































































")



 Медицина
МедицинаПохожие презентации:

")



Anémie - klasifikace. Patofyziologická kritéria
1. HEMATOLOGIE A HEMATOONKOLOGIE
Prof. MUDr. Jaroslav Čermák,CSc.2.
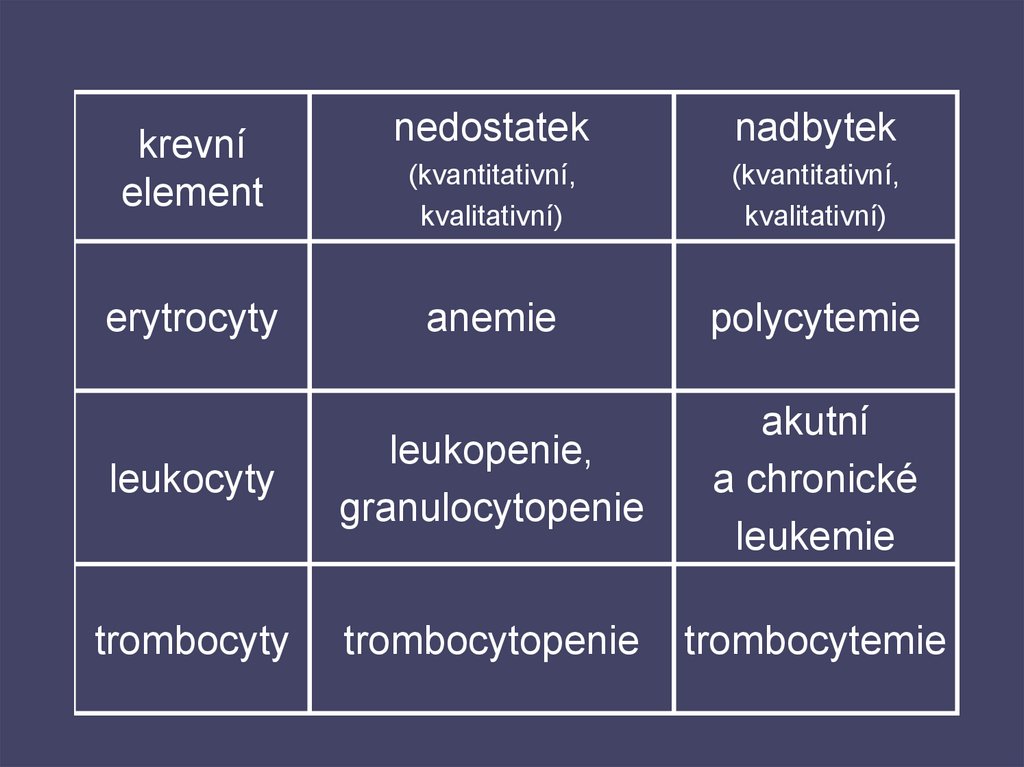
krevníelement
nedostatek
nadbytek
(kvantitativní,
kvalitativní)
(kvantitativní,
kvalitativní)
erytrocyty
anemie
polycytemie
leukocyty
leukopenie,
granulocytopenie
akutní
a chronické
leukemie
trombocyty
trombocytopenie
trombocytemie
3. ANÉMIE - KLASIFIKACE
• patofyziologická kritéria• anémie z nedostatečné či porušené
tvorby krvinek
snížená proliferace, proliferace abnormálního
buněčného klonu, porucha tvorby hemoglobinu,
chybění látek nutných k normální proliferaci a
diferenciaci.
• anémie ze zvýšeného zániku krvinek
korpuskulární a extrakorpuskulární hemolýza, ztráta
krve.
4. ANÉMIE - KLASIFIKACE
• morfologická kritéria• dle MCV:
mikrocytární, normocytární, makrocytární.
• dle MCH:
normochromní, hypochromní.
• dle počtu retikulocytů:
anémie se sníženým, normálním či zvýšeným
počtem retikulocytů.
5. MIKROCYTÁRNÍ ANÉMIE
• MCV < 80 fl, většinou hypochromní, početretikulocytů není zvýšen.
vyšetření Fe v séru a feritinu :
• anémie z nedostatku železa - ↓ Fe
• anémie při chronickém onemocnění- ↓ Fe
• thalasémie – není ↓ Fe
• sideroblastická anémie – není ↓ Fe
6. PŘÍČINY NEDOSTATKU Fe
A. NADMĚRNÉ ZTRÁTYGIT – hemeroidy,hiátová hernie,divertikly,vředy,
polypy,nádory,gastritis,M.Crohn,ulcerosní
kolitis,léky(salicyláty,antikoagulancia)
parazité.
gynekologické – menorrhagie,metrorhagie.
vylučovací ústrojí
dialýza
dárci krve
B. NEDOSTATEČNÝ PŘÍVOD
nedostatek v potravě
porucha vstřebávání – sprue,atrofická gastritis,
duodenojejunitis,resekce žaludku, parazité
C. ZVÝŠENÁ SPOTŘEBA
těhotenství
růst
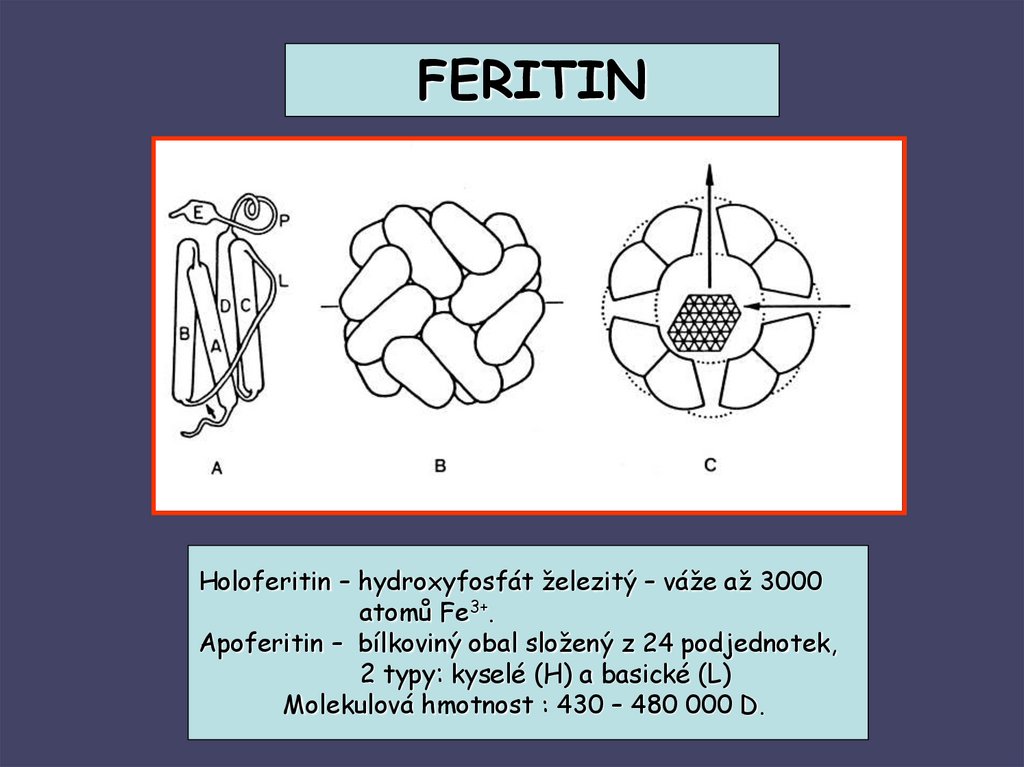
7.
FERITINHoloferitin – hydroxyfosfát železitý – váže až 3000
atomů Fe3+.
Apoferitin – bílkoviný obal složený z 24 podjednotek,
2 typy: kyselé (H) a basické (L)
Molekulová hmotnost : 430 – 480 000 D.
8.
• Feritin v séru < 10 µg/l+
• Saturace transferinu < 16 %
Diagnostika 95% nemocných
s latentním deficitem železa.
• Hladina cirkulujících
transferinových receptorů v séru :
dif.dg. mezi sideropenií - zvýšena
a anemií při chronickém onemocnění
- nezvýšena.
9.
LÉČBA ANÉMIE Z NEDOSTATKU ŽELEZAoptimální léčba vzestup hodnoty Hb o 2 g/l den.
substituce železem - musí pokrýt:
a) denní obrat
b) vzestup Hb o 2 g/l
celkem :
10 – 25 mg Fe
( dle stavu proliferace )
37,4 mg Fe
---------------------------50 - 60 mg Fe /den
Denní dávka vstřebaného Fe při léčbě rozvinuté
sideropenické anémie má činit 50 – 60 mg, to odpovídá
dávce 160 – 200 mg elementárního Fe v přípravku
podaném per os.
10.
PŘÍPRAVKY ŽELEZAnázev
(výrobce)
léková
forma
složení
Aktiferrin
(Merckle,SRN)
cps.
síran železnatý
serin
34 mg
Aktiferrin
(Merckle,SRN)
sir.
síran železnatý
serin
6,8 mg/ml
Aktiferrin
(Merckle,SRN)
gtt.
síran železnatý
serin
9,3 mg/ml
Ferrlecit
(Nattermann,SRN)
inj
komplex Fe 3+ s glukonátem
sodným
62,5 mg
Ferro-Gradumet
(Galenika,Jugoslávie)
tab
síran železnatý
105 mg
Ferronat
(Galena,ČR)
susp.
fumarát železnatý
10 mg/ml
Ferronat retard
(Léčiva,ČR)
tab
síran železnatý
105 mg
Ferrum lek i.m.
(Lek,Slovinsko)
inj
komplex hydroxidu
železitého se sacharózou
100 mg
Maltofer
(Vifor,Švýcarsko)
sir.
10 mg/ml
Maltofer
(Vifor,Švýcarsko)
gtt.
komplex Fe
3+
s polymaltosou
komplex Fe
3+
s polymaltosou
Sorbifer durules
(Egis,Maďarsko)
tab
síran železnatý
kyselina askorbová
100 mg
Tardyferon
(Robapharm,Švýcarsko)
drg
síran železnatý
mukoproteáza
80 mg
obsah látky v lékové
formě
50 mg/ml
11.
KONTROLA ÚČINNOSTI LÉČBY ŽELEZEM1. Do doby normalizace hodnoty Hb – kontrola
krevního obrazu.
2. Po normalizaci krevního obrazu – kontrola
parametrů zásob železa :
FERITIN V SÉRU
pokračování v léčbě nutno doplnit zásoby železa
( optimum = 500 mg )
kontrola : vyšetřením hladiny feritinu v séru
1 g/l feritinu v séru odpovídá 8 mg zásobního
železa.
NUTNO VŽDY ODSTRANIT VYVOLÁVAJÍCÍ
PŘÍČINU SIDEROPENIE !!
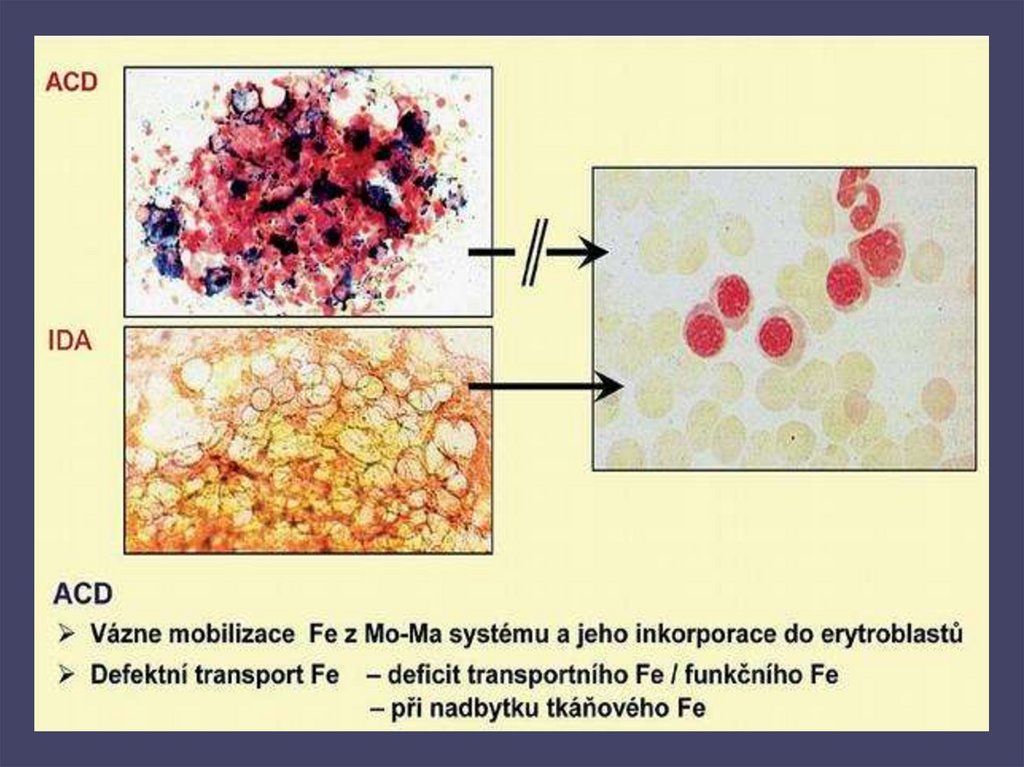
12. ANÉMIE PŘI CHRONICKÉM ONEMOCNĚNÍ
• anémie při chronických infekcích, chronických zánětech, nádorovýchonemocněních.
• anémie normocytární, normochromní, postupně hypochromie a
mikrocytóza.
• porucha distribuce Fe se sníženou utilizací v kostní dřeni při jeho
retenci v monocyto-makrofágovém systému účinkem cytokinů
(IL6, IL-1) a ↑ sekreci hepcidinu.
• Fe v séru - ↓, TIBC - ↓, Tfsat- v normě.
feritin v séru – není snížen, TfR – v normě.
kostní dřeň : sideroblasty - ↓, siderofágy - ↑.
13.
14. ANÉMIE PŘI CHRONICKÉM ONEMOCNĚNÍ
Léčba :
Základem léčby je léčba základního onemocnění.
Podávání erytropoetinu – má význam při jeho absolutním či relativním deficitu.
- anémie při chronické renální insuficienci
- kurativní radioterapie/chemoterapie u solidních nádorů
- anémie u lymfoproliferací a u časných forem MDS
Podávání inhibitorů cytokinů – nemá větší vliv na korekci anémie,
perspektivně – kombinace protilátky proti hepcidinu a EPO.
15. MAKROCYTÁRNÍ ANÉMIE
• MCV > 95 fl, počet retikulocytů nenízvýšen.
• Megaloblastové anémie
• Makrocytární anémie bez přítomnosti
megaloblastů
jaterní choroby, hypotyreoza, alkoholismus
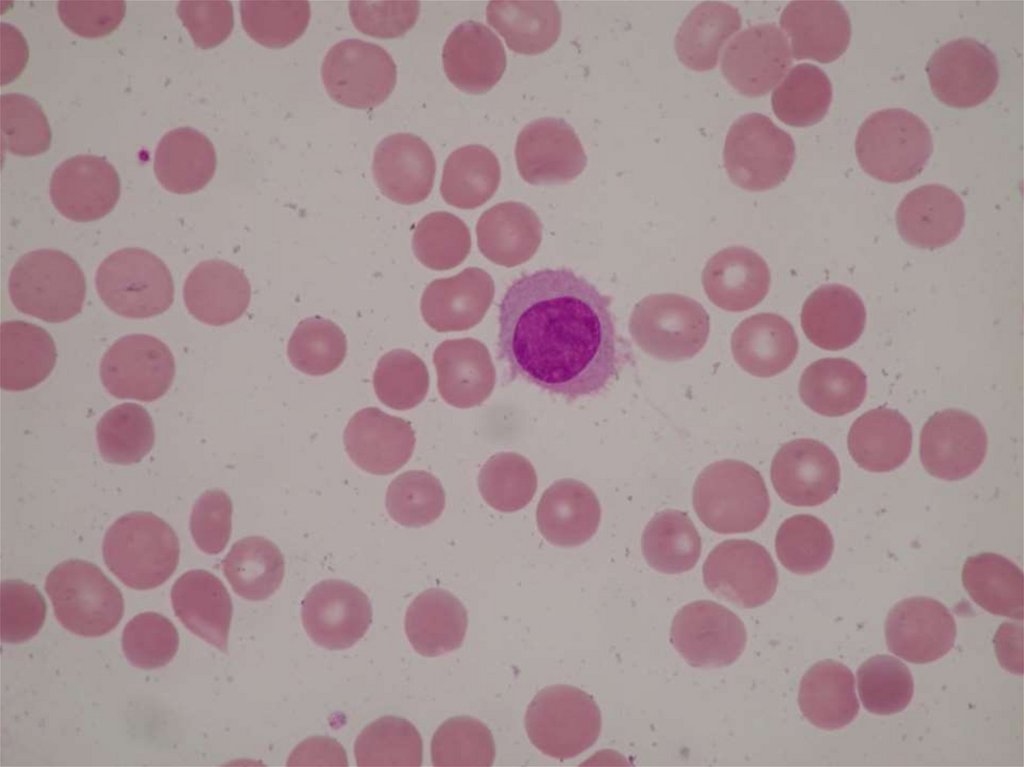
16. MEGALOBLASTOVÉ ANÉMIE
• porucha přenosu metylové skupiny sloužícík metylaci deoxyuridinu na thymidin →
porucha tvorby DNA při zachované syntéze
RNA a bílkovin.
• nedostatek vitaminu B12 či kyseliny listové
↓
MEGALOBLAST
( velký MCV s normální hemoglobinizací,
normální MCHC a zvýšený MCH ).
17.
18. MEGALOBLASTOVÉ ANÉMIE - DIAGNOSTIKA
MEGALOBLASTOVÉ ANÉMIE DIAGNOSTIKA• krevní obraz – makrocyty (↑MCV, ↑MCH,normální
MCHC), ↓RTC, megalocyty, megaloblasty,
leukocytóza s posunem doleva,trombocytopenie.
• kostní dřeň – hyperplasie erytropoezy, megaloblasty,
makrocytoza granulocytů, polyploidie mgkc.
dif.dg. oproti MDS ( cytogenetika,cytochemie )
• biochemie – ↓ B12, ↓ kys.listová, ↑ přímý i nepřímý
bilirubin, vyšetření protilátek proti vnitřnímu
faktoru a proti parietálním bb. ,normální zásoby Fe.
19. MEGALOBLASTOVÉ ANÉMIE - PŘÍČINY
• Nedostatečný přívod vitaminu B12 či kyseliny listové• Porucha vstřebávání:
a) nedostatek vnitřního faktoru, protilátky proti vnitřnímu faktoru
b) céliakální sprue, Crohnova choroba, resekce střeva
píštěle, divertikly, striktury, parasité
c) inhibitory resorpce (fenylhydantoin,PAS,pyrimidin, neomycin)
d) selektivní malabsorpce B12 s proteinurií
Vzácně :
• Poruchy transportu a resorpce při nedostatku
transkobalaminu I. a II.
• Zvýšená spotřeba (gravidita,růst,anémie s hyperplasií erytropoesy)
• Zvýšené ztráty (jaterní choroby,dialýza)
• Inhibitory dihydrofolát reduktázy (MTX,pyrimethamin)
antagonisté pyrimidinů (ARA-C) či purinů (6-MP)
20. MEGALOBLASTOVÉ ANÉMIE - LÉČBA
• Substituce - vitamin B12 300 – 1000 μg denněpostupně přechod na udržovací dávky
1 x za 2 – 4 týdny.
retikulocytární krize : 5.-7.den léčby,
vzestup počtu retikulocytů na 10-30%.
nutná kontrola parametrů zásob železa.
Dispenzarice s gastroskopickými kontrolami.
21. NORMOCYTÁRNÍ ANÉMIE
• MCV = 80-95 fl, většinou normochromní,počet retikulocytů není zvýšen.
• porucha kmenové krvetvorné buňky
- aplastická anémie, čistá aplasie červené řady
- myelodysplastický syndrom
- paroxysmální noční hemoglobinurie
• sekundární porucha kostní dřeně
- infiltrace dřeně (hematologické a jiné nádory)
- anémie při chronickém onemocnění
22. NORMOCYTÁRNÍ ANÉMIE - DIAGNOSTIKA
NORMOCYTÁRNÍ ANÉMIE DIAGNOSTIKA• Krevní obraz:
různý stupeň anemie, resp. pancytopenie, vyplavování
mladších forem
• Kostní dřeň – sternální punkce, trepanobiopsie:
- morfologie – buněčnost, dysplastické změny (porucha
proliferace a diferenciace), okrsky nezralých buněk
- cytochemie – poruchy enzymatické výbavy
mutace mitochondriální DNA (anulár.sideroblasty)
- cytogenetika – chromosomální aberace
- průtoková cytometrie – počet nezralých
prekursorů, deficit inhibitorů komplementu
- molekulární genetika – klonální povaha, exprese genů.
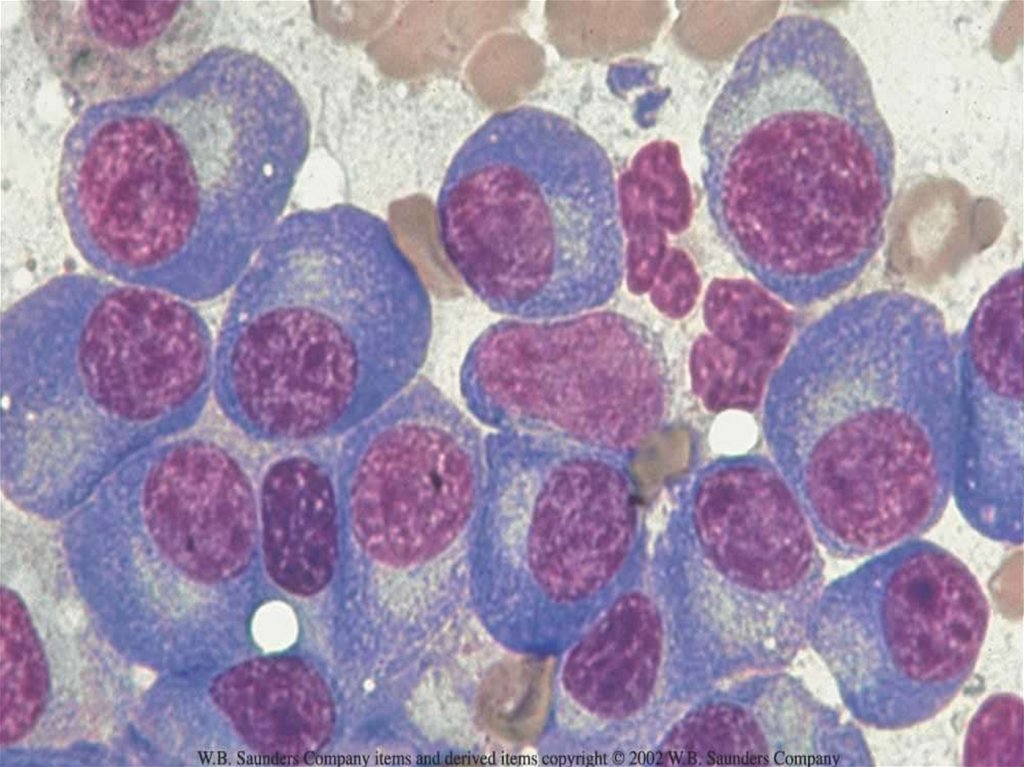
23. APLASTICKÁ ANÉMIE - DŘEŇOVÝ ÚTLUM
• Kritéria těžké aplastické anémie:- kostní dřeň < 30% buněčnosti
- krevní obraz RTC < 40 x 109/l či < 0,1%
NS < 0,5 x 109/l
PLT < 20 x 109/l
• Etiologie:
a) idiopatická (70%)
b) známá příčina
(infekce,záření,léky,cytostatika,chemic.látky)
• Patogeneze:
poškození kmenové krvetvorné buňky přímo či
aktivací imunitních mechanismů, poškození
hemopoetického stromatu.
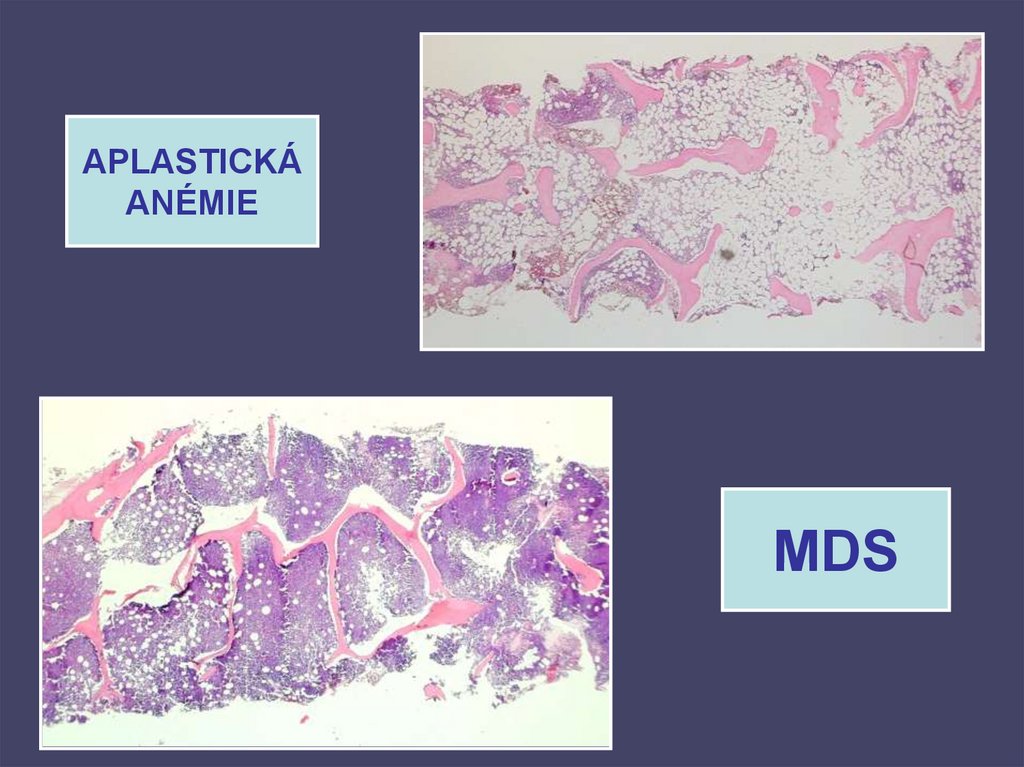
24.
APLASTICKÁANÉMIE
MDS
25. APLASTICKÁ ANÉMIE - LÉČBA
• Transplantace krvetvorných buněk:u mladších nemocných ( < 55 let ) pokud je přítomen
vhodný příbuzenský dárce.
efekt SCT – u 70-80% nemocných.
• Kombinovaná imunosuprese:
ATG
40 mg/kg 1. - 4.den
Prednison
1 mg/kg 1. - 14.den
Cyklosporin A
3 mg/kg/den
+ úprava dle hladiny CS-A – dlouhodobá udržovací léčba .
efekt u 70% nemocných.
Podávání agonistů trombopoetinového receptoru + EPO
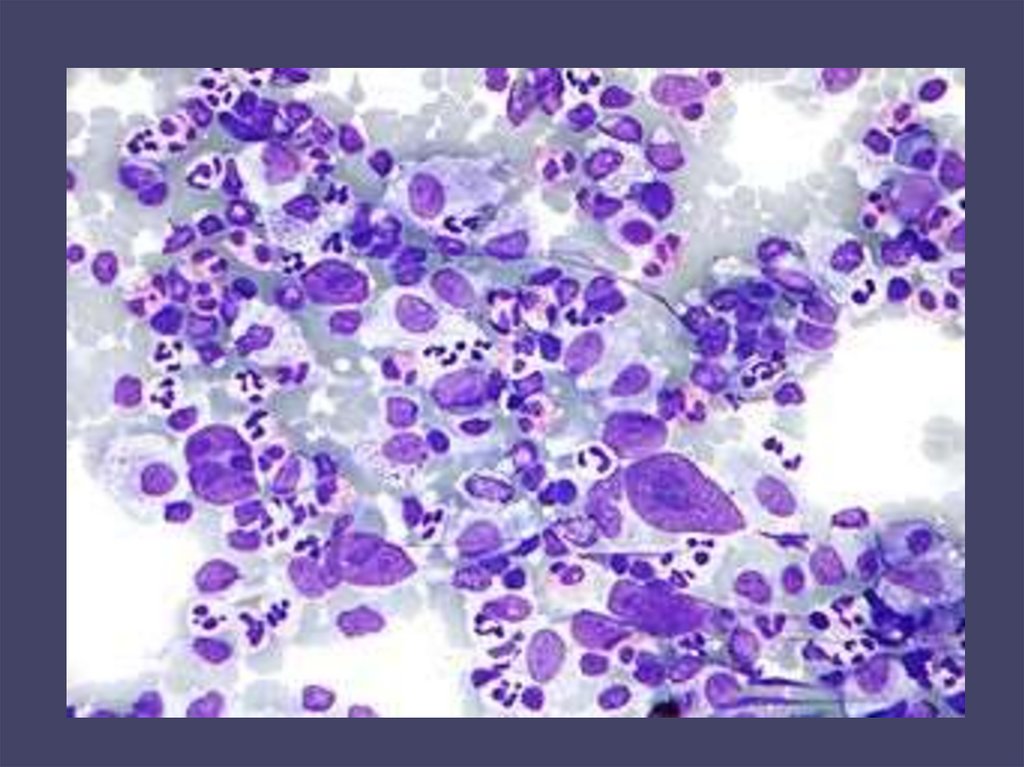
26. MYELODYSPLASTICKÝ SYNDROM
• ČASNÁ FÁZE: pancytopenie v krevním obrazepři buněčně bohaté dysplastické kostní dřeni
v důsledku proliferace patologického klonu
s vysokým stupněm apoptózy - RA,RARS,RCMD.
• POZDNÍ FÁZE: postupný nárůst nezralých
prekursorů krvetvorby náležejících
patologickému klonu, nízká apoptóza a porucha
diferenciace – leukemizace - RAEB-1, RAEB-2
(CMML,RAEB-T).
27.
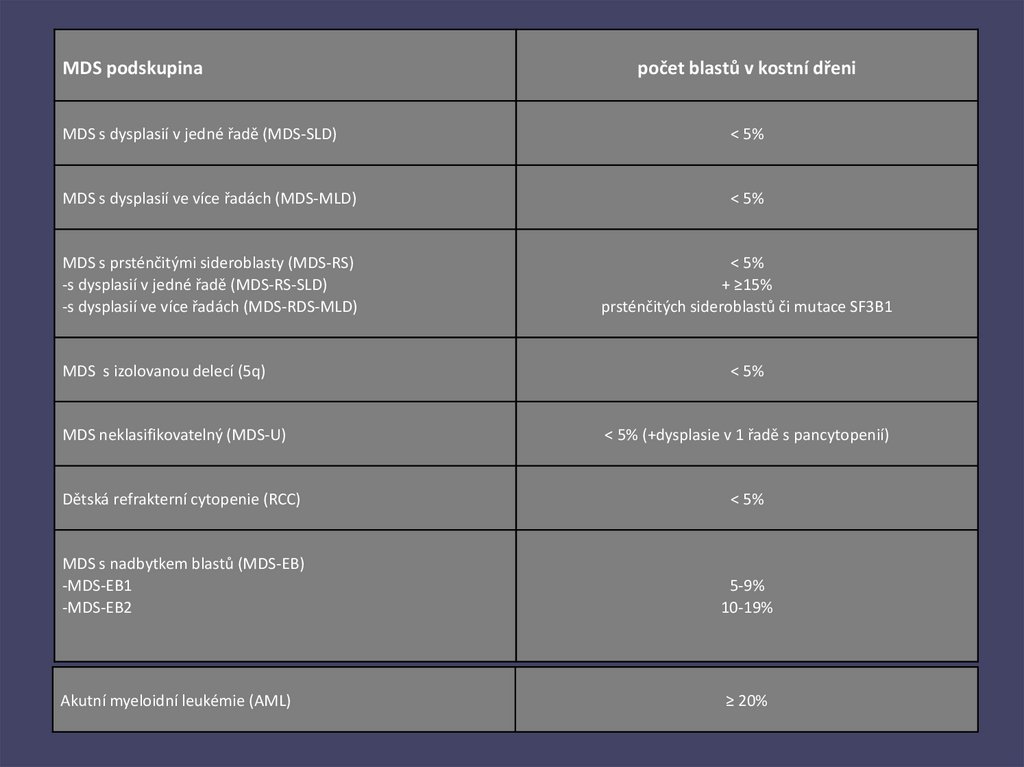
MDS podskupinapočet blastů v kostní dřeni
MDS s dysplasií v jedné řadě (MDS-SLD)
< 5%
MDS s dysplasií ve více řadách (MDS-MLD)
< 5%
MDS s prsténčitými sideroblasty (MDS-RS)
-s dysplasií v jedné řadě (MDS-RS-SLD)
-s dysplasií ve více řadách (MDS-RDS-MLD)
< 5%
+ ≥15%
prsténčitých sideroblastů či mutace SF3B1
MDS s izolovanou delecí (5q)
< 5%
MDS neklasifikovatelný (MDS-U)
< 5% (+dysplasie v 1 řadě s pancytopenií)
Dětská refrakterní cytopenie (RCC)
< 5%
MDS s nadbytkem blastů (MDS-EB)
-MDS-EB1
-MDS-EB2
5-9%
10-19%
Akutní myeloidní leukémie (AML)
≥ 20%
28.
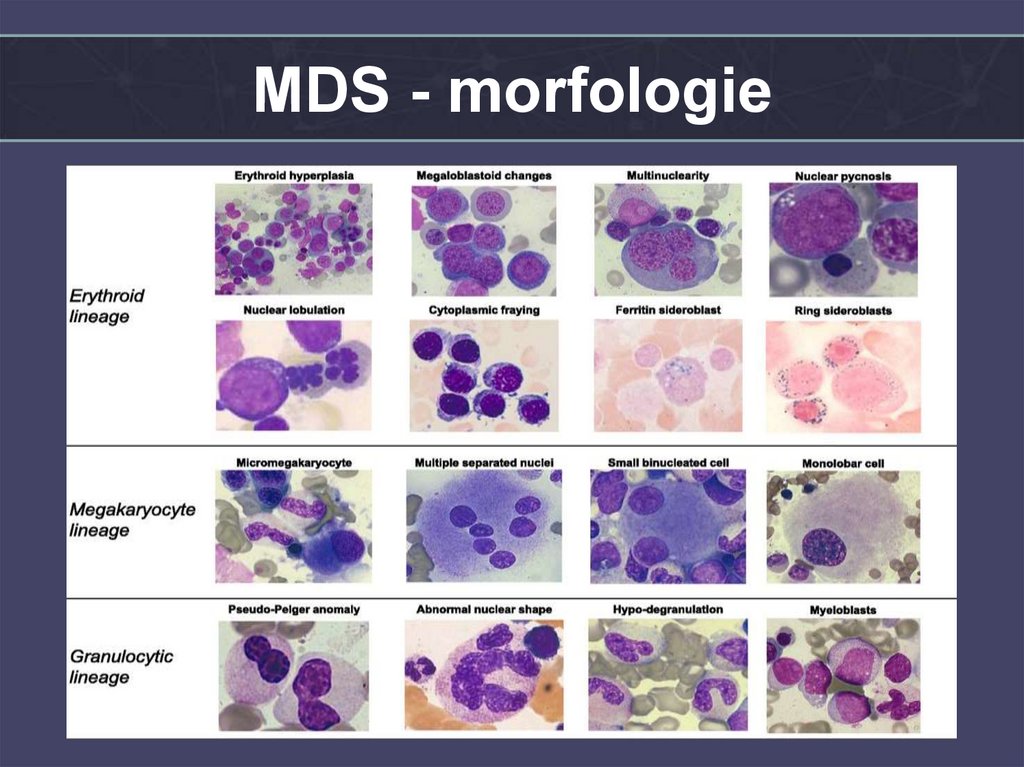
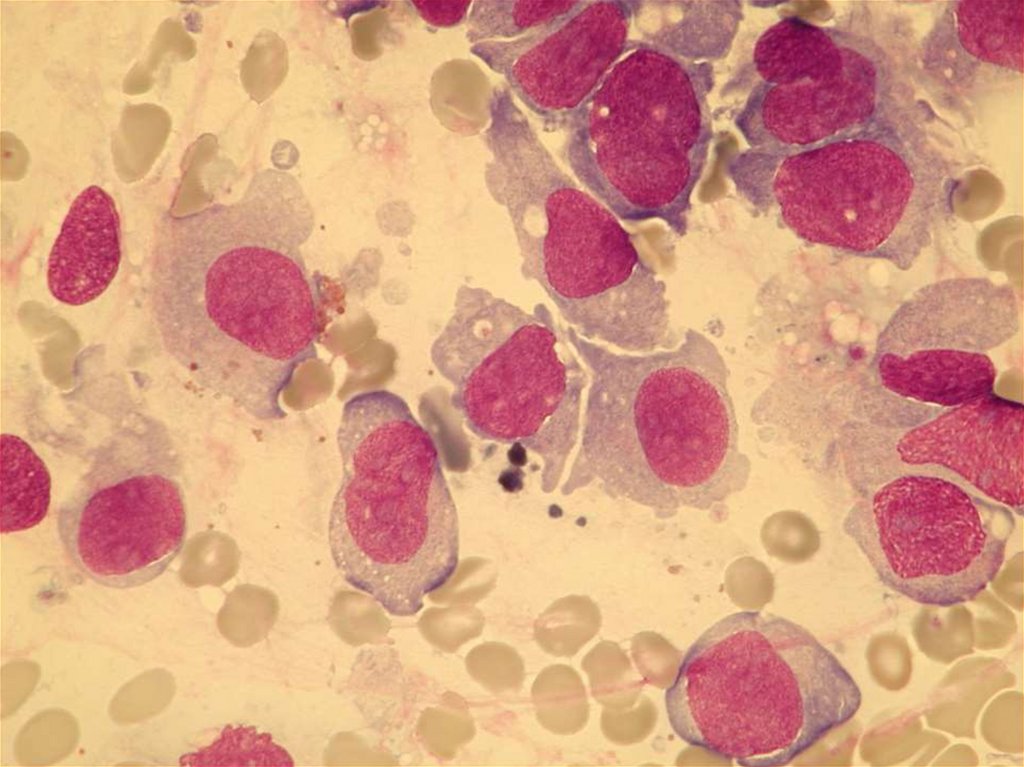
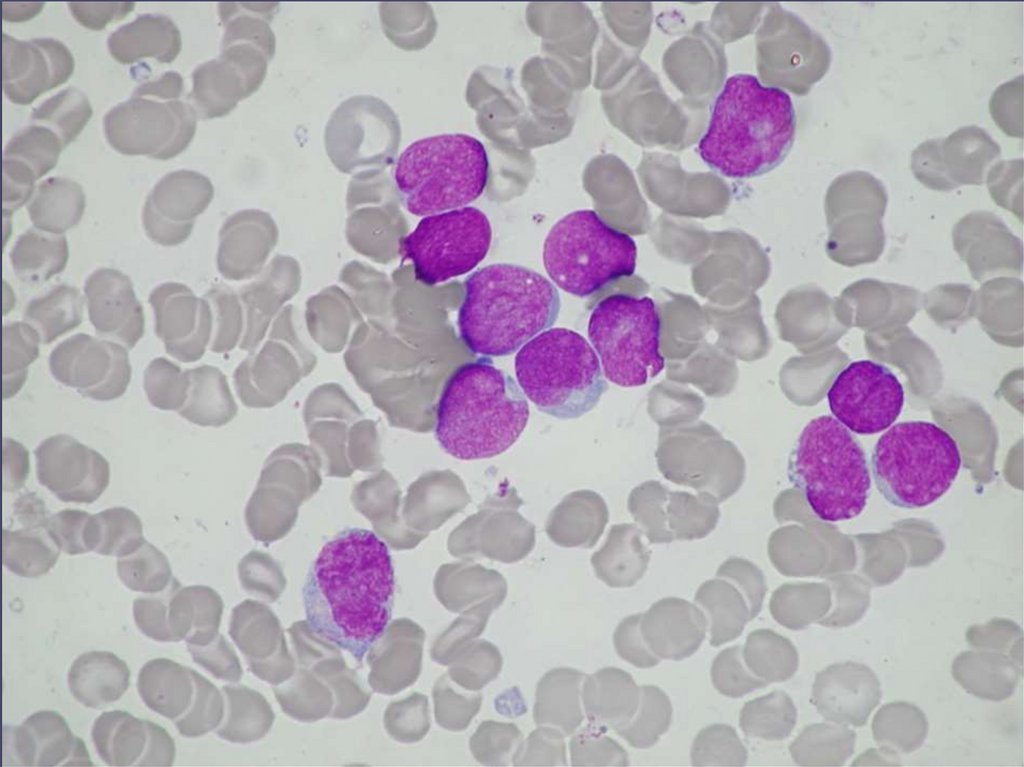
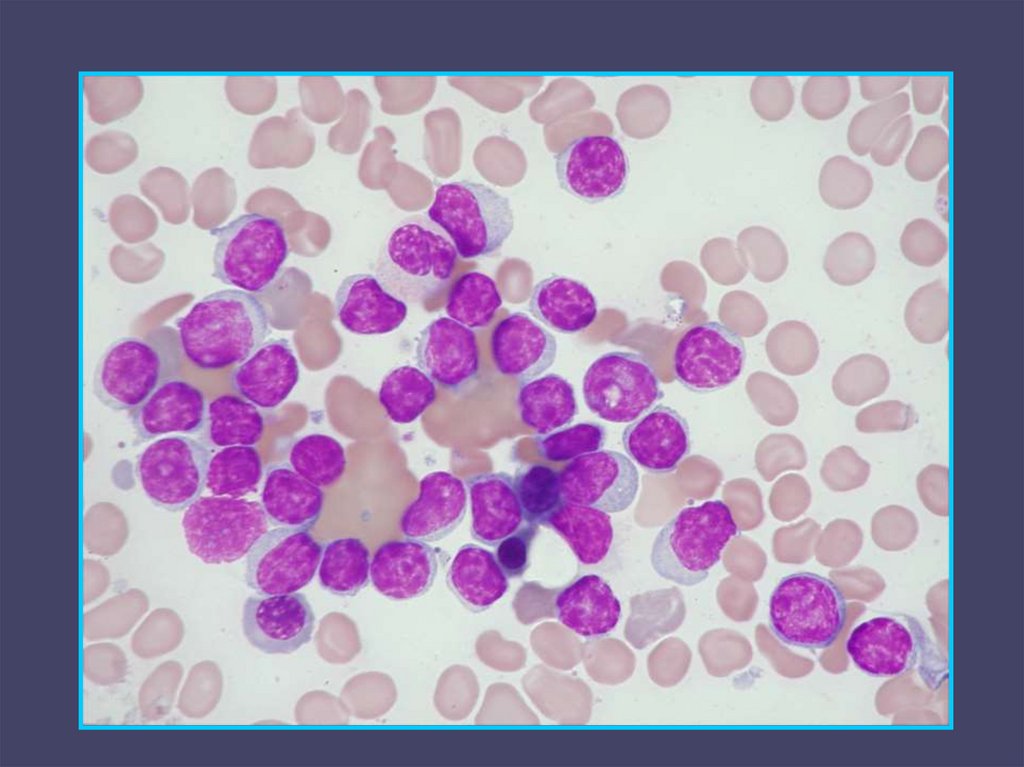
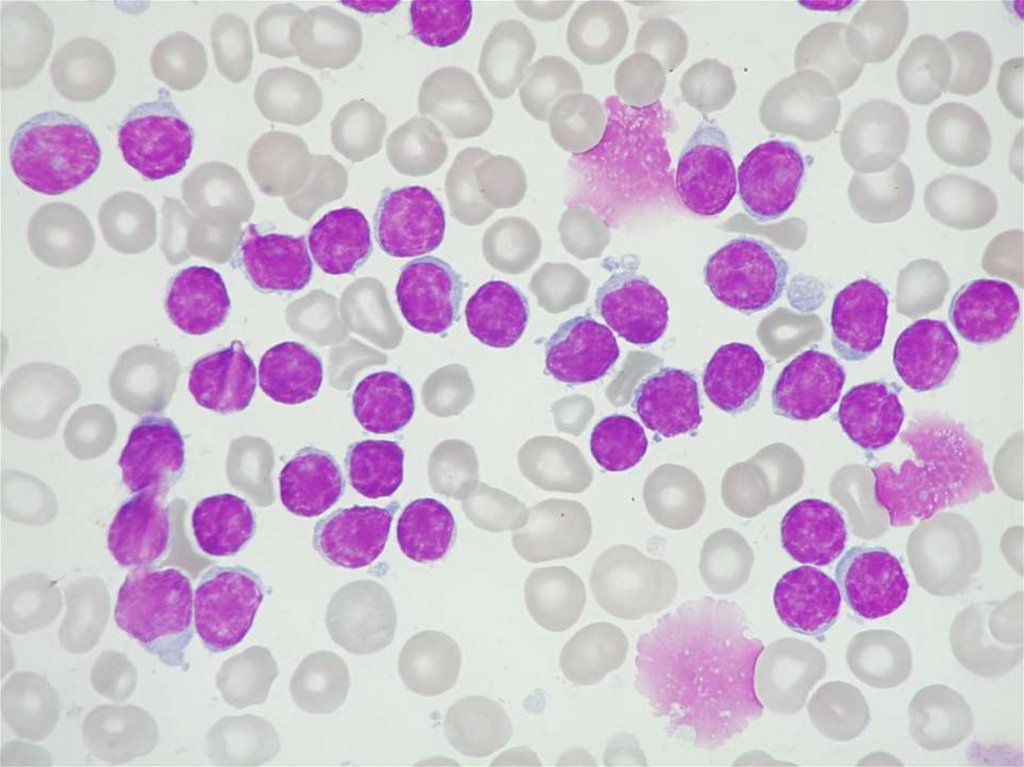
MDS - morfologie29.
30.
31.
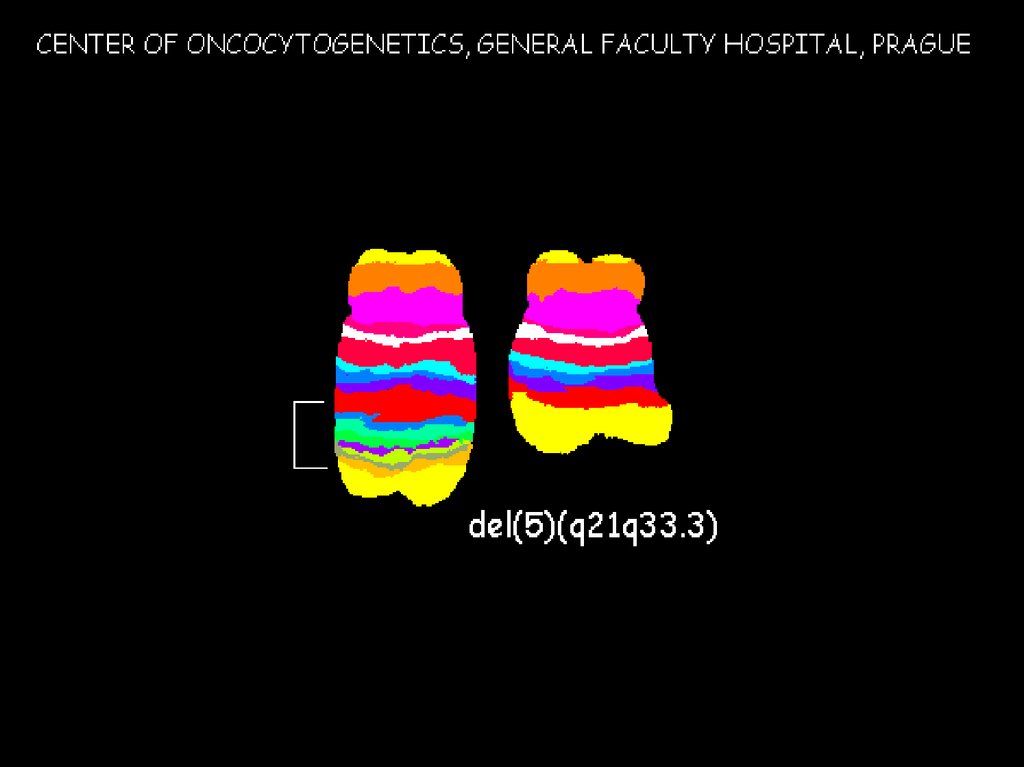
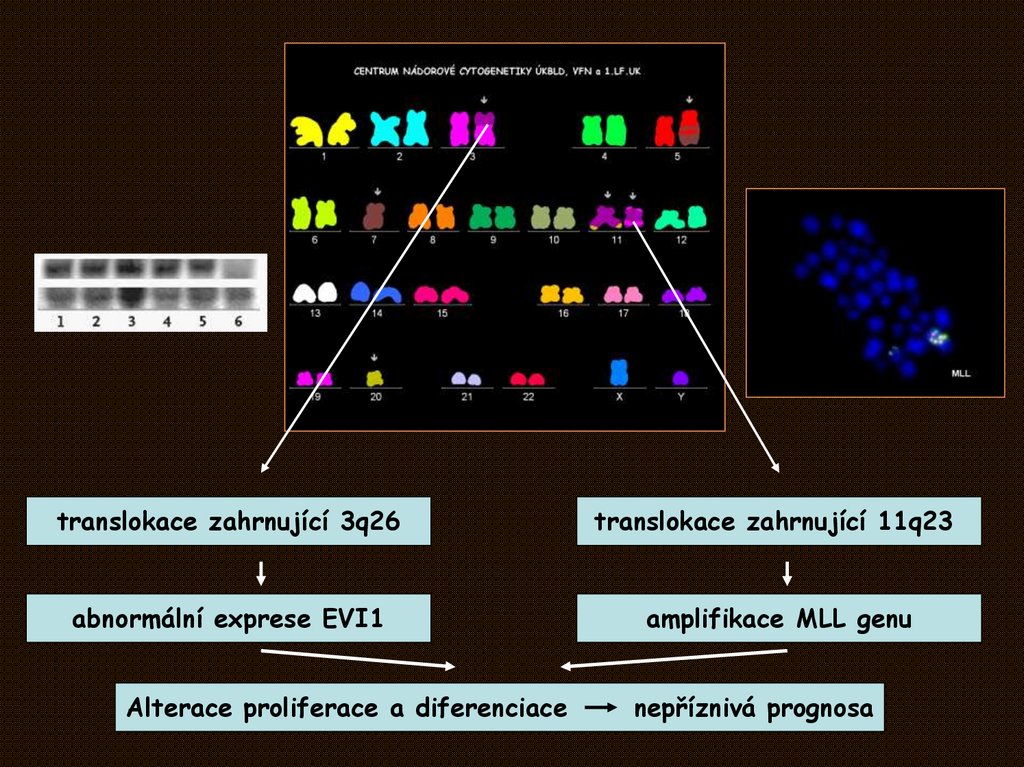
translokace zahrnující 3q26translokace zahrnující 11q23
abnormální exprese EVI1
amplifikace MLL genu
Alterace proliferace a diferenciace
nepříznivá prognosa
32. MDS - LÉČBA
• PODPŮRNÁ LÉČBA( transfuze erytrocytů a trombocytů, chelatační léčba)
• INHIBICE APOPTÓZY
( kortikosteroidy, růstové hormony, inhibitory cytokinů )
• IMUNOSUPRESE
( kortikosteroidy, cyklosporin A, ATG, revlimid )
• DIFERENCIAČNÍ LÁTKY – reverse metylace genů
( azacytidin, decitabin, inhibitory histon deacetylázy )
• CHEMOTERAPIE
( monoterapie, kombinovaná chemoterapie )
• TRANSPLANTACE KRVETVORNÝCH BUNĚK
( alogenní – příbuzenská, nepříbuzenská ).
33.
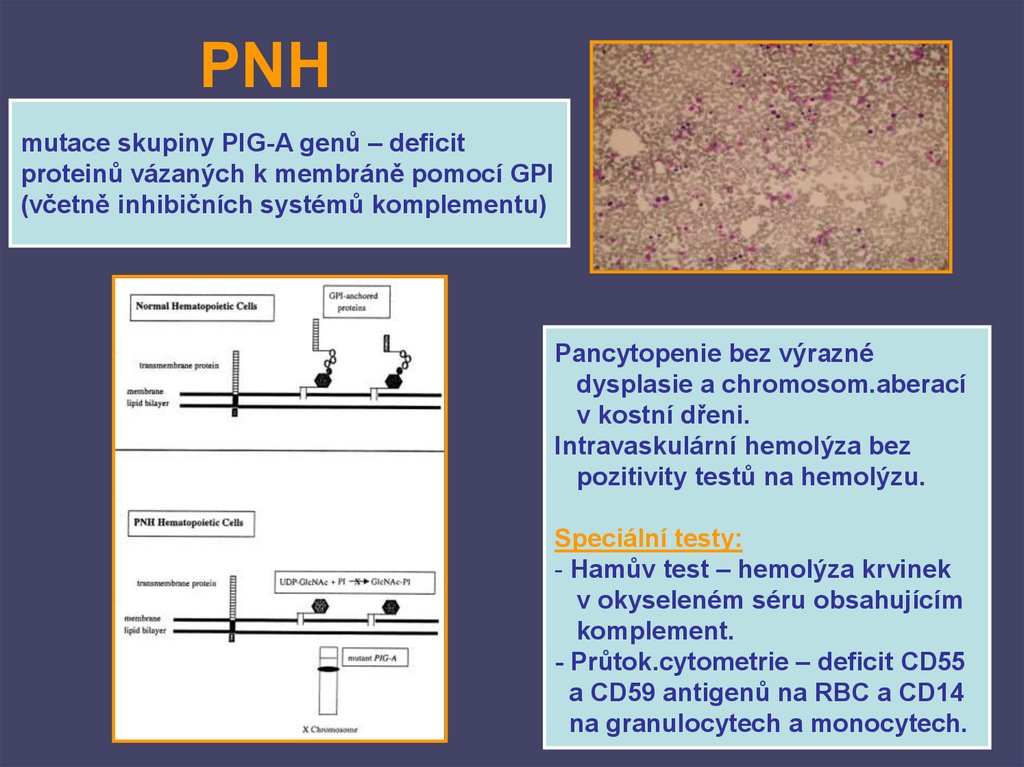
PNHmutace skupiny PIG-A genů – deficit
proteinů vázaných k membráně pomocí GPI
(včetně inhibičních systémů komplementu)
Pancytopenie bez výrazné
dysplasie a chromosom.aberací
v kostní dřeni.
Intravaskulární hemolýza bez
pozitivity testů na hemolýzu.
Speciální testy:
- Hamův test – hemolýza krvinek
v okyseleném séru obsahujícím
komplement.
- Průtok.cytometrie – deficit CD55
a CD59 antigenů na RBC a CD14
na granulocytech a monocytech.
34. Průtoková cytometrie
PNH III. erytrocytyPNH granulocyty
25%
70%
/PNH Redquant ,TM/
/PNH Cellquant, TM/
35. ANÉMIE SE ZVÝŠENÝM POČTEM RETIKULOCYTŮ
• HEMOLYTICKÉ ANÉMIE- korpuskulární
( poruchy: skladby membrány RBC,
enzymatického vybavení,
tvorby globinu )
- extrakorpuskulární
( imunitní, neimunitní )
36. KORPUSKULÁRNÍ HEMOLYTICKÉ ANÉMIE
Diagnostikakrevní obraz - normocytární anémie se
sférocyty, ↑ RTC.
biochemie - ↑ přímý a nepřímý bilirubin
speciální testy :
- autohemolýza, osmotická resistence
- DEFEKTY MEMBRÁNY.
- vyšetření enzymů ( PK,G6PD )
- ENZYMOPATIE.
- elfo Hb, DNA analýza
- HEMOGLOBINOPATIE.
37.
38. DĚDIČNÁ SFÉROCYTÓZA
• Diagnostika :↓ osmotická rezistence erytrocytů
↑ autohemolýza erytrocytů
deficit proteinu při elektroforeze
membrány erytrocytu
či pomocí FCM (EMA test)
• Léčba :
splenektomie
39.
• ERYTROCYTÁRNÍ ENZYMOPATIE• Defekt enzymů anaerboní glykolýzy
deficit pyruvát kinázy – chronická
hemolytická anémie s malým
efektem splenektomie.
• Defekt enzymů pentosového cyklu
deficit glukoso-6-fosfát dehydrogenázy
zvýšená citlivost k oxidačním
činidlům – chronická hemolýza
či hemolytické krize –
anémie s Heinzovými tělísky
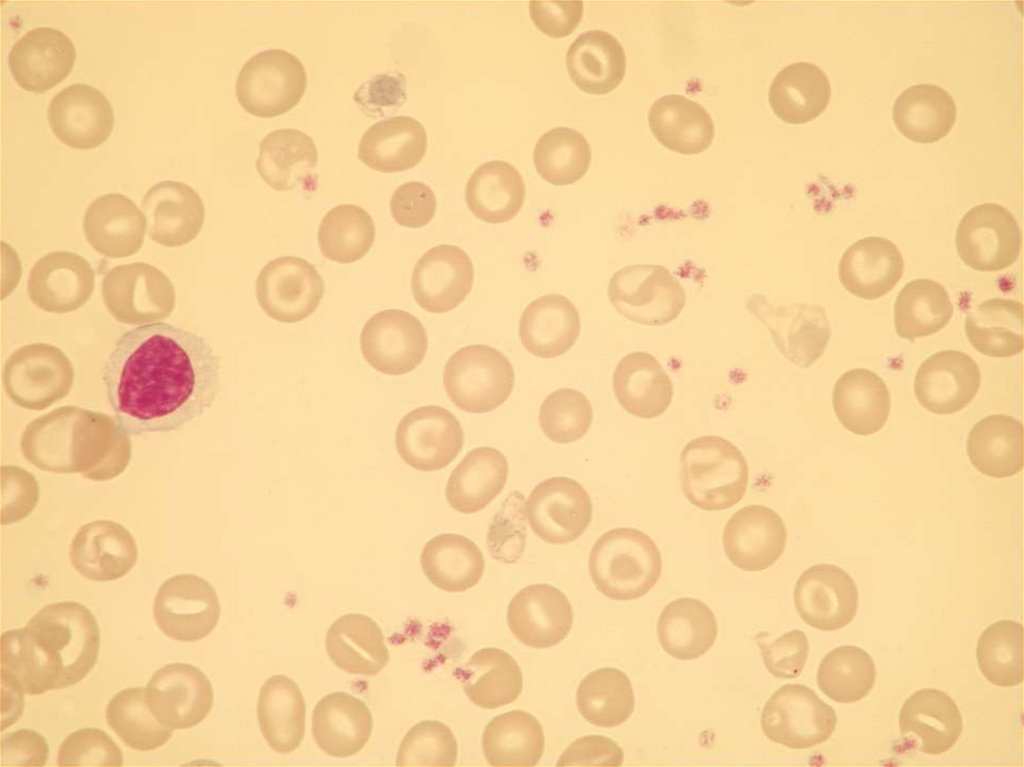
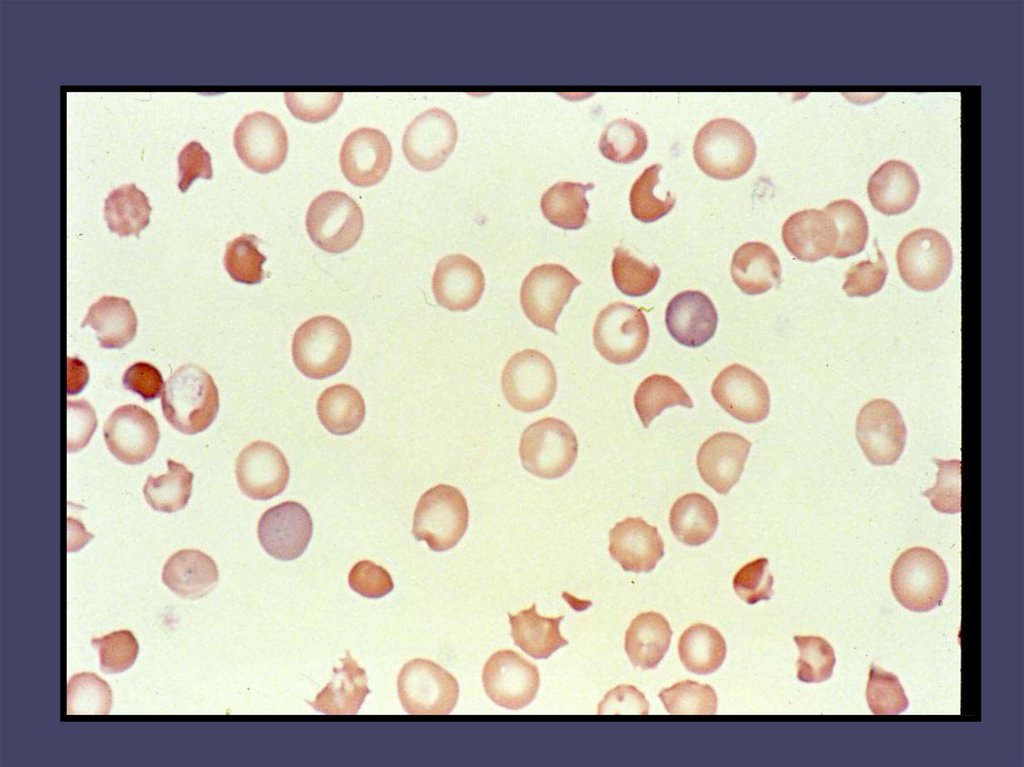
40. THALASÉMIE
• Alfa thalasémie – porucha syntézy α globinovéhořetězce
• Beta thalasémie – porucha syntézy β globinového
řetězce
Heterozygotní formy – většinou výrazná mikrocytóza
(MCV<65 fl) a hypochromie (MCH<20 pg/l) při
normálním či lehce zvýšeném počtu erytrocytů, v
krevním obraze jsou přítomny terčovité erytrocyty a
leptocyty.
Fe v séru, TIBC, satTRF, feritin v séru - nejsou sníženy.
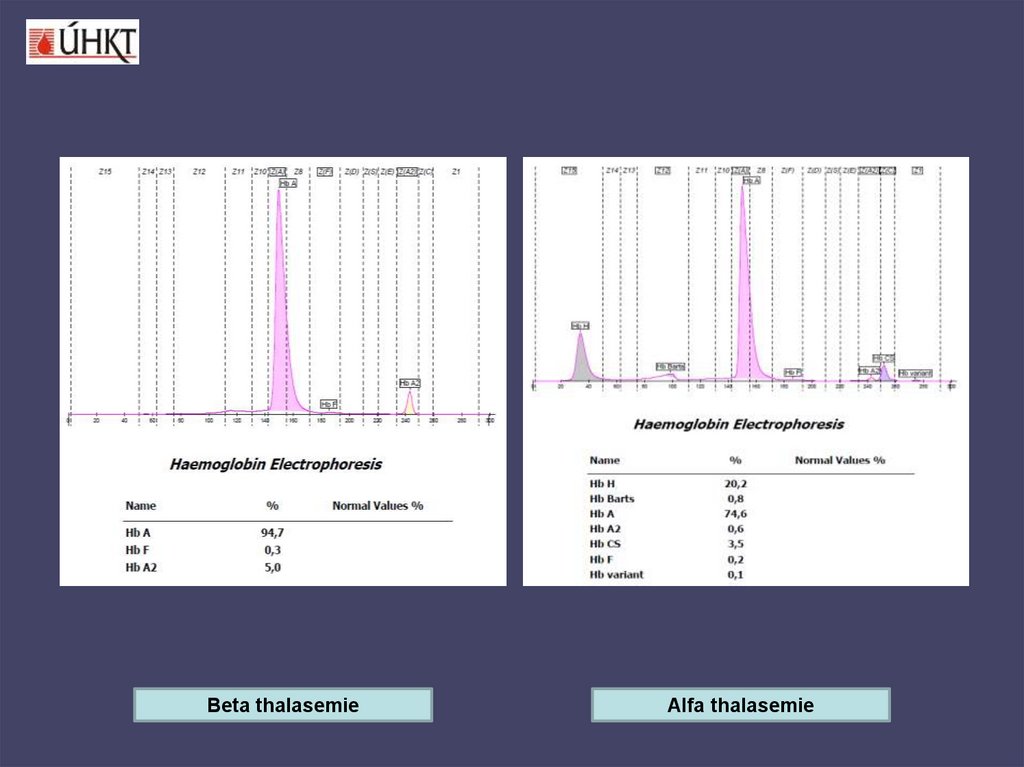
Zvýšené množství HbA2 a HbF u β-thalasémie,
určité % hemoglobinu H (β4) u α-thalasémie.
41.
42.
Beta thalasemieAlfa thalasemie
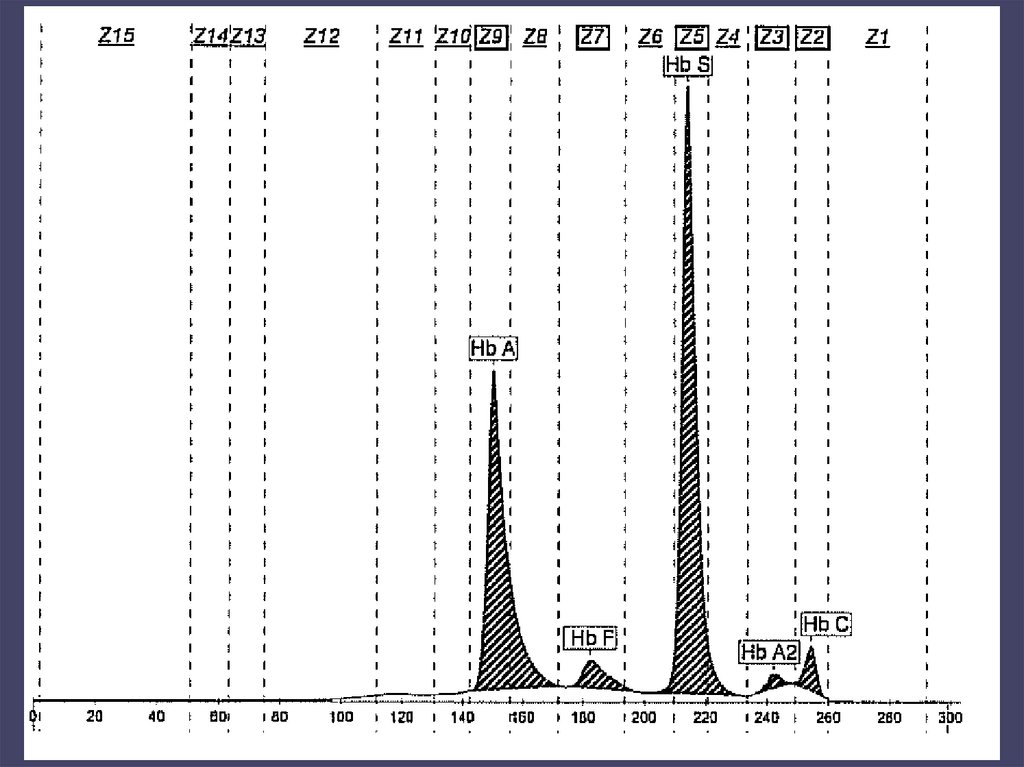
43. HEMOGLOBINOPATIE
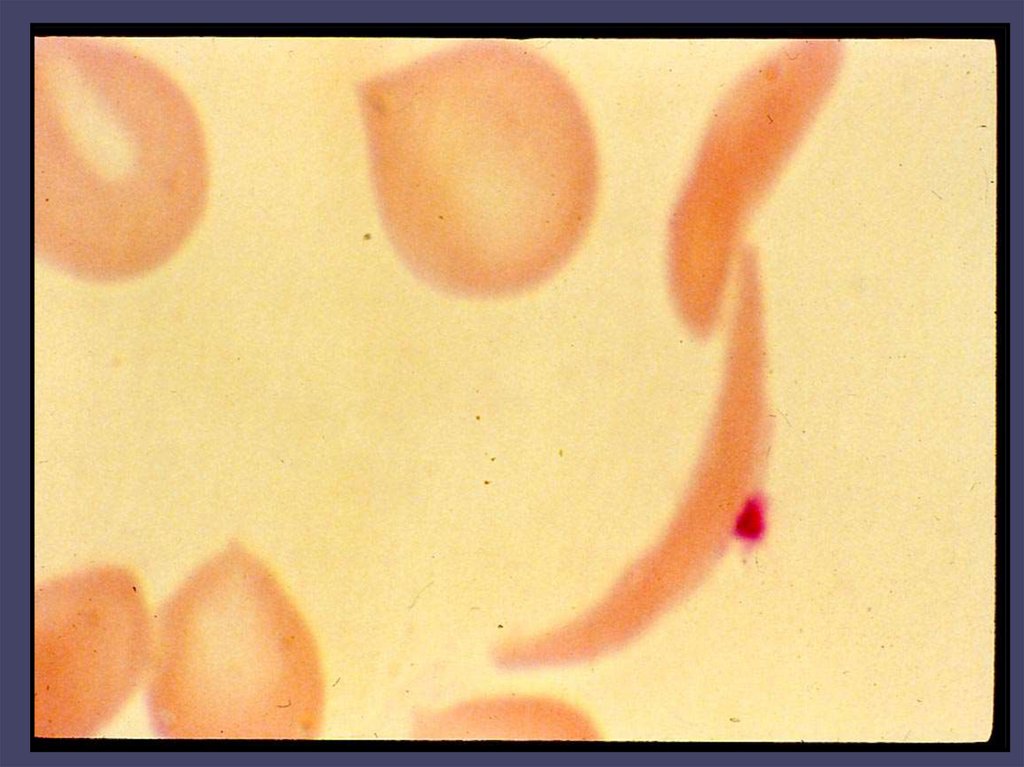
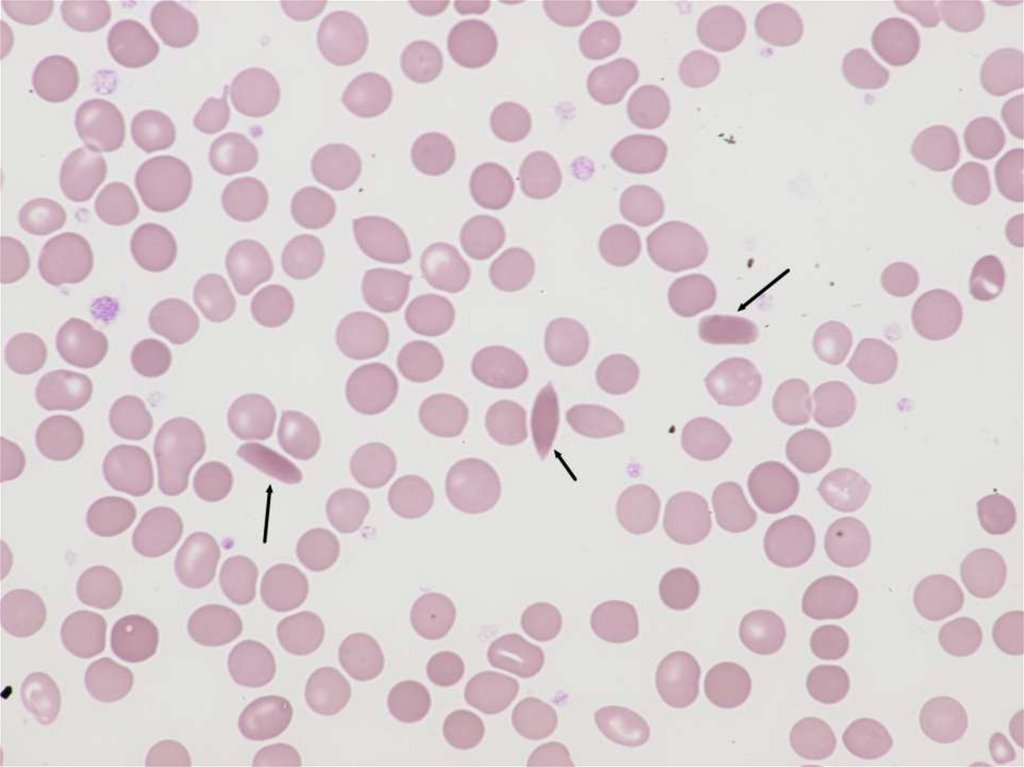
• ABNORMÁLNÍ HEMOGLOBINY( srpkovitá anemie – HbS, HbC, HbD,HbE )
• NESTABILNÍ HEMOGLOBINY
( kongenitální nesferocytární hemolytické anemie
s tvorbou Heinzových tělísek )
• METHEMOGLOBINEMIE
( stabilizace hemového Fe v trojmocné formě)
• HEMOGLOBINY SE ZVÝŠENOU AFINITOU K O2
( polycytemie – tkáňová hypoxie s cyanozou)
44.
45.
46.
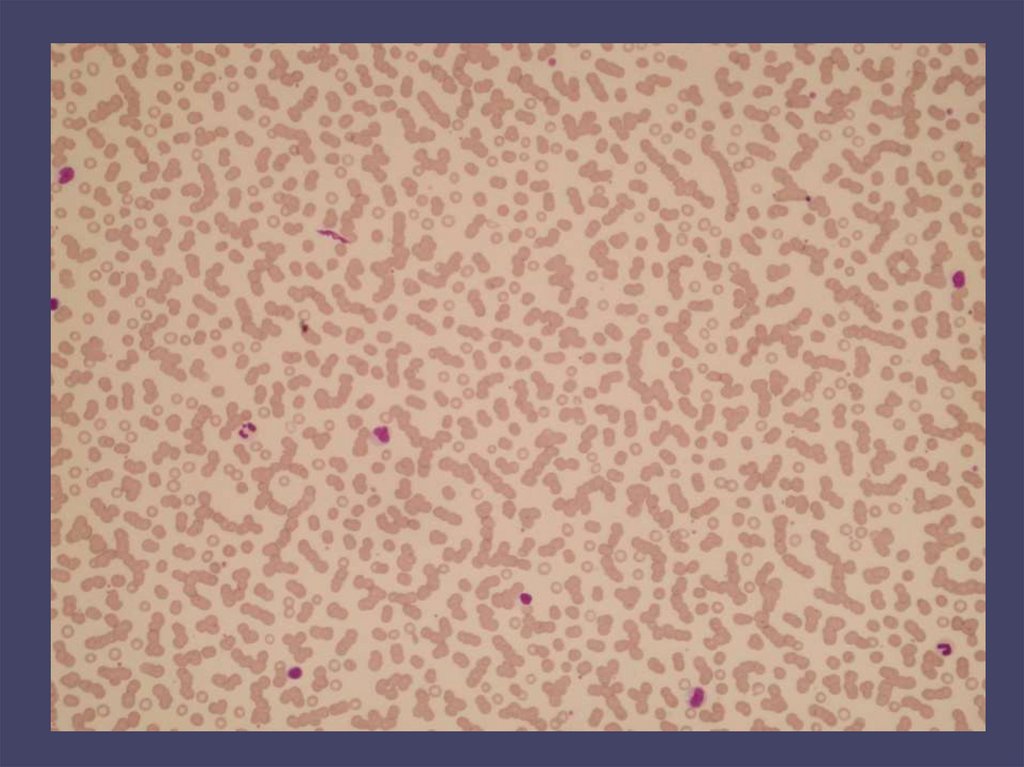
47. MIKROANGIOPATICKÁ HEMOLYTICKÁ ANÉMIE
příčina:
1. mechanická :
rozrušení integrity membrány erytrocytu mechanickou příčinou v
průsvitu cévy, nejčastěji depozity fibrinu v místě poškození endotelu
cévní stěny.
2. aktivace cytokinů :
shigatoxin – aktivace
komplementu, uvolnění
cytokinů monocyty, aktivace
endotelu, sekrece vWF a TF
– vznik destičkových trombů.
48.
49. KLASIFIKACE AUTOIMUNITNÍCH HEMOLYTICKÝCH ANÉMIÍ
• TEPELNÉ PROTILÁTKY- idiopatická
- sekundární (lymfoproliferace, jiné nádory, autoimunitní
choroby,virové infekce, imunodeficience)
- hemolytická anémie indukovaná léky
• CHLADOVÉ PROTILÁTKY
- idiopatická
- sekundární (lymfoproliferace, viry, mykoplasmata, autoimunitní
choroby)
- paroxysmální chladová hemoglobinurie (lues,spalničky aj.)
• SMÍŠENÉ CHLADOVÉ A TEPELNÉ PROTILÁTKY
50. AIHA – laboratorní nález :
• Krevní obraz:makrocytární anémie s retikulocytózou
• Biochemie:
přímý i nepřímý bilirubin,
urobilinogen v moči
• Speciální vyšetření:
přímý a nepřímý antiglobulinový (Coombsův
test)
PRŮKAZ INTRAVASKULÁRNÍ HEMOLÝZY:
volný Hb v plasmě,
hladiny haptoglobinu a hemopexinu v séru,
hemoglobinurie.
51.
52. PŘÍMÝ COOMBSŮV TEST
Erytrocyty nemocného s navázanouprotilátkou
dodáme: anti IgG
53. NEPŘÍMÝ COOMBSŮV TEST
sérum nemocného s volnými protilátkamidodáme: skupinové erytrocyty
dodáme: anti IgG či anti C3b
54. AIHA - LÉČBA :
Lehčí forma ( Hb > 80 g/l ):
PREDNISON 1 mg/kg/den 2-3 týdny
- vysazení kortikoidů až při opakované negativitě přímého
Coombsova testu.
Při nedostatečném efektu či relapsech – kombinace s :
CYKLOPHOSPHAMID 100-150 mg/den či
CYKLOSPORIN A 3 mg/kg/den.
Těžká forma ( Hb < 80 g/l ):
PREDNISON 1-2 g i.v. denně 5 dní, pak při dobrém efektu
rychlý pokles na dávku 1 mg/kg.
- kombinace kortikoidů s :
CYKLOPHOSPHAMID 200 mg i.v./den
i.v. IMUNOGLOBULINY 0,5g /kg/den
PLASMAFERESA
RITUXIMAB (anti CD20 monoklonální protilátka)
55. POLYCYTEMIE
• Primárnípolycytémia vera
primární familiární a kongenitální polycytémie
• Sekundární
snížená saturace tkání kyslíkem (plicní choroby,
cyanotické srdeční vady, hemoglobinopatie,
poškození ledvin, nádory, otravy)
56.
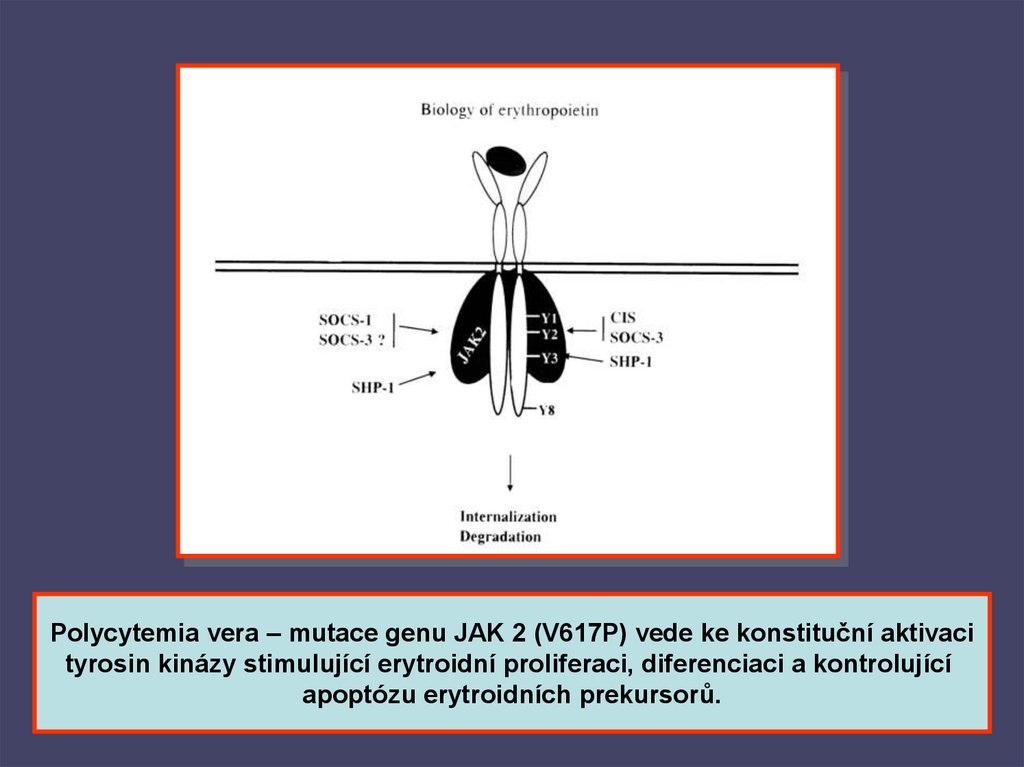
57.
Polycytemia vera – mutace genu JAK 2 (V617P) vede ke konstituční aktivacityrosin kinázy stimulující erytroidní proliferaci, diferenciaci a kontrolující
apoptózu erytroidních prekursorů.
58. POLYCYTEMIE - DIAGNOSTIKA
• krevní obraz ( ↑ HTK, ↑ trombocyty,↑ neutrofilní leuko)
• vyšetření objemu krevní masy
/krevní volum/
• autonomní růst erytroidních prekursorů
BFU-E a nízká hladina EPO
• splenomegalie
• molekulárně genetické vyšetření – mutace
JAK-2 (CALR)
59. POLYCYTEMIE - LÉČBA
• venepunkce• chemoterapie (hydroxyurea,interferon alfa)
• redukce počtu trombocytů (anagrelid)
• antiagregační a antikoagulační léčba
• alogenní transplantace krvetvorných
buněk
60. LEUKOPENIE
• kvantitativní poruchyvrozené (Kostmanův syndrom)
získané – většinou součást více
cytopenií (MDS,PNH,AA)
sekundární (systémové
choroby, chronické infekce)
• kvalitativní poruchy – syndromy
Chediak-Higashiho,Jobův.
61. LEUKÉMIE
• AKUTNÍmyeloidní a lymfoblastická
• CHRONICKÉ
chronická myeloidní leukémie
chronická myelomonocytární leukémie
chronická neutrofilní leukémie
chronická eosinofilní leukémie
chronická lymfatická leukémie → maligní
lymfomy
62. AKUTNÍ LEUKÉMIE
• DIAGNOSTIKA :sternální punkce, resp. trepanobiopsie
- morfologie a cytochemie
- průtoková cytometrie
- cytogenetika
- molekulární genetika
- lumbální punkce
63.
Akutní myeloidní leukémie s opakujícími se genetickými změnamiAML s balancovanými translokacemi/inverzemi :
AML s t(8;21)(q22;q22); RUNX1-RUNX1T1
AML s inv(16)(p13.1;q22) nebo t(16;16)(p13.1;q22); CBFB-MYH11
APL s t(15;17)(q22;q12); PML-RARA
AML s t(9;11)(p22;q23); MLLT3-MLL
AML s t(6;9)(p22;q34); DEK-NUP214
AML s inv(3)(q21;q26.2) nebo t(3;3)(q21;q26.2); RPN1-EVI1
AML /megkaryoblastová/ s t(1;22)(p13;q13); RBM15-MKL1
AML s mutacemi genů ( zatím dále nerozděleno)
Akutní myeloidní leukémie s myelodysplastickými změnami
Akutní myeloidní leukémie po léčbě (cytotoxickými látkami a/nebo radioterapií)
Akutní myeloidní leukémie jinak blíže nespecifikovaná
AML s minimální diferenciací
AML bez známek maturace (nevyzrávající)
AML se známkami maturace (vyzrávající)
Akutní myelomonocytární leukémie
Akutní monoblastická a monocytární leukémie
Akutní erytroidní leukémie
Akutní megakaryoblastická leukémie
Akutní basofilní leukémie
Akutní panmyeloza s myelofibrozou (akutní maligní myelofibroza)
Myeloidní sarkom
Myeloidní proliferace při Downově syndromu
64.
65.
66.
67.
68.
B lymfoblastická leukémie(lymfom) jinak nespecifikovanáB lymfoblastická leukémie (lymfom) a opakujícími se genetickými abnormalitami
B lymfoblastická leukémie (lymfom) s t(9;22)(q34;q11.2); BCR-ABL1
B lymfoblastická leukémie (lymfom) s t(v;11q23); s přeskupením MLL genu
B lymfoblastická leukémie (lymfom) s t(12;21)(p13;q22); TEL-AML1 (ETV6-
RUNX1)
B lymfoblastická leukémie (lymfom) s hyperdiploidií
B lymfoblastická leukémie (lymfom) s hypodiploidií (hypodiploidní ALL)
B lymfoblastická leukémie (lymfom) s t(5°14)(q31;q32); IL3-IGH
B lymfoblastická leukémie (lymfom) s t(1;19)(q23;p13.3); E2A-PBX1 (TCF3PBX1)
T lymfoblastická leukémie (lymfom)
69.
70. AKUTNÍ LEUKÉMIE - LÉČBA
• Kombinovaná chemoterapie(anthracykliny, cytosin arabinosid,
etoposid, kortikoidy,methotrexat).
Indukční a konsolidační léčba
• Transplantace krvetvorných buněk
- alogenní příbuzenská a nepříbuzenská
- autologní
Indikace podle prognostických skupin,
věku, klinického stavu, dostupnosti
dárce.
71. TRANSPLANTACE KRVETVORNÝCH BUNĚK
• dle dárce :- autologní
- syngenní
- alogenní, HLA kompatibilní dárce
příbuzenský, nepříbuzenský.
haploidentický dárce
• dle přípravného režimu :
- se standardním režimem
- s redukovaným režimem
72. TRANSPLANTACE KRVETVORNÝCH BUNĚK
• autologní :- zničení residuální nádorové populace
vysokodávkovanou chemoterapií
s následným převodem autologních
krvetvorných buněk ke zkrácení aplastické
fáze
• alogenní :
- zničení residuální nádorové populace
- nahrazení krvetvorných buněk příjemce
dárcovskými krvetvornými buňkami
73. TRANSPLANTACE KRVETVORNÝCH BUNĚK
• přípravný režim- kombinace protinádorové chemoterapie
s imunosupresí a event. celotělovým ozářením
účel: zničení vlastní krvetvorné dřeně
zničení residuální nádorové populace
navození imunotolerance k přijetí štěpu
• převod štěpu
kostní dřeň či periferní kmenové buňky
• potransplantační období
- imunosuprese nutná k přihojení štěpu
- podpůrná léčba ( transfuze, antibiotika, parenterální
výživa )
74. TRANSPLANTACE KRVETVORNÝCH BUNĚK
• INDIKACE :- nádorová onemocnění krvetvorby
akutní a chronické leukémie, lymfoproliferace
- aplastická anémie, MDS, PNH
- vrozené poruchy krvetvorby
thalasemie, srpkovitá anémie, Fanconiho
anemie, Diamond Blackfanova anemie,
mukopolysacharidosy, střádací choroby,
osteopetrosa.
- solidní nádory
vysokodávkovaná chemoterapie s podporou
autologních krvetvorných buněk
75. CHRONICKÁ MYELOIDNÍ LEUKÉMIE
76.
77. CML - DIAGNOSTIKA
• KREVNÍ OBRAZ ( leukocytoza sposunem doleva, basofilie, eosinofilie)
• STERNÁLNÍ PUNKCE
• CYTOGENETICKÉ VYŠETŘENÍ
( průkaz Ph chromosomu )
• MOLEKULÁRNÍ GENETIKA A MOLEK.
CYTOGENETIKA ( průkaz fuzního genu
BCR/ABL).
78. Léčebné možnosti u CML
• Konvenční chemoterapie(hydroxyurea, busulfan, cytosin arabinosid)
• Imunoterapie - interferon alfa (INF) v monoterapii
nebo kombinace s ARA-C, hydroxyureou
• Alogenní transplantace hemopoetických kmenových
buněk (SCT)
• Autologní transplantace hemopoetických kmenových
buněk
• STI - inhibitor BCR/ABL tyrozinkinázové aktivity
(imatinib mesylate - Glivec®, dasatinib,nilotinib)
79. Myelofibrosa
celulárníretikulinová
kolagenní
osteo-
fáze
fibróza
fibróza
skleróza
B. Bain, Bone Marrow Pathology, Blackwell 2002
80. MYELOFIBROSA – LÉČBA
• Podpůrná léčba (transfuze,kortikoidy)• Chemoterapie (Litalir)
• Ozáření sleziny, event.splenektomie
• Alogenní transplantace krvetvorných
buněk.
• Podávání inhibitorů JAK-2
81. MALIGNÍ LYMFOMY
• NEHODGKINSKÉ LYMFOMYB řada – prekursorové, zralé (periferní) –
B CLL, B PLL, lymfoplasmocytární lymfom
splenický lymfom, hairy cell leukémie,
extranodální MALT marginální, nodální marginální
folikulární lymfom, mantle cell lymfom,
DBLC lymfom, Burkittův lymtom.
Plasmocytom
T a NK řada – prekursorové, zralé (periferní)
• HODGKINŮV LYMFOM
82. MALIGNÍ LYMFOMY – DG.
• HISTOLOGICKÉ VYŠETŘENÍ (uzlina)• KREVNÍ OBRAZ, STERNÁLNÍ PUNKCE
TREPANOBIOPSIE DŘENĚ
• BIOCHEMICKÉ VYŠETŘENÍ (imunoELFO,
beta2mikroglobulin,CRP,LD,TK).
• STÁŽOVÁNÍ :
CT, NMR, PET-CT, Ga scintigrafie.
• MOLEKULÁRNĚ GENETICKÉ VYŠETŘENÍ :
bcl-2/IgH, bcl-6, c-myc/IgH
83.
84.
85.
86. MALIGNÍ LYMFOMY - LÉČBA
• Nehodgkinské lymfomy o nízké malignitě :- observace, lokalizovaná radioterapie
- kombinovaná chemoterapie (COP.CHOP,FC,
R-COP,R-CHOP)
- autologní či alogenní transplantace
• Nehodgkinské lymfomy o vysoké malignitě :
- kombinovaná chemoterapie (CHOP,R-CHOP,
DHAP,ESHAP, ALL like léčba)
- autologní či alogenní transplantace
87.
88. HODGKINŮV LYMFOM - LÉČBA
• Ozáření( jen pro časná lokalizovaná stádia )
• Kombinovaná chemoterapie
( MOPP,ABVD )
• Autologní a alogenní transplantace
89.
90. MNOHOČETNÝ MYELOM
91. MNOHOČETNÝ MYELOM - LÉČBA
• Vždy v prvé řadě vyloučit pacienty asymptomatické či minimálněsymptomatické, u kterých je indikována pouze podpůrná léčba
• 1. Melfalan/(prednison) - stále platná volba pro nemocné starší a
neindikované k SCT
• 2. Cyklofosfamid(+- kombinace)- účinná terapie, méně myelotoxická
• 3. VAD - ideální cytoredukční strategie před SCT, považována za standard
(60-70% RR)(VAMP, C-VAMP, DVD, Z-Dex, MOD-srovnatelné alternativy)
• 4. Dexamethason/kortikoidy v monoterapii- účinná metoda indukční terapie
(40-50%RR) při kontraidikacích chemoterapie
• 5. Thalidomid + Dexamethason (64% RR Mayo) nově uznávaná metoda
indukční léčby
• 6. Velcade (bortesomib) indikován v léčbě relapsů po chemoterapii a po
transplantaci, nově v kombinaci v primoléčbě
• 7. Revlimid (lenalidomid)- v kombinaci v léčbě relapsů (60% efekt)
• 8. Transplantace krvetvorných buněk autologní (1x či 2x), alogenní
• 9. Pomalidomid, carfilozumib - nové přípravky u relabujících nemocných
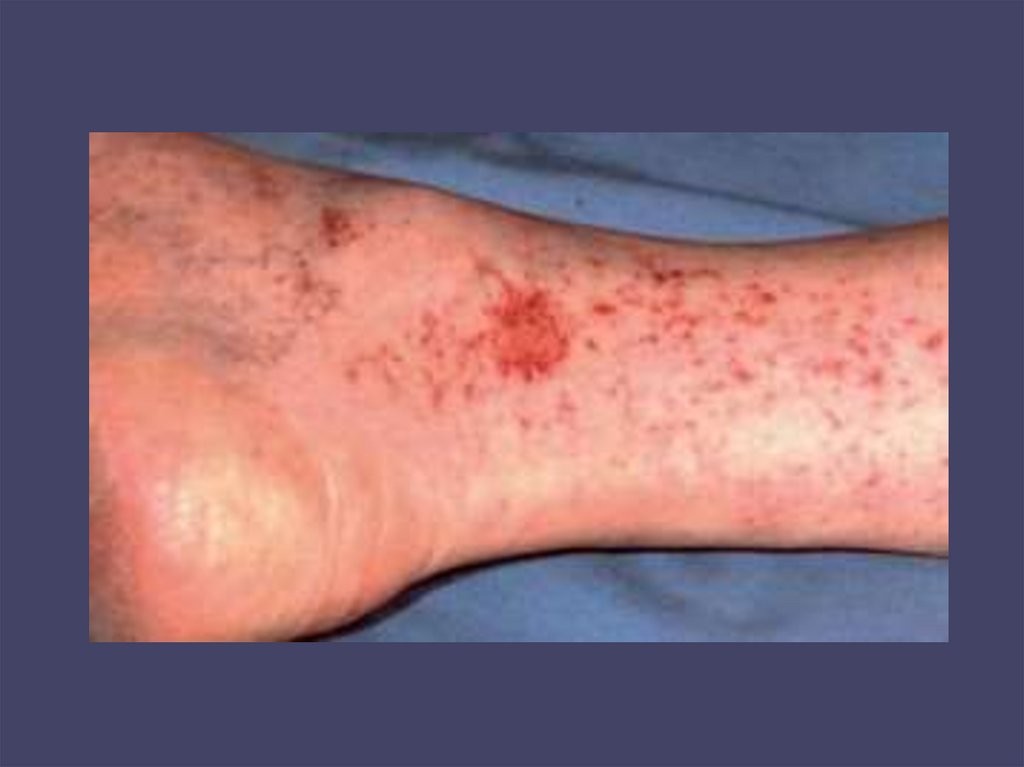
92. TROMBOCYTOPENIE
• Vrozenéporuchy adheze, aktivace, sekrece, agregace,
prokoagulační aktivity.
• Získané
a) zvýšená destrukce
imunitní – autoimunní, aloimunní
neimunitní – zvýšená konsumpe (TTP,HUS)
zvýšená destrukce
b) snížená produkce –AA.MDS,PNH,infiltrace dřeně
c) porucha distribuce (hypersplenismus)
93.
94. TROMBOCYTOPENIE – Dg.
• KREVNÍ OBRAZ, STERNÁLNÍ PUNKCE• KOAGULAČNÍ VYŠETŘENÍ
(včetně DD,FDP,AT III, lupus antikoag.)
• AGREGACE TROMBOCYTŮ
• TESTY NA SYSTÉMOVÉ CHOROBY
(ANF,anti ds-DNA,ENA,ACLA)
• CELKOVÉ VYŠETŘENÍ (včetně USG,CT)
95.
96. TROMBOCYTOPENIE - LÉČBA
• Vrozené – substituce, podáváníkoagulačních přípravků, transplantace
• Získané imunitní
kortikoidy, vysoké dávky i.v.IgG,
imunosupresiva, rituximab, agonisté
TPO receptoru
• Získané neimunitní
dle příčiny – TTP: plasmaferesa,
kortikoidy, DIC – heparin.
97. ESENCIÁLNÍ TROMBOCYTÉMIE
Dg.:KREVNÍ OBRAZ
A STERNÁLNÍ
PUNKCE + BIOPSIE
trombocytoza,
megakaryocytární
proliferace bez
fibrozy dřeně.
MOLEK.GENETIKA
mutace JAK2
98. ANAGRELID (THROMBOREDUCTIN®)
• Selektivní cytoreduktivní látka snižujícípočty trombocytů.
• Indikace: esenciální trombocytémie či
další myeloproliferativní onemocnění
provázené trombocytémií.
• Nemocní s počtem trombocytů > 1000 x
10 ꝰ/l, či nemocní s nárůstem počtu
trombo > 300 x 10 ꝰ/l během 3 měsíců či
nemocní s rizikovými faktory při
trombo < 100 x 10 ꝰ/l.
99. PORUCHY KOAGULAČNÍCH FAKTORŮ
• Nedostatek plasmatických faktorů :• Hemofilie A (deficit F VIII.)
• Hemofilie B (deficit F IX.)
• Von Willebrandova choroba (deficit vWF)
• Projevy: krvácení do kloubů
plošné krvácení
Léčba : substituce
100.
Látky nahrazující chybějící proteiny s důležitoufunkcí – koagulační faktory u hemofilie
vysoce čištěné či rekombinantní faktory :
výrazná redukce komplikací vyplývajících
z opakovaného krvácení (zejména postižení kloubů)
v důsledku profylaktického podávání dětským
nemocným a důsledné
léčby vzniklého
krvácení.
101. TROMBOFILNÍ STAVY
• Mutace faktorů krevního srážení :• F V. – Leidenská mutace
• F II. - mutace genu pro protrombin
• Projevy: trombotické komplikace ( v
mladším věku)
Léčba : antikoagulace