




























")







 Медицина
МедицинаПохожие презентации:






")



Мозжечковые атаксии
1. «МОЗЖЕЧКОВЫЕ АТАКСИИ»
Куратор: к.м.н ЛОКТИОНОВА А.ИВыполнила: ординатор ИДРИСОВА Э.А
2. МОЗЖЕЧКОВАЯ АТАКСИЯ
представляет собой симптомокомплекс, включающий специфическиенарушения статической и динамической моторики человека,
являющийся патогномоничным для любых заболеваний мозжечка.
Локализация очага:
патология мозжечка и/или его проводящих путей
поражение полушарий мозжечка приводит к развитию атаксии
гомолатеральных конечностей, поражение червя мозжечка – к
атаксии туловища.
3.
МОЗЖЕЧКОВАЯ АТАКСИЯA. Первичная (наследственные и
идиопатические, в основе которых лежит
нейродегенеративный процесс)
B. Вторичная (инфекции, опухоль,
гипотиреоз, алкоголизм,
паранеопластический синдром, побочное
действие лекарственных веществ и)
C. Наследственные нейрометаболические
расстройства
D. Аномалия развития
4.
Современная классификациянаследственных атаксий:
Врожденные (непрогрессирующие) мозжечковые атаксии
(синдром Жубера, различные варианты аплазии и гипоплазии
полушарий и червя мозжечка, гипоплазия гранулярного слоя
коры мозжечка и др.)
Аутосомно-рецессивные атаксии раннего возраста
• Атаксия Фридрейха
• Атаксии с нарушенной репарацией ДНК (атаксия
телеангиэктазия, синдром Коккейна, пигментная ксеродерма и
др.)
• Атаксии с окуломоторной апраксией (1-го и 2-го типов)
• Другие формы аутосомно рецессивных атаксий раннего
возраста (спастическая атаксия Шарлевуа–Сагенэ, синдром
Маринеско–Шегрена и др.)
Х-сцепленные рецессивные атаксии (синдром FXTAS и др.)
5.
Аутосомно-доминантные атаксии позднеговозраста:
• Прогрессирующие аутосомно-доминантные спиноцеребеллярные
атаксии (спиноцеребеллярные атаксии типов 1–28)
• Эпизодические атаксии (1-го, 2-го, 3-го типов)
• Другие редкие формы аутосомно доминантных атактических
синдромов
(синдром Герстманна–Штреуслера–Шейнкера и др.)
Метаболические атаксии:
• Митохондриальные атаксии (синдромы NARP, MERRF и др.)
• Атаксии при липидозах (абеталипопротеинемия, болезнь Ниманна–
Пика и др.)
• Атаксии при лизосомных болезнях (болезнь Тея–Сакса, дефицит
нейраминидазы и др.)
• Атаксии при наследственных аминоацидуриях и органических
ацидуриях
• Другие формы наследственных метаболических атаксий
6.
7.
8. НАСЛЕДСТВЕННЫЕ АТАКСИИ
это группа генетическиобусловленных заболеваний
двигательной
сферы,ведущей клинической
характеристикой которых
является эпизодическое или
постоянное нарушение
координации движений,
возникающее в результате
поражения мозжечка ,его
связей и/или
соответствующих сенсорных
систем.
9. Спиноцеребеллярные атаксии
Группа аутосомно-доминантных,нейродегенеративных заболеваний,сходных по
клинической картине,основным
патоморфологическим изменениям и характеру
генетического дефекта.Проявляются в возрасте
после 20-30 лет,обусловлены дегенерацией
нейрональных мозжечковых систем. При этом
различным формам характерен свой особенный
паттерн дополнительного вовлечения в
дегенеративный процесс различных отделов ЦНС и
ПНС.
В литературе описаны 28 типов СЦА.
10. Патогенез СЦА
Основная роль принадлежит увеличению количестватриплетов CAG (цитозин-аденин-гуанин).
В норме, данной последовательностью нуклеотидов
кодируется образование глутамина – аминокислоты,
необходимой для образования связей между белками
головного мозга. Из-за увеличения ее количества
происходит удлинение белковых цепей, в результате
чего начинают образовываться нерастворимые
компоненты, из-за которых нарушается
взаимодействие между отделами головного мозга и
происходит гибель некоторого количества нервных
клеток, что и способствует развитию атаксии
11. СЦА1
СЦА1 Ее причиной выступают мутации в гене ATXN1, которыйрасполагается на 6-й хромосоме. В норме данный ген имеет не более 36
CAG-повторов, на мутантных хромосомах число копий увеличено от37-83 .
Продуктом экспрессии гена ATXN1 является особый ДНК-связывающий
белок, активно участвующий в метаболизме клеток Пуркинье мозжечка –
при наличии мутантной разновидности гена это приводит к постепенной
дегенерации, что и становится причиной СЦА1.
СЦА 1 развивается преимущественно у людей в возрасте от 30- 40 лет.
Характерной особенностью заболевания является феномен антиципации.
Первыми симптомами заболевания являются неловкость, неряшливость
при совершении быстрых и интенсивных движений (например, при беге).
Со временем (обычно через несколько лет) появляются и другие симптомы
– нарушение устойчивости в позе Ромберга, дизартрия, нарушение
почерка, специфическая атаксическая походка. Параллельно с данными
симптомами могут развиваться тонические или клонические судороги,
повышение сухожильных или появление патологических рефлексов. На
более поздних стадиях заболевания могут иметь место деменция,
нарушение фонации, расстройства тазовых органов.
12. На КТ и МРТ у больных СЦА1 выявляются расширение субарахноидальных пространств полушарий и червя мозжечка, истончение средней ножки мозже
На КТ и МРТ у больных СЦА1 выявляются расширениесубарахноидальных пространств полушарий и червя мозжечка,
истончение средней ножки мозжечка с симптомом «креста»,
расширение 4желудочка, большой цистерны,цистерн ствола,в ряде
случаев – атрофические изменения больших полушарий мозга.
13. СЦА2
Причиной патологии является увеличение количества CAGповторов в гене ATXN2, локализованном на 12-й хромосоме. Вздоровом варианте гена количество вышеуказанных
последовательностей составляет от 15 до 36, тогда как при СЦА их
может быть свыше 100. Функции белка, который кодируется геном
ATXN2, на сегодняшний момент неизвестны.
СЦА2 Манифестирует в возрасте 20-40 лет, протекает практически
также, как и СЦА1. Характерными особенностями заболевания
является развитие более медленных саккад (согласованных
движений глазных яблок), а также выраженная антиципация,
особенно по мужской линии.
На КТ и МРТ у пациентов с СЦА выявляется атрофия
мозжечка,моста мозга и супратенториальных отделах.
14. Феномен “выпученных глаз”
СЦА3 Болезнь Мачадо-Джозефа,имеет довольно выраженный
полиморфизм симптомов.
Заболевание, помимо, поражения
мозжечка, может протекать и с
клиникой экстрапирамидных
расстройств (таких, как
паркинсонизм или дистония),
амиотрофией и поражением
пирамидного тракта.
Специфичными для СЦА 3
симптомами являются
офтальмоплегия, феномен
“выпученных глаз”, подергивания
окологлазных складок
СЦА 4, помимо основных
расстройств протекает с
симптомами сенсорной
невропатии.
Феномен “выпученных
глаз”
15.
СЦА 5, 6типа — САМЫЙ БЛАГОПОЛУЧНЫЙ ИСХОДСпиноцеребеллярные атаксии 5 и 6 типов отличаются
относительной благоприятностью. Они развиваются в гораздо
более позднем возрасте, а клинические проявления выражены
незначительно, что позволяет больным довольно долгое время
оставаться в социуме.
СЦА 7 типа протекает обычно с поражением сетчатки ,с ее
полным отслоением и развитием слепоты. Нарушения зрения,
зачастую, являются предвещающими симптомами и могут
значительно опережать нарушения координации.
СЦА8 типа характеризуется развитием изолированной
мозжечковой атаксии,пирамидной спастичностью,снижением
вибрационной чувствительности.Отличается медленно
прогрессирующим течением.
16. Аутосомно-рецессивный тип наследования.
Аутосомнорецессивный типнаследования.
17. ДИАГНОСТИКА
КТ и МРТ позволяют определить участки дегенерации нервныхволокон, комбинированную атрофию мозжечка и различных
уровней ствола мозга. В некоторых случаях в поздней стадии
болезни на томограммах может определяться также расширение
боковых желудочков и субарахноидальных пространств больших
полушарий мозга.
Молекулярно-генетические исследования при спиноцеребеллярной
атаксии сводятся к поиску патологически увеличенного количества
CAG-повторов в генах, ассоциированных с этим заболеванием. В
настоящее время большинство лабораторий мира осуществляет
поиск этого дефекта в генах, наиболее часто приводящих к
развитию патологии – ATXN1, ATXN2, ATXN3, ATXN7, ATXN8 и
CACNA1A.
18. АТАКСИЯ ХОЛМСА
19.
НАСЛЕДСТВЕННАЯ ОЛИВО-ПОНТОЦЕРЕБЕЛЛЯРНАЯ ДЕГЕНЕРАЦИЯ (АД)20.
Идиопатическая поздняя мозжечковаяатаксия
Группа объединяет разнообразные дегенеративные заболевания
мозжечка, манифестирующие после 20 лет и характеризующиеся
отсутствием каких-либо наследуемых генных мутаций. Клиникоморфологические проявления данных заболеваний весьма сходны с
таковыми при наследственных (чаще всего — аутосомно-доминантных)
атаксиях.
21.
ОПЦА представляет собой медленно прогрессирующеезаболевание, которое манифестирует обычно после 40—
50 лет и проявляется нарастающей мозжечковой атаксией
в сочетании с дизартрией, дисфонией, нарушением
глотания, паркинсонизмом, пирамидными знаками,
расстройством сфинктеров, реже — снижением
когнитивных функций, нарушением следящих движений
глазных яблок, снижением вибрационной
чувствительности
«Чистая» ОПЦА на фоне прогрессирования болезни
ограничивается поражением мозжечка, его связей и
структур, различных уровней ствола мозга; в этих случаях
болезнь протекает относительно благоприятно и может
растянуться на годы .
Осложненный мультисистемный вариант ОПЦА с течением
времени осложняется присоединением совокупности
симптомов поражения других областей спинного и
головного мозга(к мозжечковой стволовой дисфункции
присоединилось проявление паркинсонизма, вегетативной
недостаточности, вовлечение кортико -спинального
тракта) и эволюционирует в типичную картину
множественной системной атрофии.Является более
злокачественным.
22.
Сходная с ОПЦА симптоматика и нейроморфологияимеет место еще при двух редких
нейродегенеративных заболеваниях —
стриатонигральной дегенерации и синдроме
Шая—Дрейджера. Это позволило объединить
ОПЦА, стриатонигральную дегенерацию и синдром
Шая-Дрейджера в рамках так называемой
множественной системной атрофии. Для всех
указанных фенотипических вариантов
множественной системной атрофии характерен
поздний возраст начала болезни (обычно на 5-6-м
десятилетии жизни) и неуклонное
прогрессирование процесса, приводящее к гибели в
среднем через 6—10 лет от момента появления
первых симптомов.
23.
Паренхиматозная кортикальная мозжечковаяатрофия (болезнь Мари-Фуа-Алажуанина)- форма
идиопатической спорадической мозжечковой атаксии
начинается чаще всего на 5-м десятилетии жизни, хотя
возраст появления первых симптомов может быть
вариабельными нередко достаточно поздним (вплоть до 7-8го десятилетия).
Основные клинические симптомы — атаксия туловища в
пробе Ромберга и при ходьбе, нарушение координации
движений в ногах, дизартрия; дискоординация в руках
обычно выражена не резко и не склонна к
прогрессированию. У отдельных больных могут выявляться
также пирамидные знаки, нистагм, тремор головы и рук,
выпадение ахилловых рефлексов. Темп нарастания
симптомов весьма медленный, продолжительность болезни
может составить 20 лет и более. Таким образом,
паренхиматозную кортикальную мозжечковую атрофию
можно охарактеризовать как форму с медленным
прогрессированием мозжечковой симптоматики «в чистом
виде».
24. Нейровизуализация
МРТ-исследование, прикотором отчетливо
визуализируют
комбинированную атрофию
мозжечка и различных
уровней ствола мозга. В
некоторых случаях в поздней
стадии болезни на
томограммах может
определяться также
расширение боковых
желудочков и
субарахноидальных
пространств больших
полушарий мозга. На Т2взвешенных снимках весьма
специфичный «симптом
креста».
25.
Синдром FXTAS- нейродегенеративное заболевание позднего возраста(Х-сцепленный
аутосомно-рецессивный тип наследования). Оно развивается у мужчин
и значительно реже чем у женщин, являющихся носителями
премутации в нетранслируемой 5-области гена FMR1.
Клинически синдром FXTAS обычно манифестирует у пожилых в виде
прогрессирующей мозжечковой симптоматики (интенционный тремор,
атаксия ходьбы, дизметрия , мозжечковая дизартрия ). Характерно, что
значительная выраженность интенционного тремора не соответствует
более мягким проявлениям других мозжечковых симптомов. В 10-30%
случаев имеет место тремор покоя, а также тремор эссенциального
либо смешанного типа. Примерно у 60% больных с синдромом FXTAS
развивается паркинсонизм (брадикинезия , ригидность , реже
паркинсоновское дрожание), который обычно не чувствителен к
препаратам леводопы. Весьма типичны также вегетативные
расстройства (недержание мочи и кала, импотенция ). В ряде случаев в
структуру синдрома FXTAS могут входить когнитивные расстройства,
психиатрические симптомы (тревожность, депрессия ), периферическая
невропатия .
Наличие двусторонних очагов в белом веществе средних ножек
мозжечка характерно для синдрома FXTAS.
26. ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Проводят с РСв пользу дегенеративной природы свидетельствует неуклонно
прогрессирующее течение и отсутствие ремиссий,симметричность
симптомов,отсутствие признаков многоочагового поражния
ЦНС,сохранность брюшных рефлексов,отсутствие очаговых
изменений вещества мозга на КТ и МРТ.
27.
Дифференциальный диагноз собъемным
процессом в задней черепной ямке(в первую
очередь- с невриномой 7пары черепных
нервов),нормотензивной
гидроцефалией(триада симтомовдеменция,атаксия,недержание мочи),не вызывает
серьезных затруднений. Он базируется на данных
нейровизуализации с особенностяими
течения заболевания и семейного анамнеза,
а также-в ряде случаев-на специфической
серологической
(нейросифилис,нейроборрелиоз)или
биохимической диагностике(наследственные
метоболические)
28. Вертебро-базилярная недостаточность
При ВБН у больного обычно имеется большое числоспецифических субъективных
жалоб(головокружение,тошнота,ощущение
проваливания,наличие повторных острых эпизодов
ТИА ),не характерных для аутосомно-доминантных
атаксий с их неуклонно прогрессирующим
нарастанием, координаторных расстройств.Более
того для ВБН не типично наличие относительно
атрофии мозжечка на КТ, требует четкой
верификации патологии сосудов мозга с помощью
ультразвуковых и ангиографических методов
исследования.
29.
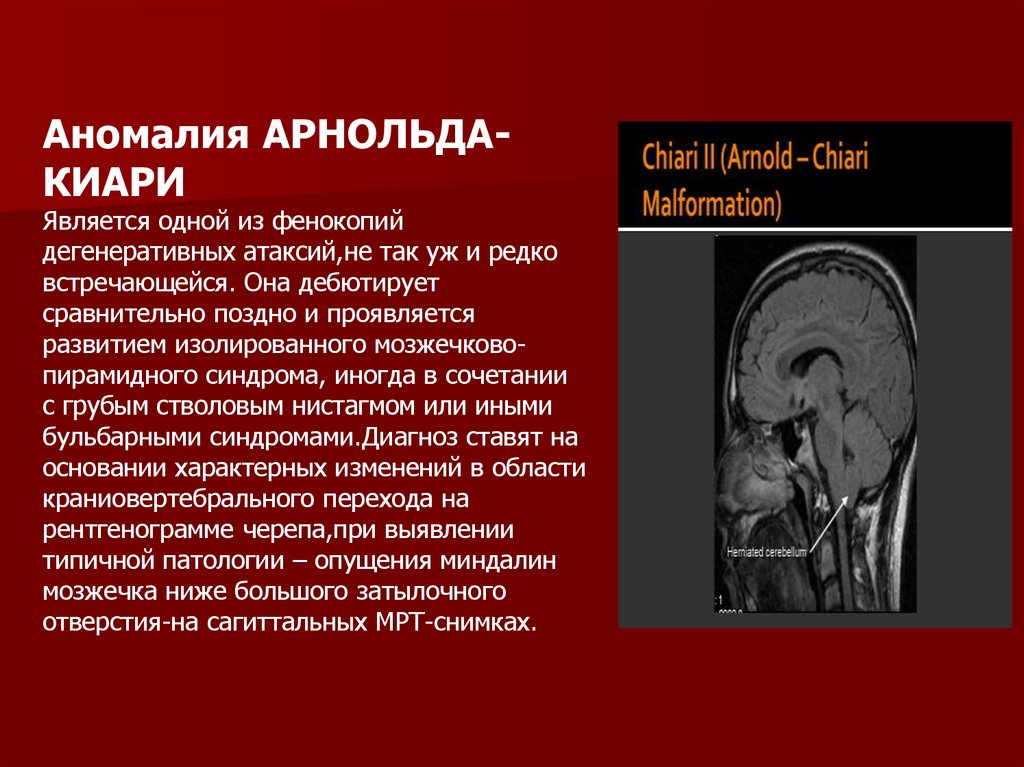
Аномалия АРНОЛЬДАКИАРИЯвляется одной из фенокопий
дегенеративных атаксий,не так уж и редко
встречающейся. Она дебютирует
сравнительно поздно и проявляется
развитием изолированного мозжечковопирамидного синдрома, иногда в сочетании
с грубым стволовым нистагмом или иными
бульбарными синдромами.Диагноз ставят на
основании характерных изменений в области
краниовертебрального перехода на
рентгенограмме черепа,при выявлении
типичной патологии – опущения миндалин
мозжечка ниже большого затылочного
отверстия-на сагиттальных МРТ-снимках.
30.
Глютеновая атаксия – прогрессирующаяатаксия, развивающаяся в результате
непереносимости глютена,содержащегося в
некоторых злаках –
пщенице,ячмени,ржи.Характерно развитие
атаксии туловища,в меньшей степени
конечностей,к которым у ряда больных позднее
присоединяются дисфагия,дизурия,нарушения
глубокой чувствительности,угнетение
ахилловых рефлексов,амиотрофии и другие.На
КТ и МРТ выявляется изолированная атрофия
полушарий и червя мозжечка,на ЭНМГ
признаки аксональной невропатии.ДИАГНОЗ
потверждают при обнаружении в крови
больных антиглиадиновых антител.
31. ЛЕЙКОДИСТРОФИИ И ДРУГИЕ НАСЛЕДСТВЕННЫЕ НАРУШЕНИЯ ОБМЕНА(ГАНГЛИОЗИДЫ,ДЕФИЦИТ ВИТ.Е И ДР)
ДЛЯ дифференцирования аутосомнодоминантых атаксий с указаннымисиндромами большое значение имеет,еще до
проведения биохимической
диагностики,качественный генеалогический
анамнез:отмеченные метоболические болезни
как правило наследуются по аутосомнорециссивному или Х-сцепленному
рецессивному типам.
32.
Поздняя симптоматическая мозжечковаяатрофия , ассоциированная с
гипотиреозом.
Характеризуется поздним спорадическим началом
болезни, преимущественным поражением церебеллярных
структур, дегенеративным характером патологического
процесса и низким содержанием тиреоидных гормонов
щитовидной железы в сыворотке крови. Кроме нарушений
в виде атаксии ходьбы, дискоординации в конечностях и
нарушениях речи наблюдаются клинические проявления
гипотиреоза (слабость, сонливость, утомляемость,
постоянное чувство холода, изменение голоса, нарушения
слуха, одутловатость лица и отеки конечностей –
микседема).
33.
Синдром кортикальной мозжечковой атрофии приалкоголизме.
Клиническая картина такого синдрома весьма близка к
проявлениям паренхиматозной кортикальной мозжечковой
атрофии и включает выраженную шаткость походки, резкое
нарушение координации и интенционный тремор в ногах
(функция рук страдает в значительно меньшей степени),
негрубую дизартрию, признаки полинейропатии (угнетение
сухожильных рефлексов, чувствительные расстройства, боли и
др.), тремор пальцев рук. Характерно подострое развитие
симптоматики, нередко приуроченное к очередному запою, после
чего в дальнейшем заболевание прогрессирует очень медленно и
не влияет на продолжительность жизни. Обычно после
прекращения приема алкоголя нарастание симптомов
приостанавливается, однако существенного уменьшения
проявлений болезни не отмечается. При КТ- и МРТ-исследовании
у таких пациентов, как правило, обнаруживается не только
атрофия червя и полушарий мозжечка, но и заметное
расширение субарахноидальных пространств больших полушарий
мозга.
34. ЛЕЧЕНИЕ
Эффективное патогенетическое лечение донастоящего времени не разработано. Возможности
симптоматической терапии также достаточно
ограничены и не оказывают существенного влияния на
прогноз заболевания. Новые подходы к лечению
атаксий могут быть связаны с достижениями
молекулярной биологии и раскрытием интимных
механизмов дегенерации нейронов на белковом уровне,
однако это направление находится пока лишь на
начальном этапе исследований и практической
разработки.
35. Симптоматическая терапия:
ноотропные средства: холин-альфосцерата (глиатилина 400 мг3р\д в течение 2-3 мес), ноотропил,церебролизин и другие
предшественники ацетилхолина.
витамины группы В, Е(1000-1600мг\сут в теч 1-2мес)
препаратов метаболического ряда (эссенциале, коэнзим Q10,
милдронат ,тиоктацид и др)
при наличии спазмов мышц хороший эффект оказывает
баклофен 5-15мг\сут или сирдалуд 4-6мг\сут
При поражении периферических нервов и мышц используют
фосфаден (аденил), рибоксин, ретаболил.
При возникновении непроизвольных движений назначают
клоназепам, циклодол (акинетон), тиаприд, галоперидол
назначают массаж, ЛФК, электростимуляцию мышц
36.
Лечебная физкультура (вестибулярнаягимнастика) и проведение сеансов баланстренинга с использованием специальных
стабилометрических платформ.
Указанные физические методы лечения должны
применяться с особой осторожностью, с
исключением резких изменений положения тела,
наклонов туловища и головы, с постоянным
контролем уровня АД.
37.
Прогноз неблагоприятный – выраженное прогрессирующеетечение, со временем приводит сначала к инвалидизации, а
затем к смерти больного. Однако в конкретном случае
прогноз может быть и менее негативным – например, при
развитии патологии в пожилом возрасте и своевременно
начатом поддерживающем лечении большинство тяжелых
симптомов попросту не успеет проявиться.
Если проявляется в молодом или детском возрасте,
продолжительность жизни таких больных даже при
интенсивном лечении и тщательном уходе будет резко
снижена.
Профилактика осуществляется методом медикогенетического консультирования родителей, наследственный
анамнез которых отягощен по этому состоянию, и
генетической пренатальной диагностики, при этом
необходимо учитывать характер наследования атаксии.