

















 Медицина
МедицинаПохожие презентации:



")





 желтуха")
Функциональные гипербилирубинемии. Синдром Жильбера
1.
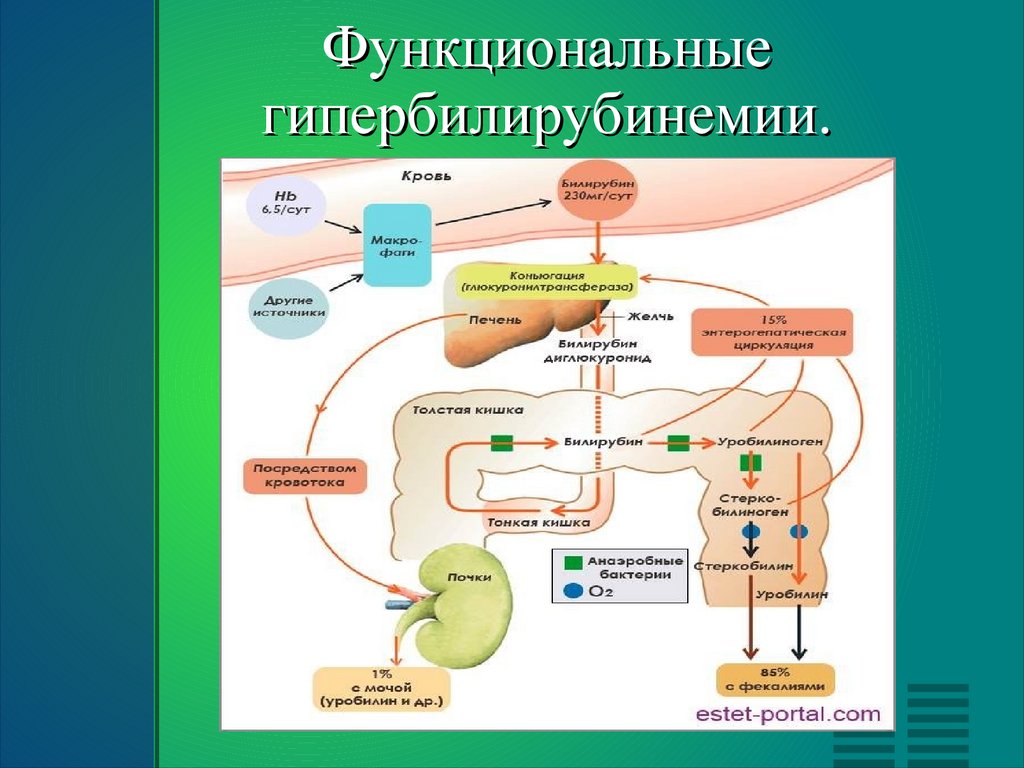
Функциональныегипербилирубинемии.
2.
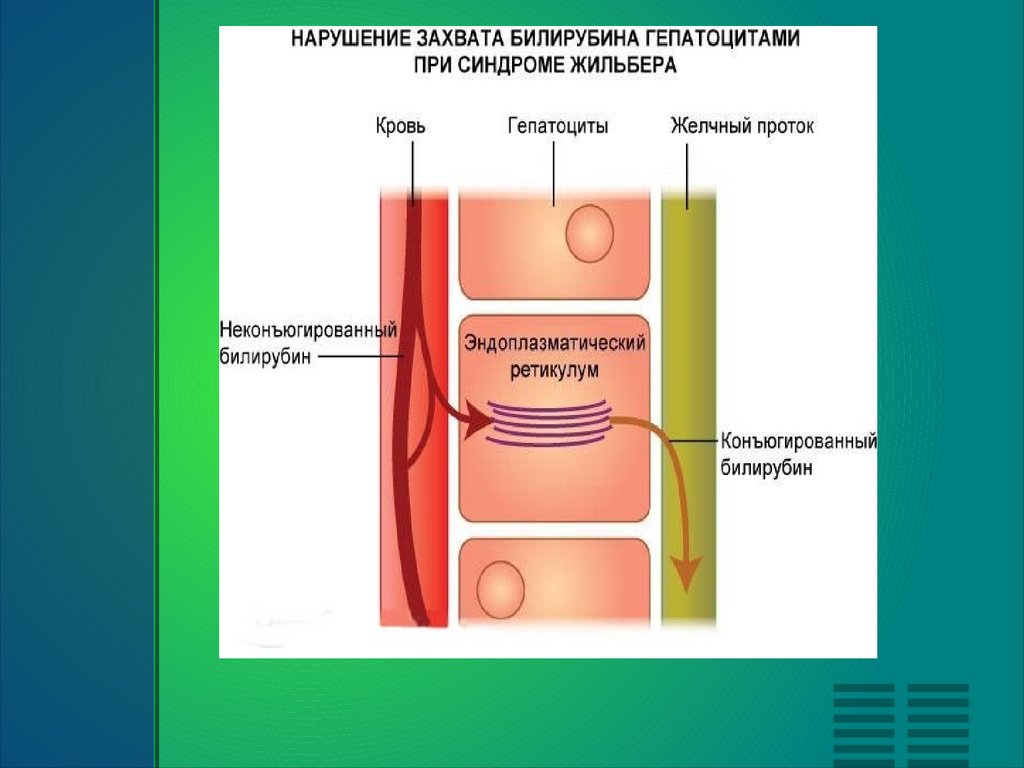
Синдром ЖильбераГенетически обусловленное нарушение обмена билирубина, возникающее
вследствие дефекта микросомальных ферментов печени и приводящее к
развитию доброкачественной неконьюгированной гипербилирубинемии. В
большинстве случаев синдрома Жильбера отмечается интермиттирующая
желтуха различной степени выраженности, тяжесть в правом подреберье,
диспепсические и астеновегетативные расстройства; возможно появление
субфебрилитета, гепатомегалии, ксантелазм век. Диагноз синдрома
Жильбера подтверждают данные клинической картины, семейного
анамнеза, лабораторных и инструментальных исследований,
функциональных проб. При синдроме Жильбера показано соблюдение
режима питания, прием индукторов микросомальных ферментов,
гепатопротекторов, желчегонных трав, энтеросорбентов, витаминов.
Синдром Жильбера впервые был
описан французским
гастроэнтеролог - Августином
Николя Гилбертом (1958-1927) и
его коллегами в 1901 году.
3.
Самая частая форма наследственногопигментного гепатоза, которая выявляется у
1-5% населения. Синдром распространён
среди европейцев (2-5%), азиатов (3%) и
африканцев (36%). Заболевание впервые
проявляется в юношеском и молодом
возрасте, в 8-10 раз чаще у мужчин.
Морфологические изменения в печени характеризуются
жировой дистрофией гепатоцитов и накоплением
желтовато-коричневого пигмента липофусцина в
печёночных клетках, чаще в центре долек по ходу
желчных капилляров.
4.
5.
Различают 2 варианта синдрома Жильбера: «врожденный» - проявляющийся безпредшествующего инфекционного гепатита (большинство случаев) и манифестирующий после
перенесенного острого вирусного гепатита. Причем постгепатитная гипербилирубинемия может
быть связана не только с наличием синдрома Жильбера, но и с переходом инфекции в
хроническую форму.
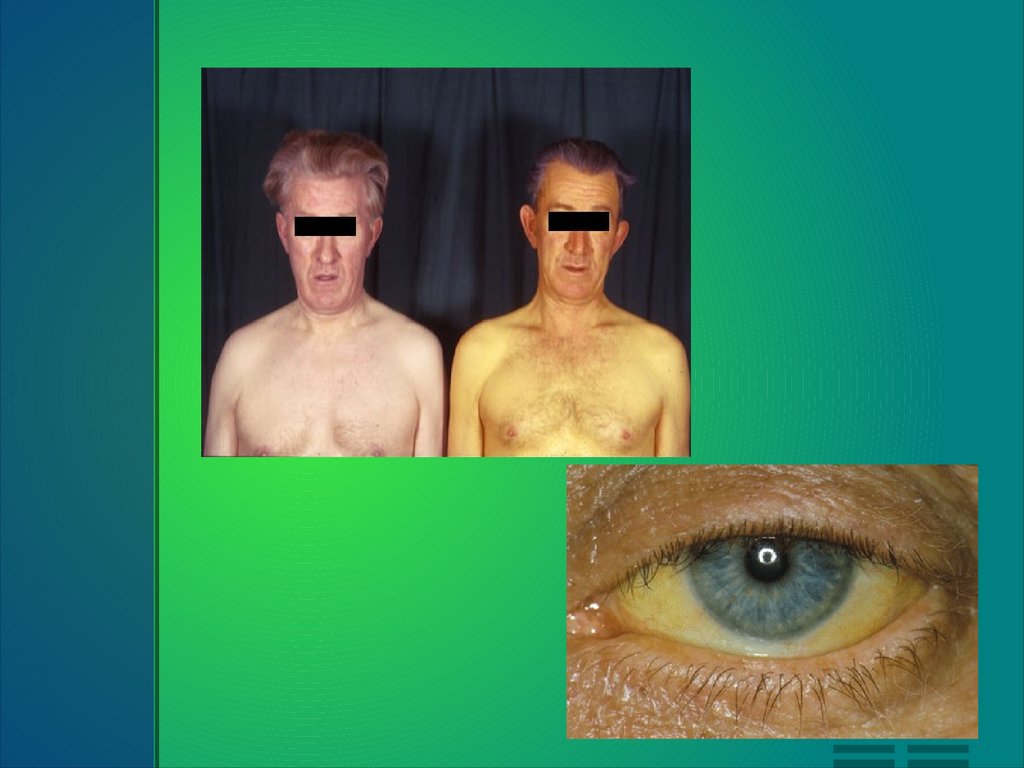
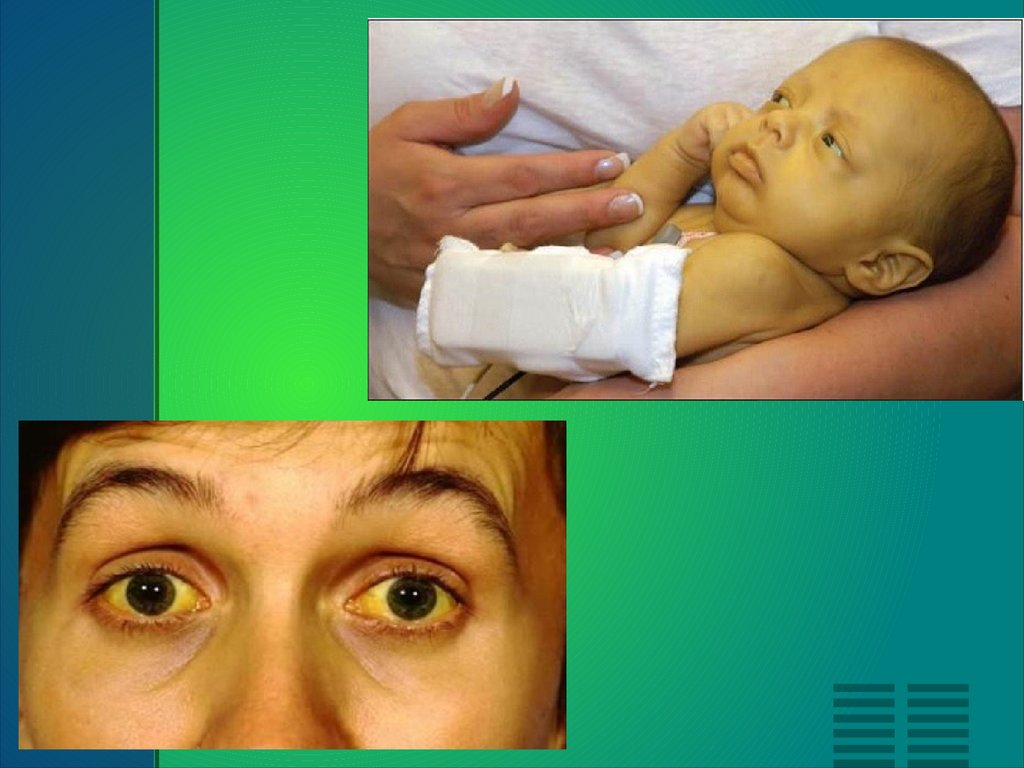
Клинические симптомы синдрома Жильбера обычно развиваются у детей в возрасте 12 лет и
старше, течение заболевания волнообразное. Для синдрома Жильбера характерна
интермиттирующая желтуха различной степени выраженности - от субиктеричности склер до
яркой желтушности кожных покровов и слизистых оболочек. Желтушное окрашивание может быть
диффузным или проявляться частично в области носогубного треугольника, на коже стоп,
ладоней, в подмышечных впадинах. Эпизоды желтухи при синдроме Жильбера возникают
внезапно, усиливаются после воздействия провоцирующих факторов и разрешаются
самостоятельно. У новорожденных детей симптомы синдрома Жильбера могут иметь сходство с
транзиторной желтухой. В отдельных случаях при синдроме Жильбера могут возникать единичные
или множественные ксантелазмы век.
Большинство больных синдромом Жильбера предъявляют жалобы на тяжесть в правом
подреберье, чувство дискомфорта в брюшной полости. Наблюдаются астеновегетативные
расстройства (быстрая утомляемость и подавленность, плохой сон, потливость), диспепсические
явления (отсутствие аппетита, тошнота, отрыжка, метеоризм, нарушения стула). В 20% случаев
синдрома Жильбера имеется незначительное увеличение печени, в 10% - селезенки, может
наблюдаться холецистит, дисфункция желчного пузыря и сфинктера Одди, повышается риск
развития холелитиаза.
Примерно у трети пациентов с синдромом Жильбера жалобы отсутствуют. При незначительности
и непостоянстве проявлений синдрома Жильбера заболевание долгое время может оставаться
незамеченным для больных. Синдром Жильбера может сочетаться с дисплазией соединительной
ткани – часто встречается при синдромах Марфана и Элерса-Данлоса.
6.
7.
ДиагностикаКлинически проявляется не ранее, чем в возрасте 20 лет.
Часто больной не подозревает о том, что страдает
желтухой, пока она не обнаружится при клиническом
осмотре или при проведении лабораторных исследований.
Физикальные методы обследования:
1) опрос — указание в анамнезе на периодические
эпизоды умеренной желтухи, возникающей чаще после
физического перенапряжения или инфекционного
заболевания, в том числе гриппа, после продолжительного
голодания или соблюдения низкокалорийной диеты, однако
у больных с гемолизом уровень билирубина при голодании
не повышается;
2) осмотр — субиктеричность (легкая желтизна)
слизистых и кожных покровов.
8.
Лабораторные исследованияОбязательные:
1) общий анализ крови;
2) общий анализ мочи;
3) уровень билирубина в крови — повышение уровня общего билирубина за счет непрямой
фракции;
4) проба с голоданием — повышение уровня билирубина на фоне голодания - В течение 48
часов больной получает питание энергетической ценностью 400 ккал/сутки. В первый день
пробы натощак и спустя двое суток определяют билирубин сыворотки крови. При подъеме его
на 50 — 100% проба считается положительной.
5) проба с фенобарбиталом — снижение уровня билирубина на фоне приема фенобарбитала
за счет индуцирования конъюгирующих ферментов печени;
6) проба с никотиновой кислотой — в/в введение вызывает повышение уровня билирубина за
счет уменьшения осмотической резистентности эритроцитов;
7) анализ кала на стеркобилин — отрицательный;
8) молекулярная диагностика: анализ ДНК гена УДФГТ (в двух аллелях обнаруживается
мутация - увеличение количества ТА повторов больше 6ТА/6ТА);
9) ферменты крови: АсАТ, АлАТ, ГГТП, ЩФ — как правило, в пределах нормальных значений
или незначительно повышены.
При наличии показаний:
* белки сыворотки крови и их фракции — может наблюдаться увеличение общего белка и
диспротеинемия;
*протромбиновое время — в пределах нормы;
* маркеры вирусов гепатита B, С, D — отсутствие маркеров;
* бромсульфалеиновая проба — снижение выделения билирубина на 20 %.
9.
Инструментальные и другиеметоды исследования
Обязательные:
1) УЗИ органов брюшной полости — определение размеров и состояние
паренхимы печени; размеров, формы, толщины стенок, наличие конкрементов в
желчном пузыре и желчных протоках.
При наличии показаний:
*чрескожная пункционная биопсия печени с морфологической оценкой биоптата
— для исключения хронического гепатита, цирроза печени.
Консультации специалистов Обязательные:
*терапевт.
При наличии показаний:
*клинический генетик — с целью верификации диагноза.
10.
ЛечениеИндукторы ферментов монооксидазной системы
гепатоцитов: фенобарбитал и зиксорин (флумецинол) в
дозах от 0,05 до 0,2 г в сутки в течение 2-4нед. Под их
влиянием снижается уровень билирубина в крови и
исчезают диспептические явления. В процессе лечения
фенобарбиталом иногда возникают вялость, сонливость,
атаксия. В этих случаях назначаются минимальные
количества препарата (0,05 г) перед сном, что позволяет
принимать его длительное время. При приеме зиксорина
отмечаются хорошая переносимость препарата, отсутствие
каких-либо побочных действий.
По поводу Зиксорина есть сомнения: с 1998г. его
распространение в России запрещено, а компания
производитель (Гедеон Рихтер) его больше не производит.
Можно применять кордиамин по 30-40 капель 2-3 раза в
день в течение недели. В связи с тем что у значительной
части больных наблюдается развитие холецистита и
желчнокаменной болезни, рекомендуются прием настоев из
желчегонных трав, периодическое проведение тюбажей из
сорбита (ксилита), карловарской соли и соли "Барбара".
Если билирубин достигает 50 мкмоль/л и сопровождается
плохим самочувствием, то возможен приём фенобарбитала
коротким курсом (30-200 мг/сут. в течение 2-4 недель).
Фенобарбитал входит в состав таких препаратов, как
барбовал, корвалол и валокордин, поэтому некоторые
предпочитают применять эти капли (20-25 капель 3 раза в
день), хотя эффект от такого лечения отмечается лишь у
малой части пациентов.
11.
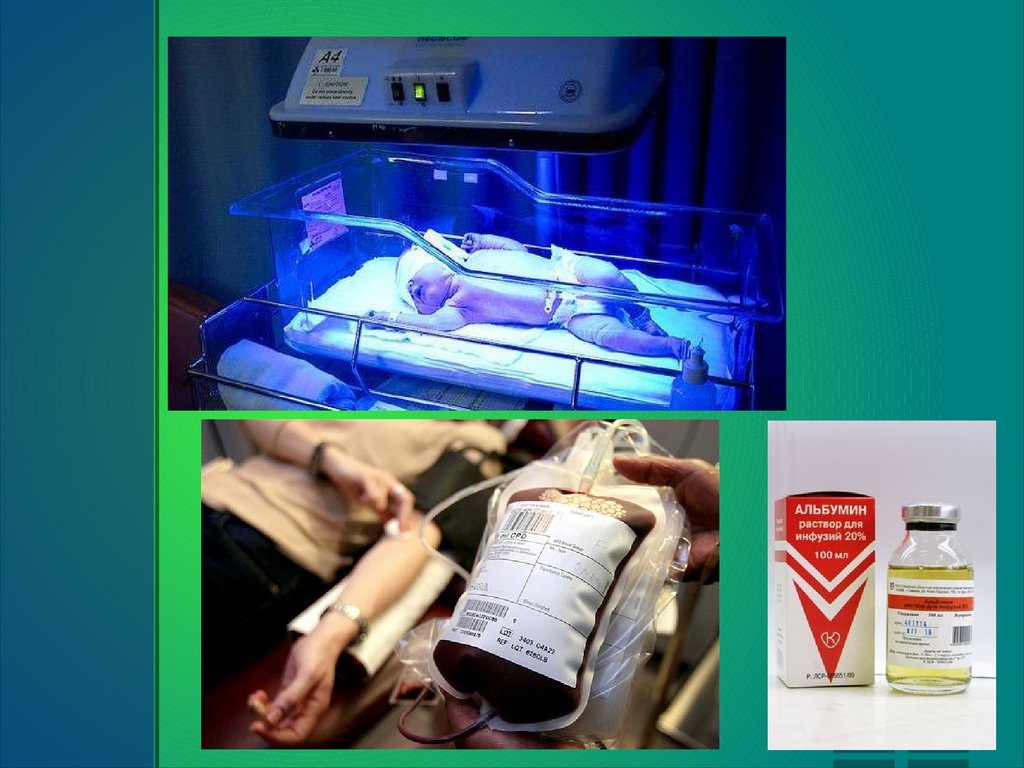
Выведение конъюгированного билирубина (усиленный диурез,активированный уголь как адсорбент билирубина в кишечнике);
Связывание уже циркулирующего билирубина в крови (введение альбумина в
дозе 1 г/кг массы в течение 1 часа). Особенно целесообразно введение
альбумина перед заменным переливанием крови;
Разрушение билирубина, фиксированного в тканях, тем самым
освобождаются периферические рецепторы, которые могут связать новые
порции билирубина, предотвращается его проникновение через
гематоэнцефалический барьер. Достигается это посредством фототерапии.
Максимальный эффект наблюдается при длине волны 450 нм. Лампы с синим
светом более эффективны, однако они затрудняют наблюдение за кожей
ребенка. Фотоисточник помещают на расстоянии 40 — 45 см над телом. Глаза
необходимо защитить.
Стремление избежать провоцирующих факторов (инфекции, физические и
психические нагрузки, употребление алкоголя и гепатотоксичных лекарств)
Противопоказана инсоляция
Диета с ограничением тугоплавких жиров и продуктов, содержащих
консерванты.
Витаминотерапия - особенно витамины группы В.
Санация хронических очагов инфекции и лечение имеющейся патологии
желчевыводящих путей.
В критических случаях — обменное переливание крови.
Возможен курсовой приём гепатопротекторов: Бонджигар, карсил, легалон,
хофитол, ЛИВ-52.
Желчегонные средства в период обострений
Для снижения уровня свободного билирубина целесообразно применять
препараты урсодезоксихолиевой кислоты.
12.
13.
Прогноз благоприятный, зависит от того как протекаетболезнь. Гипербилирубинемия сохраняется пожизненно,
однако не сопровождается повышением смертности.
Прогрессирующие изменения в печени обычно не
развиваются. При страховании жизни таких людей их относят
к группе обычного риска. При лечении фенобарбиталом или
кордиамином уровень билирубина снижается до нормы.
Необходимо предупредить больных, что желтуха может
появиться после интеркуррентных инфекций, повторных рвот
и пропущенного приёма пищи. Отмечена высокая
чувствительность больных к различным гепатотоксическим
воздействиям (алкоголь, многие лекарства и др.). Возможно
развитие воспаления в желчевыводящих путях,
желчнокаменной болезни, психосоматических расстройств.
Родители детей, страдающих этим синдромом, должны
проконсультироваться у генетика перед планированием
очередной беременности. Аналогичным образом следует
поступать, если у родственников семейной пары,
собирающейся иметь детей, диагностирован синдром.
14.
Синдром Криглера-НойяраВрождённая наследственная злокачественная неконъюгированная
гипербилирубинемия, характеризующаяся желтухой и тяжёлым поражением нервной
системы. Тип наследования аутосомно-рецессивный. С равной частотой встречается
у мальчиков и девочек.
Гипербилирубинемия является следствием нарушения конъюгации в печени
билирубина с глюкуроновой кислотой, обусловленного отсутствием или значительной
недостаточностью фермента глюкуронилтрансферазы.
Выделяют два варианта синдрома:
1-й тип: полное отсутствие активности глюкуронилтрансферазы. Обусловлен
мутациями в кодирующей последовательности гена UGTIAI, что приводит к
образованию неполноценного фермента уридиндифосфатглюкуронидазы, который
разрушается. В связи с чем реакции глюкунизации билирубина не происходит и
непрямой билирубин накапливается в организме, в том числе ядрах серого вещества,
обуславливая тяжёлую клинику заболевания.
2-й тип: синдром Ариаса — активность фермента менее 20 % от нормальной.
Заболевание также обусловлено мутациями в кодирующей последовательности гена
UGTIAI. Больные часто являются компаундгетерозиготами, имеющими в одной
хромосоме инсерцию в промотере, а в другой миссенс-мутацию в экзоне. Кроме того
описаны пациенты несущие инсерции в промоторной области гена UGTIAI в
гомозиготном состоянии в сочетании со структурной мутацией в экзоне.
15.
16.
Синдром Люси-Дрисколредкий вариант наследственного пигментного гепатоза, который
именуют «транзиторной семейной гипербилирубинемией
новорожденных» (transient familial neonatal hyperbilirubinemia). Описан в
1960 г. J.F. Lucey и Y.M. Driskoll. Синдром Люси-Дрисколла развивается
уже в первые дни жизни ребенка, проявляясь интенсивной, неуклонно
нарастающей желтухой ядерного типа с накоплением в крови
неконъюгированного билирубина, что в части случаев может обусловить
развитие «билирубиновой энцефалопатии» и летальный исход.
Установлено, что развитие синдрома Люси-Дрисколла объясняется
наличием в молоке матери ингибиторов конъюгации свободного
билирубина с глюкуро-новой кислотой, которые передаются ребенку при
грудном вскармливании. Попадая в общий кровоток, а затем в печень,
они блокируют микросомальный фермент УДФ-ГТ и синтез связанного
билирубина. В настоящее время эти ингибиторы идентифицированы:
они представляют собой прегнан-3в и 2Оа-диол. При своевременном
установлении истинной причины развившейся желтухи и переводе
ребенка на искусственное вскармливание неконъюгированная
гипербилирубинемия постепенно уменьшается и исчезает; в течение 1-2
мес наступает выздоровление.
17.
Синдром Дабина-Джонсонанаследственно детерминированная конъюгированная гипербилирубинемия с
аутосомно-доминантным типом наследования. Синдром Дабина-Джонсона
развивается вследствие несостоятельности АТФ-зависимой канальцевой
транспортной системы, локализованной в билиарной мембране гепатоцита, что
затрудняет экскрецию связанного билирубина в желчь и вызывает его частичное
поступление (рефлюкс, регур-гитацию) из гепатоцита в кровь. Как установлено, в
части случаев cиндрома Дабина-Джонсона наблюдается также затруднение при
захвате и переносе свободного билирубина из крови в гепатоцит, поэтому
гипербилирубинемия часто имеет смешанный характер: повышается
одновременно содержание и связанного, и свободного билирубина.
Гипербилирубинемия достигает уровня 100 мкмоль/л и выше, в основном за счет
конъюги-рованной фракции.
Клинически cиндром Дабина-Джонсона проявляется повышенной
утомляемостью, абдоминальными болями, вплоть до коликообразных,
локализующимися в правом подреберье, тошнотой, кожным зудом, иногда диареей, а также симптомами вегетативной дистонии. В редких случаях
возможен субфебрилитет. 20-30% пациентов cиндромом Дабина-Джонсона
жалоб не предъявляет. В небольшой части случаев увеличены размеры печени,
очень редко - селезенки. У 50% пациентов кал ахоличен, наблюдается
билирубинурия, а также повышенная экскреция копропорфиринов с мочой,
причем 50-80% из них составляет изомер I типа.
Функциональные пробы печени, как правило, не изменены.
18.
При визуальном осмотре через лапароскоп печень при cиндроме ДабинаДжонсона окрашена в коричнево-черный цвет (black liver disease).Морфологическое исследование биоптатов печени обнаруживает в
цитоплазме гепатоцитов аморфные пигментные включения,
локализованные перибилиарно, преимущественно в центре долек,
которые обычно не затрагивают купферовские клетки. Они представляют
собой округлые грубозернистые включения желтовато-коричневого цвета
(«шоколадная печень»), содержащие липофусцин (вещество из группы
хромолипоидов). Их появление связывают с нарушением секреции
анионных метаболитов некоторых аминокислот (тирозина, триптофана,
фенилаланина). При электронной микроскопии биоптатов печени на
билиарном полюсе гепатоцитов полностью отсутствуют микроворсинки
или их количество резко снижено. Пероральная и внутривенная холецистография не выявляет тени желчного пузыря («отрицательная
холецистография»), так как контрастное вещество в желчь не поступает.
Однако признаков холестаза нет.
Характерны результаты бромсульфалеиновой пробы. После
внутривенного введения красителя его концентрация в период с 20-й по
45-ю мин снижается, а с 90-й по 120-ю мин возрастает. Задержка
индикатора в печени достигает 7-10 ч. Радионуклидная проба с
бенгалроз-13 обнаруживает удлинение периода полувыведения
радионуклида до 7 ч. Течение cиндрома Дабина-Джонсона
волнообразное. Прогноз благоприятный. Лечение не разработано.
Рекомендуют витамины, соблюдение диеты, отказ от приема алкоголя.
19.
Синдром Роторавпервые описан A.B. Rotor et al. в 1948 г. под названием «семейная
негемолитическая конъюгированная желтуха».
Это редкая, наследственно детерминированная, преимущественно
конъюгированная гипербилирубинемия с аутосомно-доминантным типом
наследования. Манифестирует обычно с момента рождения ребенка,
иногда - в пубертатном периоде, чаще у детей мужского пола. Клинически
проявляется хронической или интермиттирующей нерезко выраженной
желтухой. Иногда гипербилирубинемия достигает 100 мкмоль/л, главным
образом за счет связанного билирубина, с преобладанием
билирубинмоноглюкуронидов. У значительной части пациентов с
синдромом Ротора наблюдается также умеренное повышение фракции
свободного билирубина. Часть больных с синдромом Ротора предъявляет
диспепсические жалобы, испытывает болевые ощущения в правом
подреберье, указывает на снижение аппетита, повышенную утомляемость,
однако их общее состояние нарушается мало. Возможно и бессимптомное
течение синдрома Ротора. Иногда определяется небольшое увеличение
размеров печени, но селезенка остается в пределах нормы. Функции
печени, как правило, не нарушены. Содержание стеркобилина в кале
периодически снижено, и он становится гипохоличен. В моче определяется
билирубинурия; содержание копропорфиринов в моче незначительно
повышено, преобладает изомер-I. Признаков гемолиза нет.
20.
Бромсульфалеиновая проба регистрирует задержку выделениякрасителя до 45 мин, но отсутствует характерный для синдрома ДабинаДжонсона вторичный подъем («пик») концентрации индикатора в крови.
Лапароскопия не выявляет коричнево-черной окраски печени, а при
гистологическом исследовании ее биоптатов в гепатоцитах отсутствуют
зерна темного пигмента, но в части случаев определяется
мелкокапельная жировая дистрофия по ходу желчных канальцев.
При внутривенной холецистографии желчный пузырь не
визуализируется, но пероральная холецистография выявляет тень
желчного пузыря. В основе синдрома Ротора лежит врожденное
ослабление АТФ-зависимой транспортной системы, специфичной для
некоторых мультивалентных анионов, в том числе для связанного
билирубина. Частично нарушен и транспорт свободного билирубина из
крови в гепатоцит. В результате возникает затруднение экскреции
связанного билирубина из гепатоцита в желчь на уровне аппарата
Гольжди, в периканикулярной зоне и/или на билиарном полюсе
гепатоцита с последующим рефлюк-сом (регургитацией) связанного
билирубина в кровь. По своим клиническим проявлениям и механизму
развития синдрома Ротора близок к синдрому Дабина-Джонсона, но не
идентичен ему. Общее состояние пациентов страдает мало; прогноз
благоприятен. Лечение не разработано.
21.
22.
23.
Выполнила сдудентка 604 группы, КоротееваВ.А.