
")




































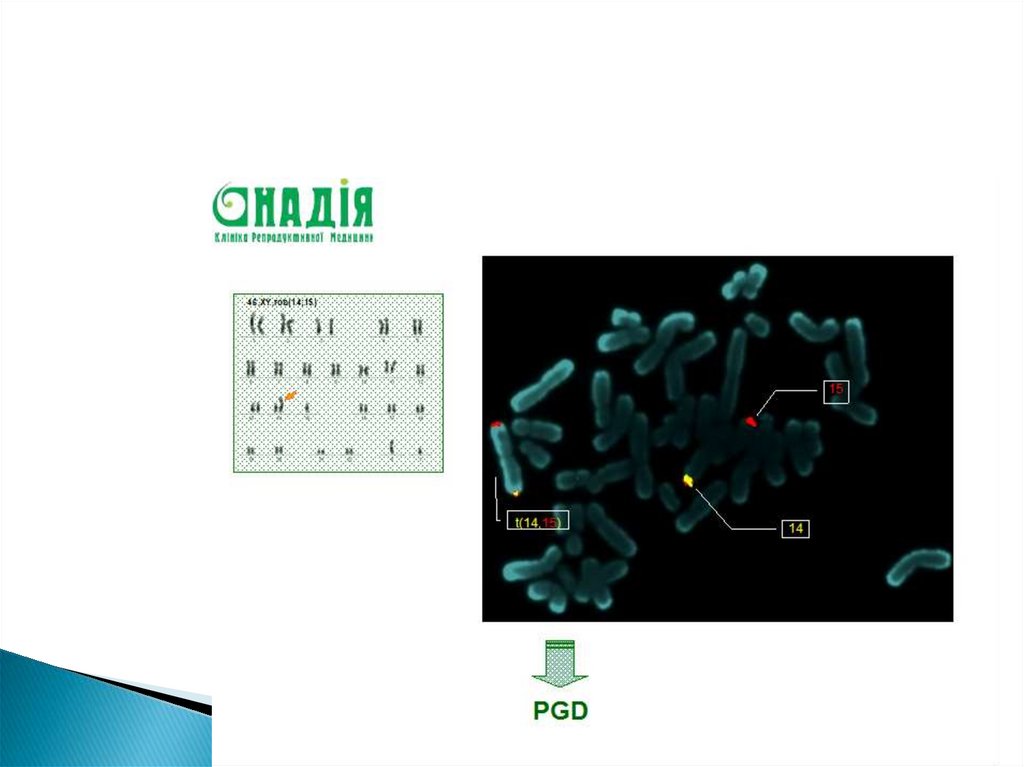
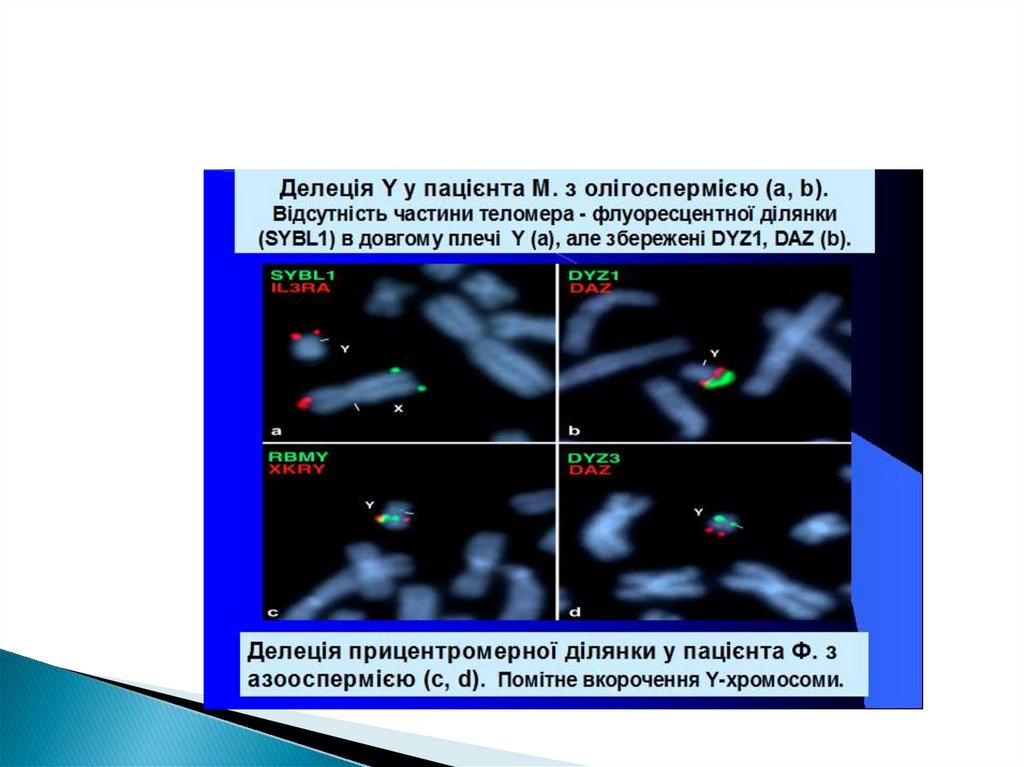













")
")
")




")







")
")







")





, синдром Мартина-Бела")






 Медицина
МедицинаПохожие презентации:










Хромосомные болезни. Количественные и структурные аномалии
1. Хромосомные болезни
Количественныеи
структурные
аномалии
хромосом
2. Хромосомные болезни (хромосомные синдромы)
-это группа врожденных наследственныхзаболеваний, вызванных числовыми или
структурными аберрациями хромосом,
видимыми в световом микроскопе и
характеризующихся множественными
врожденными пороками развития
3. Числовые изменения хромосом:
Полиплоидия- увеличение количествахромосом, кратное гаплоидному набору
Анеуплоидия – увеличение или уменьшение
количества хромосом в наборе, не кратное
гаплоидному.
Нарушения обоих типов возникают как
следствие ошибок в мейозе и митозе.
4. Структурные нарушения хромосом
Инсерция – вставка генетическогоматериала вследствие его перемещения
Делеция – отсутствие участка хромосомы
в одной из гомологичных хромосом
(частичная моносомия в этом участке)
Инверсия – изменение линейной
последовательности генов на хромосоме
вследствие разворота на 180 с
последующим воссоединением разрывов
с другими частями хромосомы
5. Структурные нарушения хромосом
Дупликация – удвоение генетическогоматериала (участка хромосомы),
возникает из-за неравномерного
кроссинговера между сестринскими
хроматидами и сопровождается
патологическими фенотипами
Кольцевая хромосома – делеция концевых
участков хромосомы с двух сторон,
приводящая к слипанию оставшихся
концов хромосомы
6. Структурные нарушения хромосом
Дицентричная хромосома имеет 2центромеры которые появляются
вследствие изохроматидного разрыва. При
этом теломерный акроцентрический регион
теряется, а сестринские хроматиды
объединяются
7. Структурные нарушения хромосом
Изохромасома – содержит или удвоенноекороткое или удвоенное длинное плечо.
Формируется в мейозе из-за разрыва
хромосомы в области центромеры поперек,
а не обычного деления вдоль.
8. Структурные нарушения хромосом
Транслокации хромосомвозникают
после разрыва и обмена материалом
между двумя или несколькими
хромосомами. Выделяют:
Сбалансированные реципрокные (обмен
участков двух негомологичных хромосом) не
связаны с потерей генетического материала,
фрагменты хромосом воссоединяются в новых
комбинациях, кариотип 46. Могут приводить к
умственной отсталости, фенотипическим
проявлениям
Несбалансированные транслокации
9. Структурные нарушения хромосом
Робертсоновские транслокации – обменмежду двумя акроцентрическими
хромосомами с утратой ими коротких
плеч, что приводит к образованию одной
метацентрической хромосомы вместо
двух акроцентрических. Носители имеют
моносомию по коротким плечам
акроцентрических хромосом, но они
здоровы, т.к. утрата коротких плеч
компенсируется работой таких же генов в
других 8 акроцентрических хромосомах.
10. Робертсоновские транслокации
У носителей робертсоновскихтранслокаций может образовываться 6
типов гамет, в которых нулисомные гаметы
приведут к моносомии по аутосомам в
зиготе, но такие зиготы не развиваются.
11. Хромосомные аберрации
Мозаичные формы ХБ связаны снеправильным делением клеток на
различных стадиях эмбрионального
развития. Характеризуются более легким
течением, отсутствием отдельных
симптомов при наличии доминирующих
признаков патологии индивидуально
вовлеченной хромосомы, что связано с
присутствием клона нормальных клеток,
частично компенсирующих генный
дисбаланс аберрантных клеток.
12. Геномный импритинг
- это процесс,который обеспечивается
специфическим метилированием
цитозиновых оснований ДНК и включает
транскрипцию генов
Может проявляться не только на уровне
гена или кластера генов. Он может
затрагивать целую хромосому и даже
геномы
13. Геномный импритинг
ГИ- это специфические структурно-молекулярные изменения отдельных
участков хромосом, происходящие во
время образования мужских и женских
половых клеток, что приводит к различной
степени проявления (экспрессии)
гомологичных генов у потомства.
14. Геномный импритинг
В участках генома,подверженныхимпритингу, обнаруживается
моноаллельная (а не биаллельная)
экспрессия генов, т.е. если импритирован
материнский ген, экспрессируется только
отцовский, и наоборот.
15. Геномный импритинг
Известно около 30-ти генов в геномечеловека, подверженных импритингу и
имеющих тканеспецифическую
моноаллельную эксперессию, а также 3
кластера генов, локализованных в
хромосомах 7q32, 11p15, 15q 11.2-13
(опухоли, синдром Прадера -Вилли,
Ангельмана)
16. Последствия разных вариантов геномного импритинга
Триплоды, у которых материнский иотцовский гаплоидные наборы
представлены в соотношении 1:2
характеризуются задержкой развития
(андроид- большая голова плода,
маленькое веретенообразное тело )и
частичным пузырным заносом, а
триплоды с соотношением 2:1
отличаются резкой гипоплазией
плаценты(эмбрион и плод не
развиваются)
17. Последствия разных вариантов геномного импритинга
При удвоении отцовского гаплоидногонабора и отсутствии материнского ранний эмбриогенез нормальный. Далее
ткани собственно эмбриона не
формируются.бурно разрастается
трофобласт с образованием полного
пузырного заноса
Следовательно, развитие плаценты
обеспечивается геномом отца, а раннее
развитие эмбриональных структур
обеспечивается геномом матери
18. Однородительская дисомия
ОРД- число хромосом нормальное, ноодна из пар представлена хромосомами
одного и того же родителя, вследствие
нерасхождения хромосом во время
мейоза.
Если в геноме одного из родителей, чья
хромосома имеет дисомию, есть
рецессивный признак, он удвоится,
произойдет гомозиготизация и разовьется
рецессивная наследственная болезнь
19. Болезни геномного импритинга
Эффект ОРД может выразиться вопределенном синдроме:
при ОРД 6-й хромосомы отцовского
происхождения может развиться
транзиторный неонатальный сахарный
диабет
При ОРД 7-й хромосомы (материнской)синдром Рассела-Сильвера или
внутриутробная гипотрофия
20. Болезни геномного импритинга
ОРД 11-й хромосомы (отцовской) связанас синдромом Беквита-Видемана
Синдром Прадера-Вилли с материнским
ГИ (20-25% случаев) характеризуется
ожирением, мышечной гипотонией,
гипогонадизмом, умственной
отсталостью, низким ростом,
акромикрией. В 75-85% случаев связаны с
делецией сегмента 15q11- q13 отцовской
хромосомы
21. Болезни геномного импритинга
Синдром Энгельмана («счастливой куклы») в70% случаев обусловлен делецией 15q11q13 материнской хромосомы и
характеризуется глубокой умственной
отсталостью, судорожными «кукольными»
движениями. Лишь в 2% случаев ОРД 15
имеет отцовское происхождение.
22. Болезни геномного импритинга
ГИ имеет отношение к опухолевому росту.Некоторые эмбриональные опухоли
возникают в результате потери
гетерозиготности материнского
происхождения по хромосоме 11 и 13
(опухоль Вильямса, остеосаркома,
ретинобластома).
23. Клинические проявления ХБ
Чернепно-лицевые дизморфииВрожденные пороки развития внутренних
органов
Нарушения роста и развития
Задержка психомоторного развития
Нарушение функций нервной, эндокринной
и иммунной систем
24. Клиническая картина ХБ зависит от :
Генотипа организмаИндивидуальности вовлеченной в аберрацию
хромосомы или ее участка (набор генов)
Типа аберрации
Размера недостающего (при делеции) или
избыточного (при частичной трисомии)
материала
Степени мозаичности организма по аберрантным
клеткам
Условий среды
Стадии онтогенеза или возраста больных
25. Диагностические признаки ХБ
1-я группа – комплекс признаков,позволяющих заподозрить хромосомную
аномалию (физическое недоразвитие,
дизморфии мозгового и лицевого
черепа), косолапость,клинодактилия
мизинцев,ряд пороков внутренних
органов
26. Диагностические признаки ХБ
2-я группа –комплекс признаков, которыевстречаются при определенных
хромосомных заболеваниях:
- при трисомии 18- долихоцефалия,
флексорное положение кистей, короткий
и широкий палец (большой) стопы
- при трисомии13 – расщелина верхней
губы и неба, флексорное положение
кистей, косоглазие, дефект скальпа
27. Диагностические признаки ХБ
3-я группа – признаки, характерные толькодля данной патологии - кошачий крик при
5р-, алопеция при синдроме 18р-.
ХБ характеризуются клиническим
полиморфизмом, при этом более тяжелая
манифестация наблюдается при
аутосомных аберрациях
28. Патологические эффекты ХА
ХА вызывают нарушение генетическогобаланса. Проявляются на всех этапах
онтогенеза (проэмбриональном,
эмбриональном, фетальном и
постнатальном) в виде:
Летальности
Врожденных пороков развития
29. Патологические эффекты ХА
Летальный эффект проявляется на стадиизиготы
Суммарный вклад ХА во внутриутробную
гибель составляет 45%
У 2-4 нед ХА обнаруживаются в 60-70%,
в 1 триместре беременности у 50%
абортусов, во - втором – у 25-30%, у
плодов, погибших после 20 нед – в 7%
случаев.
30. Патологические эффекты ХА
ХА в соматических клетках могут вызыватьразличные последствия:
остаться нейтральными для клетки
обусловить гибель клетки
активизировать деление клетки
изменить функцию
являться причиной злокачественного
роста
(tr 9-22 вызывает миелолейкоз)
31. Патологические эффекты ХА
Облучение и химические мутагеныиндуцируют ХА
ХА накаливаются с возрастом
Критический возраст по ХА:
- период новорождённости
- подростковый период (16 лет)
- старческий
32. Патогенез ХБ
Патогенез остается неяснымВыделяют три типа генетических
эффектов:
специфические
полуспецифические
неспецифические
33. Патогенез ХБ
Специфические связаны с изменениемчисла структурных генов , кодирующих
синтез белка. Так, при трисомии 21
обнаружено у 50% больных повышение
активности надпероксиддисмутазы, гена
локализованного на хромосоме 21
Но ни в одном случае не обнаружено
белка-маркера при ХБ
34. Патогенез ХБ
Полуспецифические эффектыобусловлены изменением числа генов:
рибосомные и транспортные РНК,
гистоновые и рибосомные белки,
сократительные белки актина и тубулина,
которые в норме контролируют ключевые
этапы метаболизма клетки, процессов ее
деления, межклеточных взаимодействий.
Фенотипические эффекты дисбаланса
этих генов пока неизвестны.
35. Патогенез ХБ
Неспецифические эффекты ХА связывают сизменением содержания гетерохроматина в
клетке
Патогенез ХБ развертывается в раннем
внутриутробном развитии и продолжается в
постнатальном периоде
36. Диагностика ХБ
Цитогенетический метод – позволяетсудить о кариотипе больного – числе и
структуре хромосом
Молекулярно-цитогенетический метод –
использование различных ДНК-зондов,
позволяющих маркировать отдельные
хромосомы. ДНК-зонды представляют
собой клонированные фрагменты генома
человека
37. Диагностика ХБ
Флюоресцентная гибридизация in situ(FISH- Fluorescence In Situ Hubridization) –
включает применение специально
приготовленных
(флюоресцирующих)ДНК-проб для
выявления генетических дефектов на
хромосомном уровне и проведения
молекулярно-цитогенетической
диагностики с использованием
флюоресцентного микроскопа.
38.
39.
40.
41. Трисомия хромосомы 21
Синдром ДаунаВстречаемость
1 : 700-800 – 1000
новорожденных
Кариотип
Полная форма 47,хх + 21 , 47, ху + 21
Транслокационная 46, хх,tr(13-21) (4%)
Мозаицизм: 47,ху + 21/ 46, ху
(2%)
42. Трисомия хромосомы 21
Частота СД не имеет какой-либовременной, этнической или
географической разницы
Частота СД зависит от возраста матери и
в меньшей степени отца
Частота в возрасте матери 45 лет
составляет 1:12
Риск увеличивается если отцу более 50, а
матери 40 лет
43. Трисомия хромосомы 21
Соотношение мальчиков и девочек –1:1
критический сегмент, отвечающий за
фенотипические проявления расположен в
участке 21q22
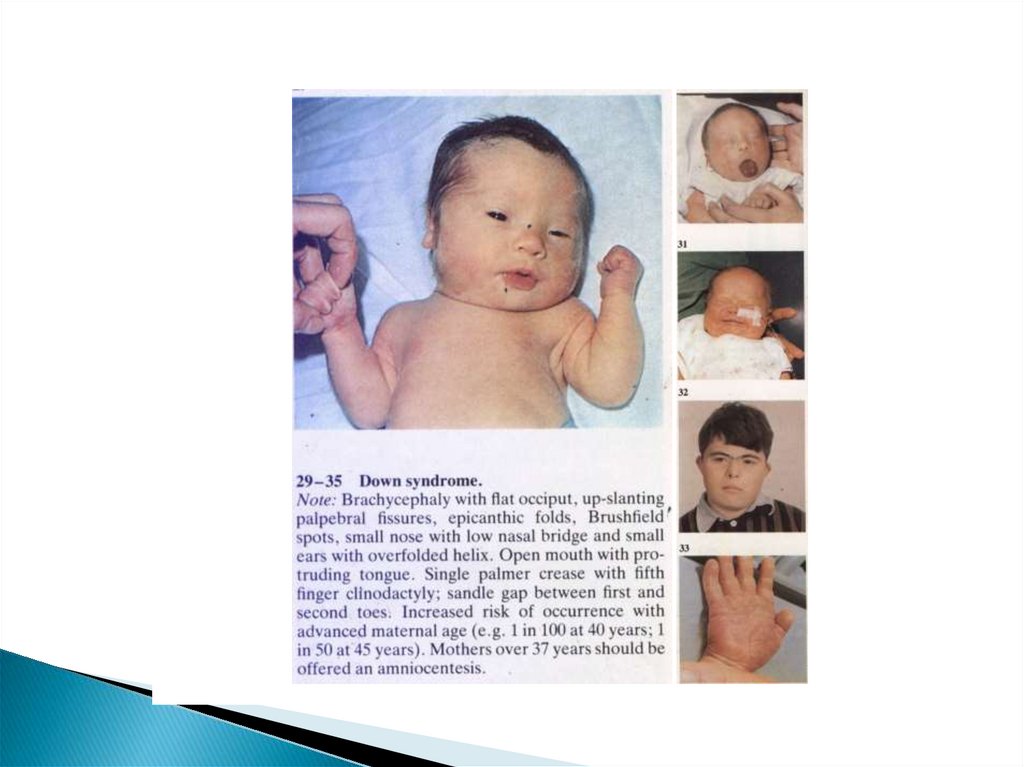
44. Клинические проявления СД
Основные признаки:Умственная отсталость
Мышечная гипотония
Брахицефалия
Эпикант и монголоидный разрез глазных
щелей
Катаракта
45. Клинические проявления СД
Пятна Брушвильда (белого цвета очаги награнице наружной и средней трети
радужки)
Косоглазие, реже помутнение роговицы и
хрусталика
Толстые губы
Утолщенный язык с
бороздами(«складчатый язык»)
Плоская спинка носа
46. Клинические проявления СД
Узкое небоДеформированные ушные раковины
Избыток кожи на шее
Разболтанность суставов
Поперечная линия ладони («обезьянья
линия»)
Клинодактилия мизинца
47. Клинические проявления СД
Дети рождаются в срок с умеренновыраженной пренатальной гипоплазией
(масса тела при рождении 3200-3500г)
Аномалии внутренних органов:
Пороки сердца (дефекты перегородок в
сочетании с аномалиями крупных сосудов)
48. Клинические проявления СД
Желудочно-кишечный тракт : атрезия илистеноз 12п.к., реже атрезия прямой
кишки, заднего прохода, пищевода
Мочевая система: гипоплазия или
дисплазия почек, кортикальные кисты,
гидроуретер, гидронефроз
Мозга: гипоплазия верхней височной
извилины, укорочение ножек мозга и
моста, вентральное смещение мозжечка
49. Клинические проявления СД
Нарушение психической деятельности:хуже выполняют вербальные задания
Дети послушны, подражают, повышена
внушаемость
Лечение: успешно разрабатываются
принципы коррекции, позволяющие
социально адаптировать их к жизни.
50.
51. Трисомия хромосомы 13
Синдром ПатауЧастота встречаемости 1 : 6000
Соотношение полов 1 : 1
Средний возраст матерей – 32, отцов – 34
Дети рождаются с пренатальной
гипоплазией (масса тела до 2500)
Осложнение беременности – многоводие
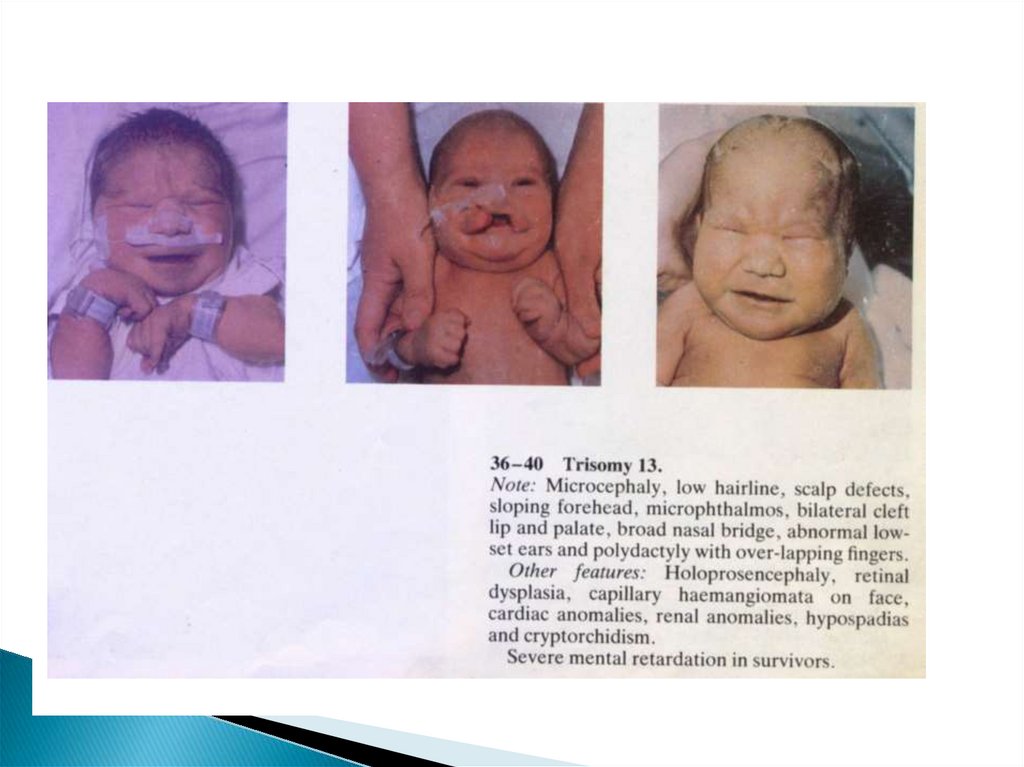
52. Клинические проявления
МикроцефалияГипертелоризм
Тригоноцефалия
Микрофтальмия
Низкий скошенный
Колобома радужки
лоб
Широкий нос с
запавшим
переносьем
Узкие глазные
щели
Помутнение
хрусталика
Расщелина
верхней губы и
неба
Полидактилия
кистей и стоп
53. Клинические проявления
Флексорноеположение кистей
«стопа качалка»
Крипторхизм
Гипоспадия
Рипоплазия
полового члена
Удвоение матки и
влагалища
Пороки
ц.н.с.(аринэнцефа
лия,
голопроэнцефалия
, аплазия и
гипоплазия
мозолистого тела,
гипоплазия
мозжечка, аплазия
и гипоплазия
зрительных
нервов)
54. Основные признаки СП
Расщелина губы и небаДефекты скальпа, которые имеют круглую
форму до 1-1,5 см в диаметре с гладким
дном, представленным апоневротическим
шлемом
Продолжительность жизни – чаще
умирают до 1 года из-за пороков не
совместимых с жизнью
Дети, которые живут до 2-3 лет, страдают
глубокой идиотией
55.
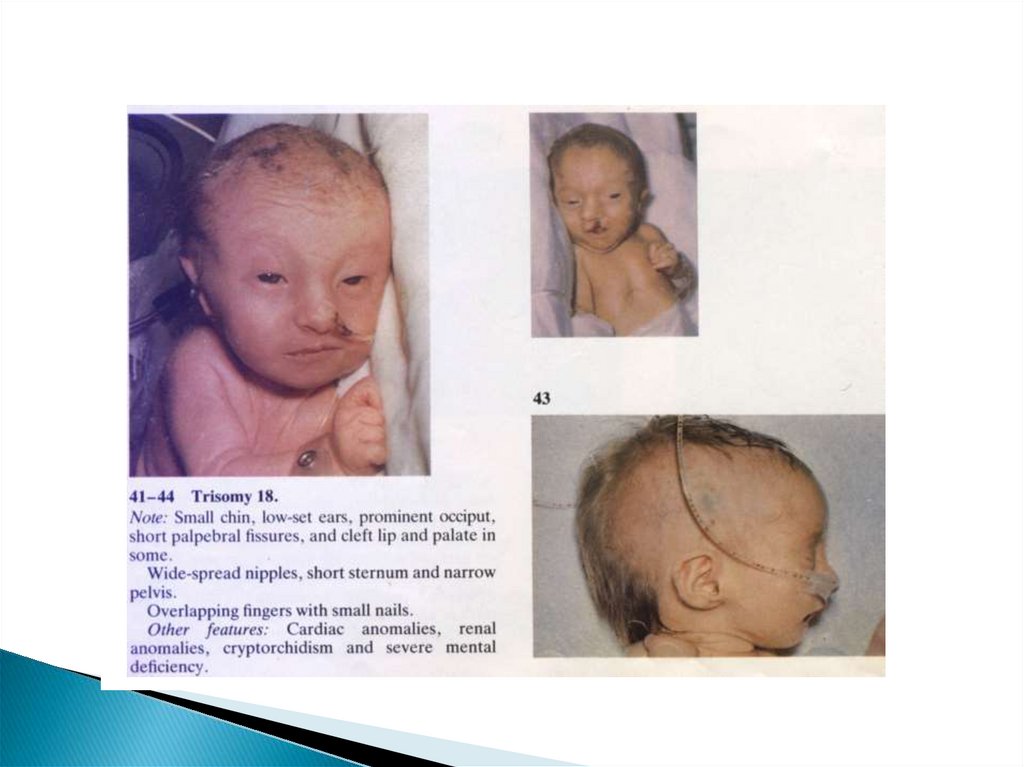
56. Трисомия 18 (Синдром Эдварса)
Частота 1: 7000Девочки страдают значительно чаще, чем
мальчики
Дети рождаются с пренатальной
гипоплазией (масса тела до 2200), от
доношенной беременности, осложненной
многоводием
Фенотипические проявления характерны и
многообразны
57. Клиника трисомии 18 (СЭ)
ДолихоцефалияМикрофтальмия
Низкорасположен.
ушные раковины
Высокое небо
Микрогнатия
Микростомия
Гипертрофия клитера
гипоспадия,
крипторхизм
Аномалии
конечностей (кожная
синдактилия и др.)
Дефекты внутренних
органов
Дефекты развития
ц.н.с. (гипоплазия
мозжечка и
мозолистого тела)
Спиномозговые грыжи
Дети умирают на
первом году жизни
58.
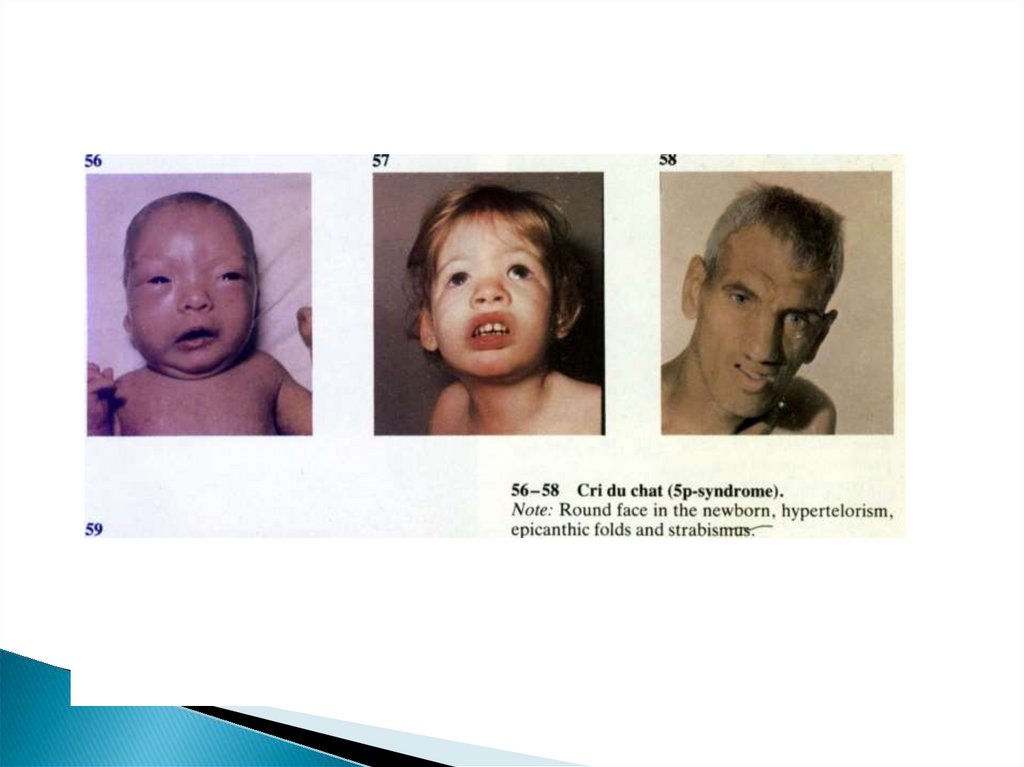
59. Моносомия короткого плеча хромосомы 5 (синдром «кошачьего крика» «5р-»)
Частота 1 : 45000 рожденийЦитогенетический полиморфизм- от утраты
трети до половины длины короткого плеча
хромосомы, реже всего плеча
Редко встречается кольцевая хромосомаr(5), изредка мозаицизм
60. Синдром «кошачьего крика»
Нет корреляции частоты рождения детейс синдромом и возрастом родителей
Продолжительность жизни не установлена
Дети рождаются с массой тела 2500
Генетический прогноз в семье зависит от
цитогенетических данных родителей, при
сбалансированных транслокациях с
участием хромосомы 5 он высок
61. Клиника синдрома 5р-
Характерный признак – специфическийплач (крик кошки), что связано с
изменением гортани (сужение, мягккость
хрящей, отечность или необычная
складчатость слизистой, уменьшение
надгортанника)
Отставание в умственном и физическом
развитии
микроцефалия
62. Синдром кошачьего крика
Низкорасположенные
деформированные
ушные раковины
Микрогения
Лунообразное
лицо
Эпикант
косоглазие
Гипертелоризм и
антимонголоидны
й разрез глазных
щелей
Гипотония мышц
Может быть
атрофия
зрительного нерва
Пороки
внутренних
органов
63. С возрастом
Постепенноисчезают:
Лунообразное
лицо
«кошачий крик»
Мышечная
гипотония
Прогрессируют:
Микроцефалия
Косоглазие
Отставание
умственного и
физического
развития
64.
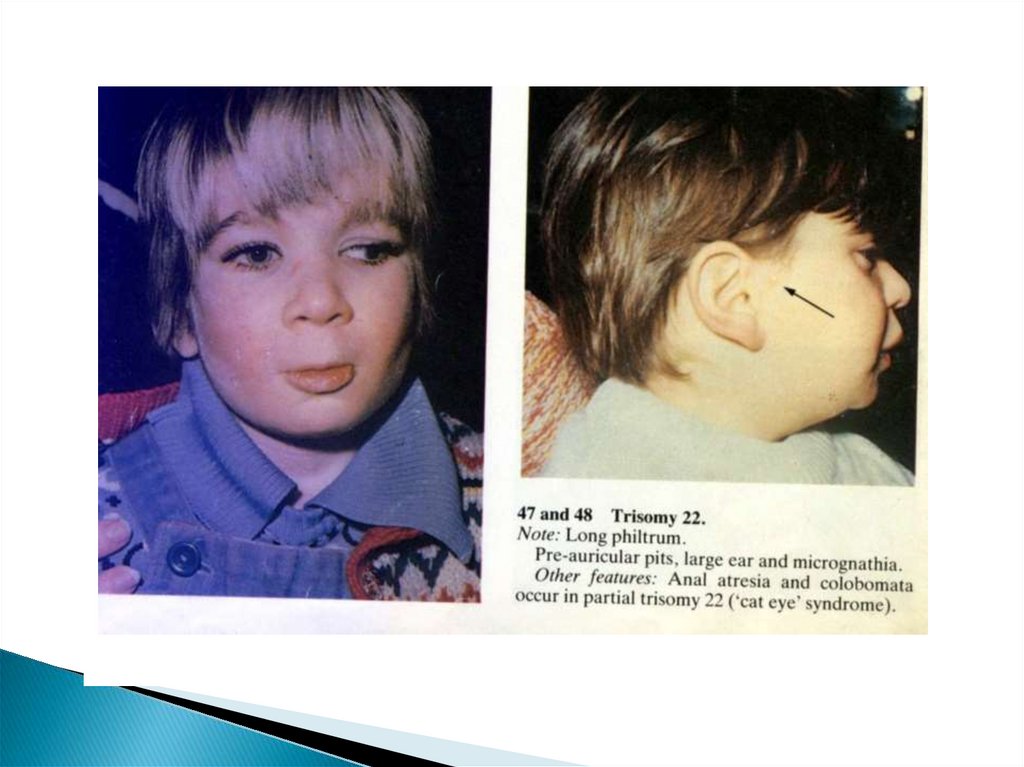
65. Трисомия хромосомы 22 (синдром «кошачьих глаз»)
Синдром назван из-за вертикальнойлокализации колобомы радужки у этих
больных
Клинически полный синдром обусловлен
трисомией участка 22pter- 22q11
В популяции встречается редко
Чаще мозаичные формы заболевания,
когда нерасхождение хромосом
происходит в митозе, а не в мийозе
66. Синдром «кошачьих глаз»
Дети рождаются с выраженнойпренатальной гипоплазией (до 2800г) при
нормальной продолжительности
беременности
Постоянные признаки заболевания:
Умственная отсталость(глубокая
олигофрения)
Задержка физического развития
Микроцефалия
Экзофтальм, колобома радужки,
косоглазие
67. Постоянные признаки заболевания
Удлиненный фильтрРасщелина верхнего неба
Микроретрогения
Преаурикулярные ямки
Гипоплазия большого пальца
Крипторхизм, гипоспади
Мышечная гипотония
Пороки сердца, почек, ж.кт (атрезия
ануса)
68.
69. Трисомия 8
Хромосома 8 – средней величинысубметацентрическая хромосома, одна из
крупных хромосом в группе G
Идентифицируется только при
дифференциальном окрашивании
Известны случаи частичных и полных
трисомий, кольцевых хромосом, а также
частичных моносомий
Описано более 100 случаев
70. Трисомия 8
Клинических различий между полнойформой и мозаичной нет
Популяционная частота не известна
Чаще преобладают лица мужского пола
Дети рождаются доношенными с
нормальной массой тела
Не отстают в росте
Умеренная задержка умственного
развития
71. Трисомия 8
Частый признак – поражение головногомозга (агенезия мозолистого тела,
гидроцефалия)
Продолжительность жизни до 12 (иногда
17) лет
72. Специфические признаки
Выпуклый лобвывернутая
нижняя губа
Аплазия
надколенника,
контрактуры
глубокие
борозды между
межпальцевыми
подушечками
Пороки мочевой
системы
(гидронефроз)
73. Трисомия 8
Трисомию 8 считают четко очерченнымхромосомным синдромом с момента
первых случаев описания
74. Аномалии половых хромосом (гоносом )
Синдромы связаны с численныминарушениями хромосом X и Y(полными
моносомиями, трисомиями, дисомиями), а
также структурными изменениями (без
участия аутосом)
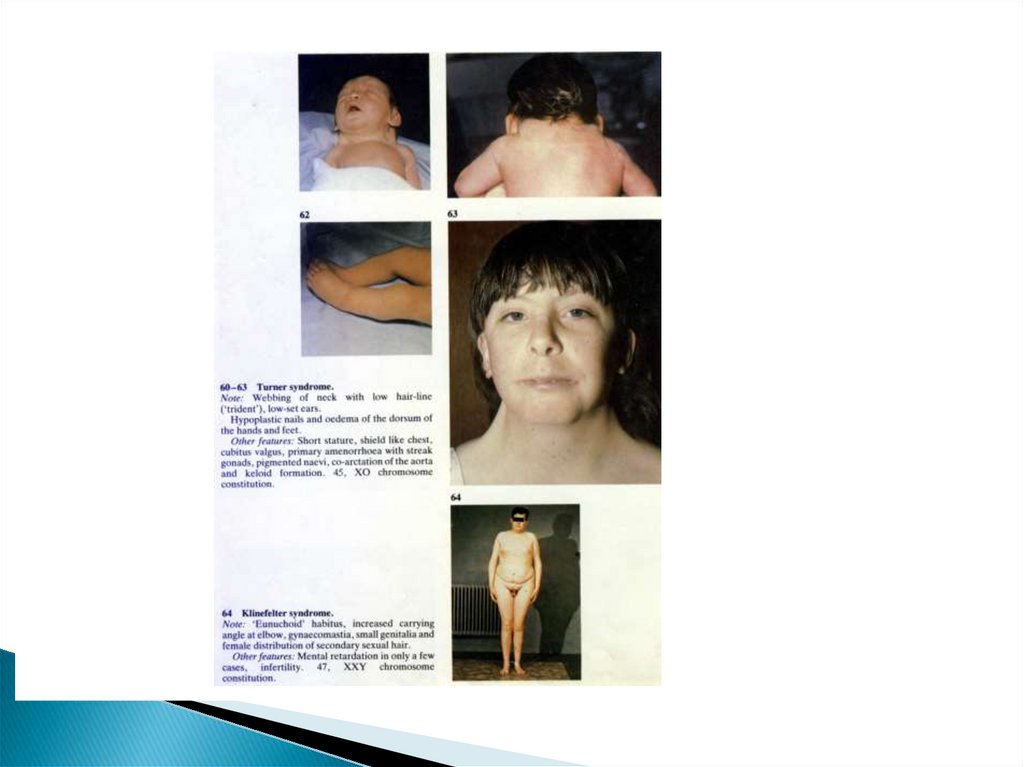
75. Моносомия Х (синдром Шерешевского- Тернера)
Единственная моносомия уноворожденных – 45, ХО
Частота в популяции – 1: 2000 девочек 1: 6000
Мозаичные формы рассматриваются
отдельно (кариотип 45,ХО/46,ХХ)
При структурных аномалиях наблюдается
клиническая картина синдрома
Шерешевского-Тернера
76. Моносомия Х
Структурные изменения хромосомы ввиде- делеции короткого или длинного
плеча хромосомы, изохромасомы,
кольцевой хромосомы.
Мозаицизм 45,Х/46,XY (2-5% от общего
числа СШТ) характеризуется широким
диапазоном клинических признаков (от
типичного СШТ до нормального мужского
фенотипа) в зависимости от соотношения
клеточных клонов.
77. Моносомия Х
Девочки со структурными и численнымианомалиями хромосомы Х клинически не
отделимы друг от друга
Клинические проявления:
- гипогонадизм: недоразвитие половых
органов и вторичных половых признаков
- врожденные пороки развития
(коарктация аорты, подковообразная
почка,гидронефроз)
- низкий рост
78. Моносомия Х
Фенотипические проявления:Антимонголоидный разрез глазных
щелей, эпикант, птоз, высокий и широкий
лоб, ретрогения, деформированные низко
расположенные ушные раковины,короткая
шея с крыловидной складкой, низкий рост
волос на шее, широкая грудная клетка,
вальгусное положение локтей,
клинодактилия мизинца.
79. Фенотипические проявления
Гонады больных представленысоединительнотканными тяжами, в
которых находятся
недифференцированные клетки или
рудименты женских гонад без
овариальных элементов, отсутствуют
примордиальные фолликулы
Дети рождаются в срок с пренатальной
гипоплазией (масса тела до 2500) и
ростом до 45 см
80. Фенотипические проявления
С возрастом у детей начинает выявлятьсянизкорослость
Вторичные половые признаки плохо
развиты
Интеллект больных приближается к
норме. Характерно недоразвитие
эмоционально-волевых проявлений:
подчиняемость, узость интересов,
бедность абстракций, незначительная
продуктивность мышления
81. Фенотипические проявления
При мозаичных формах наблюдаетсястертость клинической картины: развитие
половых признаков, регулярные
менструации
82. Лечение
В допубертатном периоде назначаютанаболические стероиды, которые ускоряют
рост
В пубертатном периоде – эстрогены, затем
– гестагены. Такая терапия способствует
появлению вторичных половых признаков,
но большинство больных остается
бесплодными.
83.
84. Дисомия Х при мужском кариотипе (синдром Клайнфельтера)
Наиболее частаягоносомная патология у
мужчин
Частота встречаемости в популяции – 1,2 на
1000 новорожденных
Одна из форм первичного мужского
гипогонадизма
Кариотип 47,ХХУ
85. Синдром Клайнфельтера
Клинические признаки появляются впрепубертатном и пубертатном периодах
Характерные признаки:
Евнухоидное телосложение с длинными
клнечностями, узкими плечами и широким
тазом
Наружные половые признаки
сформированы по мужскому типу, но
гипоплазированы. Микроорхизм – один
из важных признаков
86. Клинические признаки синдрома
ГинекомастияУмственная отсталость (в стадии легкой
дебильности), но не всегда
IQ в некоторых случаям достигает 80
Отмечается аутизм, мнительность,
склонность к алкоголизму, асоциальному
поведению
87. Клинические проявления
Признаки, не имеющие диагностическогозначения:
Катаракта, деформация ушных раковин,
прогнатизм, алопеция, поперечная
ладонная складка, сколиоз,
неврологические нарушения, пороки
сердца.
Дальнейшее увеличение числа хромосом
Х в кариотипе ведет к большей
умственной отсталости
88. Полисомия Х
Комплексное название синдрома:трисомия (ХХХ), тетрасомия Х (ХХХХ),
пентасомия Х(ХХХХХ
Трисомия Х – 47,ХХХ
Женщины имеют в основном нормальное
психическое и физическое развитие,
нормальную плодовитость, но риск
хромосомных нарушений у потомства и
спонтанных абортов повышен,
89. Полисомия Х
Дополнительная хромосома с возрастомувеличивает 2 раза риск заболевания
каким-либо психозом, склонность к
эпилепсии и шизофрении. У женщин с
тетра- и пентосомией описаны отклонения
в умственном развитии.
90. Синдром Х-ломкой хромосомы (синдром фрагидной хромосомы Х), синдром Мартина-Бела
Синдром умственной отсталости,сцепленной с ломкой хромосомой Х или
синдром FRAX
Частота в популяции 1 : 4000 среди лиц
мужского пола и 1: 6000- женского
91. Диагностически значимые признаки
Оттопыренные низко расположенныеушные раковины
Увеличение тестикул или макроорхизм,
нередко тотальное увеличение гонадмакрогенитализм с гиперпигментацией
мошонки
Основной симптом- умственная отсталость
92. Диагностические признаки
Существует вариабельностькоэффициента интеллектуальности (IQ) у
больных в пределах от 13 до75 единиц
У детей наблюдается аутизм с
гиперактивностью, двигательной
расторможенностью, неадекватным
поведением, что может быть связано со
структурными изменениями мозжечка и
дефектами лобных долей головного
мозга
93. Синдром ломкой хромосомы
Определен ген синдрома FRAX-FMR-1(Fragile X Mental retardation –1), который
включает необычные CGG-повторы,
кодирующие 30 аргининовых остатков.
CGG- повторы являются компонентом
ломкого сайта хромосомы Х и могут быть
участком, за счет которого идет
амплификация ДНК при мутации в гене
94.
95. Дисомия У
Кариотип 47,ХУУЧастота встречаемости 1,5 на 1000
новорожденных
Отсутствуют обязательные признаки
синдрома
Характерны:
Высокий рост
Психопатические черты характера,
импульсивность,отсутствие сильных
привязанностей
96. Дисомия У
Плохое владение собой по поводупримитивных эмоций
Среди физических аномалий:
макроцефалия, увеличение конечностей,
прогнатизм, выступающие надбровные
дуги, высокое небо, гипертрофия языка,
грубые черты лица
Нарушение половой дифференцировки
(крипторхизм,гипогонадизм, дисплазия
гениталий), но мужчины фертильны.