагидрофолат-редуктазы уровень гомоцистеина может быть
снижен благодаря употреблению фолиевой кислоты по 5 мг/день.
Витамин В12 также оказывает положительное влияние.
Одновременно назначается диета со сниженным содержанием
метионина, что достигается специальным подбором продуктов,
бедных этой аминокислотой.