


















 Медицина
МедицинаПохожие презентации:










Синдромы частичных моносомий, возникших в результате делеций различных участков в хромосомах
1.
Государственное бюджетное образовательное учреждениевысшего образования
«Тюменский государственный медицинский университет»
Министерства здравоохранения Российской Федерации
Синдромы частичных моносомий,
возникших в результате делеций
различных участков в хромосомах
4, 9, 11, 13,18, 21 и 22.
Выполнила: Захарова Анастасия,
Студентка 211 группы
Проверила: ассистент кафедры
Баянова Анна Евгеньвена
Тюмень, 2021
2.
Цель и задачиЦель: Определить такое понятие как,
синдромы частичных моносомий.
Задачи:
1. Рассмотреть понятие делеции.
2. Рассмотреть делеций различных
участков в хромосомах 4, 9, 11, 13,18, 21
и 22.
3.
Моносомия – это наличие всего одной из парыгомологичных хромосом. В случае обширной
делеции в какой-либо хромосоме иногда
говорят о частичной моносомии.
Делеция (от лат. deletio — уничтожение) потеря участка хромосомы. Делецией называют
потерю одной или более нуклеотидных позиций
в последовательности.
4.
Некоторые причины появленияделеций
Делеции подразделяются на интерстициальные (потеря
внутреннего участка) и терминальные (потеря концевого
участка)
Причины возникновения делеций:
1. Делеция может быть следствием разрыва хромосомы.
2. В результате неравного кроссинговера в мейозе.
3. В результате проскальзывания при репликации. Оно
происходит если в последовательности содержится много
коротких повторов.
4. Проскальзывание при участии коротких повторов может
происходить и в нормальной не реплицирующейся хромосоме.
Тогда образуются петли и ферменты репарации удаляют обе
или одну из них. Это также приводит к разнообразным
мутационным событиям.
5.
Синдром Вольфа—ХиршхорнаВпервые синдром Вольфа—Хиршхорна
был зарегистрирован в 1961 г. Это
больные с комплексом врожденных
аномалий и задержкой психомоторного
развития в результате делеции или
потери короткого плеча хромосомы 4.
Размеры делеции колеблются от
небольших
терминальных
до
занимающих
около
половины
дистальной части короткого плеча.
Отмечается, что в большинстве случаев
делеции возникают de novo, 13%
являются следствием транслокаций у
родителей.
6.
Синдром Вольфа—ХиршхорнаЧастота встречаемости синдрома 1:50 000—1:100 000
новорожденных независимо от расовой принадлежности.
В 2 раза чаще поражаются девочки. Заболевание отличается
высокой летальностью, особенно в раннем возрасте; в
первые 2 года жизни умирают около 35% пациентов.
Выжившие дети имеют грубую задержку в развитии.
Максимальная продолжительность жизни пациентов с этим
синдромом — 26 лет.
Для диагностики заболевания используется цито-генетический
метод. Лечение больных симптоматическое. Родители нуждаются в
медико-генетическом консультировании
7.
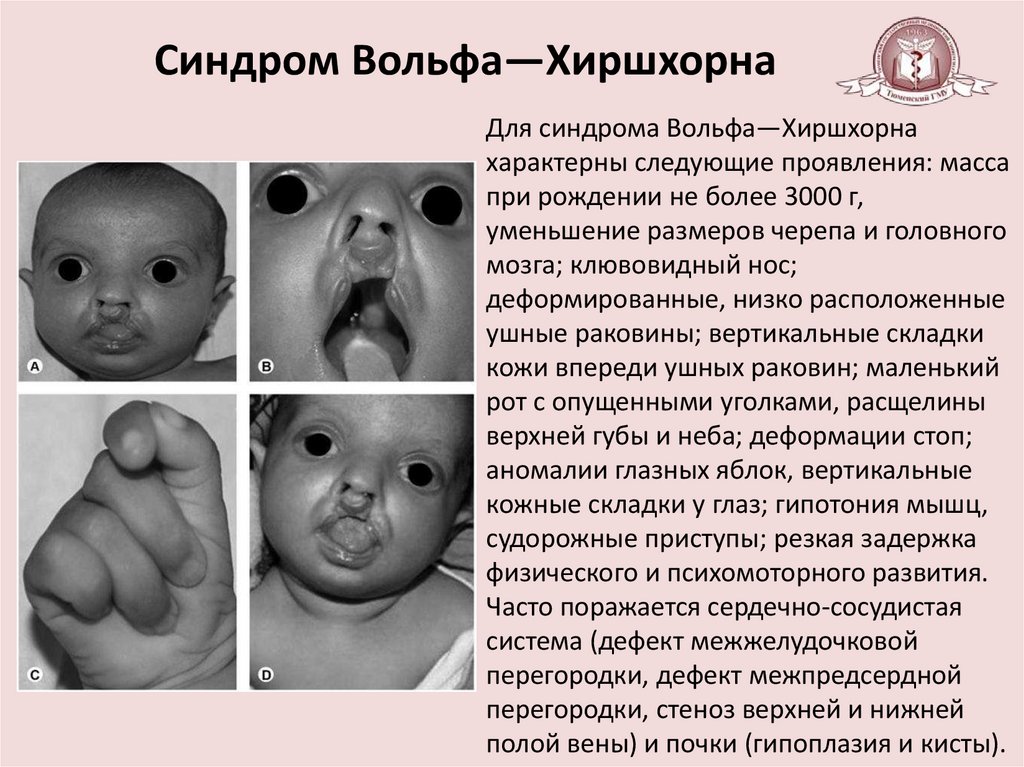
Синдром Вольфа—ХиршхорнаДля синдрома Вольфа—Хиршхорна
характерны следующие проявления: масса
при рождении не более 3000 г,
уменьшение размеров черепа и головного
мозга; клювовидный нос;
деформированные, низко расположенные
ушные раковины; вертикальные складки
кожи впереди ушных раковин; маленький
рот с опущенными уголками, расщелины
верхней губы и неба; деформации стоп;
аномалии глазных яблок, вертикальные
кожные складки у глаз; гипотония мышц,
судорожные приступы; резкая задержка
физического и психомоторного развития.
Часто поражается сердечно-сосудистая
система (дефект межжелудочковой
перегородки, дефект межпредсердной
перегородки, стеноз верхней и нижней
полой вены) и почки (гипоплазия и кисты).
8.
Синдром АльфиВпервые описан в 1973 году. Цитогенетические
варианты могут быть различны: частичная делеция
короткого плеча 9 хромосомы, транслокации.
Долгое время не существовало доступного способа
определения микроделеции 9p24, поэтому мы до сих
пор не знаем, как часто встречается это
отклонение. Вполне возможно, что существуют
тысячи людей с такой же делецией, но диагноз им
поставлен не был.
9.
Синдром АльфиНекоторые мальчики с делецией 9p24 родились с аномалиями
развития половых органов.
Аномалии развития внешних половых органов могут быть
относительно небольшими, такими как гипоспадия, при
которой отверстие мочеиспускательного канала смещено вниз,
или неопущение яичек в мошонку при рождении. В некоторых
случаях последствия могут быть более серьезными, включая
реверсию (переопределение) пола, неопределенные или
напоминающие женские половые органы. Внутренние половые
органы тоже могут развиваться неправильно. Эти аномалии
встречаются только у мальчиков. У девочек внешние половые
органы развиваются нормально и практика показывает, что
у этих девочек происходит нормальное половое созревание в
обычном возрасте.
10.
Синдром Альфи11.
Синдром Альфи12.
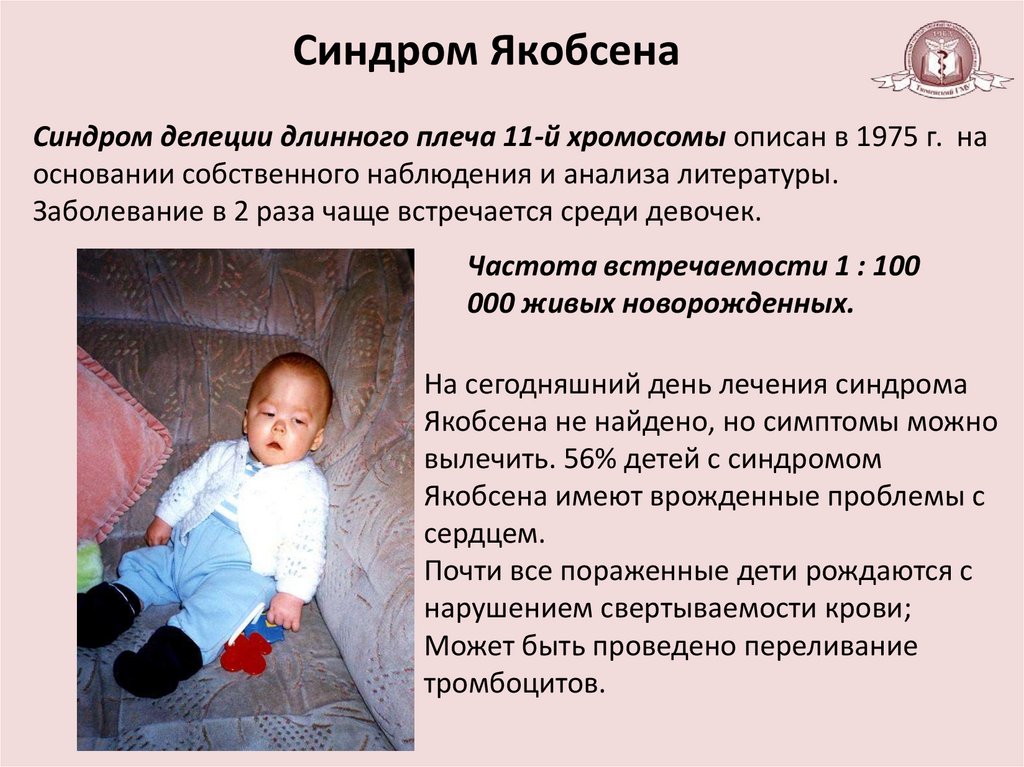
Синдром ЯкобсенаСиндром делеции длинного плеча 11-й хромосомы описан в 1975 г. на
основании собственного наблюдения и анализа литературы.
Заболевание в 2 раза чаще встречается среди девочек.
Частота встречаемости 1 : 100
000 живых новорожденных.
На сегодняшний день лечения синдрома
Якобсена не найдено, но симптомы можно
вылечить. 56% детей с синдромом
Якобсена имеют врожденные проблемы с
сердцем.
Почти все пораженные дети рождаются с
нарушением свертываемости крови;
Может быть проведено переливание
тромбоцитов.
13.
Синдром ЯкобсенаНаиболее общими клиническими признаками
СЯ являются: пре- и постнатальная задержка
физического развития, психомоторного
развития, характерный лицевой дисморфизм,
тромбоцитопения. Пациенты имеют
деформации сердца, почек, желудочнокишечного тракта, гениталий, ЦНС и/или
скелета. Также могут присутствовать
нарушения слуха, зрения, гормональные и
иммунологические проблемы.
Таким образом, выраженность клинических
особенностей разнообразна. Описаны случаи
сочетания СЯ с такими ортопедическими
патологиями, как плоскостопие, большой и
длинный 1-й палец стопы, клинодактилия,
брахидактилия, синдактилия 2-го и 3-го
пальцев стоп.
14.
Синдром ЯкобсенаВ рамках довольно разнообразной картины, у некоторых детей
обнаруживают уязвимость к поведенческим расстройствам.
Некоторые дети ведут себя вызывающе и привлекают внимание. У
некоторых детей обнаруживают демонстративные эмоциональные
вспышки, но их частота, как и частота всех проявлений агрессии
обычно уменьшаются после развития речи. У некоторых детей
развивается компульсивное поведение (как например, измельчение
разных предметов). У многих детей диагностируют синдром
дефицита внимания с гиперактивностью (ADHD). В целом, поведение
детей, вероятно, улучшается в структурированной среде, и
существует предположение, что такие дети более склонны к
контакту со взрослыми, чем с детьми своего возраста. Семьи,
имеющие отношение к указанной проблеме, в случае если ребенок
начинает бить или кусать других или проявляет признаки
навязчивого поведения, должны обращаться за своевременной
поддержкой.
15.
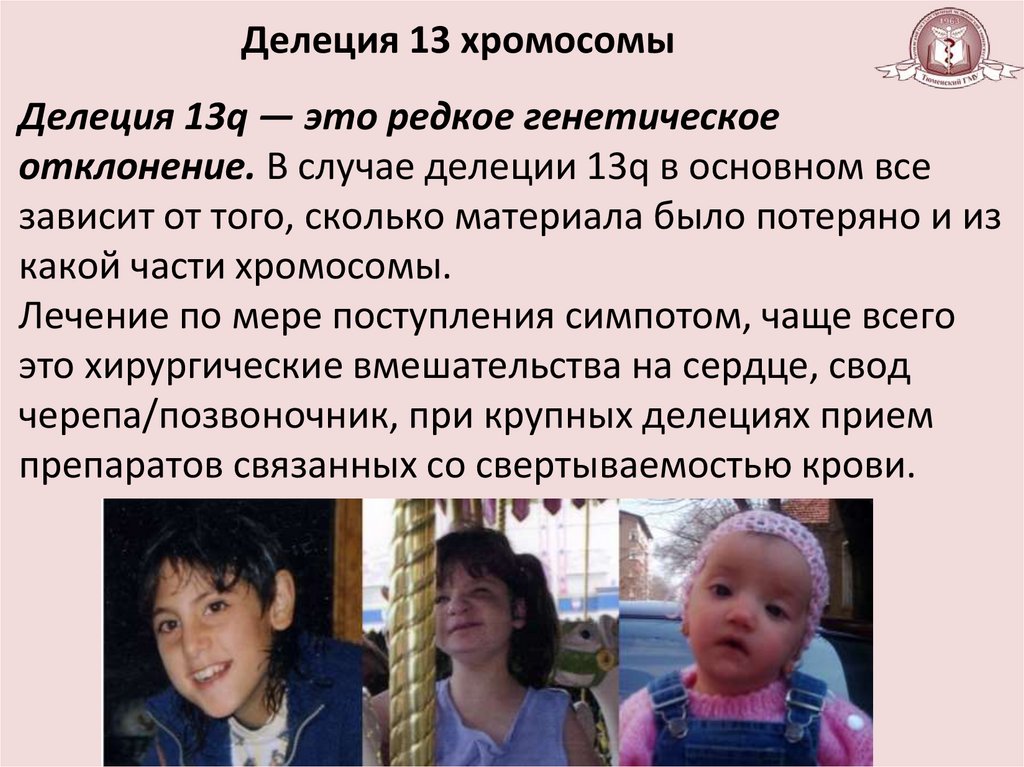
Делеция 13 хромосомыДелеция 13q — это редкое генетическое
отклонение. В случае делеции 13q в основном все
зависит от того, сколько материала было потеряно и из
какой части хромосомы.
Лечение по мере поступления симпотом, чаще всего
это хирургические вмешательства на сердце, свод
черепа/позвоночник, при крупных делециях прием
препаратов связанных со свертываемостью крови.
16.
Делеция 13 хромосомыУ всех детей были обнаружены задержки в развитии разной степени выраженности.
К характерным особенностям делеции хромосомы 13q относятся: нарушение
формирования мягких тканей свода черепа, при котором область, не являющаяся
типичным родничком, не покрывается костью,дефект межпредсердной
перегородки (ДМПП) или дефект межжелудочковой перегородки (ДМЖП) – которое
может зарасти само или нуждаться в хирургической коррекции; низкий уровень
определенных химических веществ крови, в частности факторов свертывания крови
VII и X, но обычно недостаточно низкий, чтобы привести к появлению
кровоподтеков или кровотечений; нарушение формирования отдельных позвонков
(врожденное недоразвитие половины позвонка. Другие известные отличительные
черты – микроцефалия (маленький размер головы).
17.
Синдром де ГрушиСиндром терминальной делеции
длинного плеча хромосомы 18,
впервые описанный в 1964 г.
Клинический фенотип характеризуется
широким варьированием
диагностических признаков в
зависимости от генного состава
утраченного фрагмента хромосом.
Включает краниофациальный
дисморфизм, задержку физического
развития, аномалии зрительного
анализатора, половых органов, а
также разнообразную
психоневрологическую патологию,
эпилептоидные проявления и
глубокую умственную отсталость.
18.
Синдром де ГрушиУ небольшой части людей делеция может иметь очень
лёгкие проявления. У одной матери с делецией 18q нет
проблем, связанных с делецией, в то время как у её дочери,
которая унаследовала её, имеется общая задержка
развития, трудности в обучении и нарушение слуха. У 23летней женщины с делецией 18 отмечалась задержка в
развитии, но в пубертатном периоде она нагнала своих
сверстниц. У неё нормальные интеллект и способности к
работе, а навыки устной речи выше средних.
Половая зрелость наступает как обычно. Наблюдения
группы Unique показали, что у девушек половая зрелость
наступает между 11 и 17 годами жизни, чаще всего около 13
лет. О половой зрелости мальчиков известно очень мало
19.
Делеция 21 хромосомыСиндром делеции длинного плеча 21 хромосомы (синдром 21 q-).
Выделен в отдельную нозологическую группу в 1970 году.
Цитогенетически характеризуется частичной делециеи длинного плеча
хромосомы 21, а также кольцевой хромосомой. Отмечены мозаичные
формы этих хромосомных нарушений. Диагностическими признаками
заболевания являются: низкая масса тела при рождении,
микроцефалия, антимонголоидный разрез глаз, эпикант, широкая
переносица, большие низко посаженные деформированные уши с
расширенным слуховым каналом. К основным признакам относятся
также сколиоз, паховая грыжа, косолапость. Их пороков внутренних
органов наблюдаются пороки сердца и патология почек (гидронефроз,
удвоение почечных лоханок). Отмечена задержка психомоторного
развития. При грубых пороках развития дети умирают в первые недели
жизни, хотя описаны больные в возрасте 50 лет.
Лечение по мере поступления симптомов.
20.
Синдром Ди ДжорджиСиндром кольцевой хромосомы 22. Выделен в самостоятельный синдром в 1972
году. Цитогенетические варианты могут быть различны: кольцевая хромосома 22,
делеции длинного плеча этой хромосомы и мозаичные варианты. В 90% случаев
данные хромосомные нарушения являются мутациями de novo. Основными
диагностическими признаками заболеванияя вляются: микроцефалия, эпикант,
крупные выступающие глаза ("глаза лани"), расщелина язычка и неба, густые брови
и ресницы. Кроме того, отмечается дисплазия тазобедренных суставов. Умственная
отсталость проявляется как легкой, так и выраженной олигофренией. Больные легко
возбудимы, отмечается частая смена настроения, нарушение координации, а также
грубое недоразвитие речи, вплоть до ее отсутствия.
21.
Делеция 22 хромосомыУчитывая, что в 1% случаев у пациентов с
полной делецией хромосомы 22
обнаруживается тяжелый Т-клеточный
иммунодефицит с очень низким или
полным отсутствием Т клеток,
диагностика иммунных нарушений у таких
пациентов является неотъемлемой частью
алгоритма ведения.
В алгоритм диагностики и лечения
больных с делецией хромосомы 22
входит обязательное исследование
функции тимуса . К основным
диагностическим исследованиям
относят:
• ультразвуковое исследование
• развернутый анализ крови с
дифференцированным подсчетом
лейкоцитов в абсолютном значении;
• определение концентрации
иммуноглобулинов М, А, G;
• фенотипирование лимфоцитов
• определение общего числа Т клеток
определение пролиферативной
активности лимфоцитов на
фитогемагглютинин при низком числе Т
клеток;
• оценка иммунного ответа на
вакцинацию столбнячным анатоксином
и/или НШ-вакциной.
22.
Список использованных источников1. Лечение врожденной косолапости в сочетании с синдромом
Якобсена при помощи метода Понсети.- И.Ю. Круглов, Н.Ю.
Румянцев, Г.Г. Омаров, Н.Н. Румянцева-2018
2. Атипичный случай синдрома делеции длинного плеча 11-й
хромосомы.- Э.И. Костырко, Ж.В. Прокопцева, Ким Ми Дя-2007.
3. Алгоритм ранней диагностики и лечения синдрома делеции
хромосомы 22 (22q11. 2).- Л.С. Намазова-Баранова, О.В. Гинтер,
Т.А. Полунина, И.В. Давыдова,К.В. Савостьянов, А.А. Пушков1, Н.В.
Журкова, Т.Я. Мосьпан-2017.
4. Случай делеции длинного плеча хромосомы 18 у ребенка 2
месяцев.- Е.М. Камалтьтова, О.А. Салюкова, О.С. Федорова, С.Л.
Вовк, Е.Л. Тимошина, Л.М. Огородова-2006.
5. Особенности диагностики и тактики ведения пациента с
синдромом Ди Джорджи.- Коренюк Е.С., Ярошевская Т.В.,
Самойленко И.Г-2016.