















 Медицина
МедицинаПохожие презентации:




")

")

")

Фенилкетонурия (ФКУ, фенилпировиноградная олигофрения, болезнь Феллинга). Классическая фенилкетонурия
1.
ФенилкетонурияВЫПОЛНИЛА: ТАКИУЛЛИНА
К.Н. 414 ГР ЛЕЧ. ФАК-ТЕТ.
2.
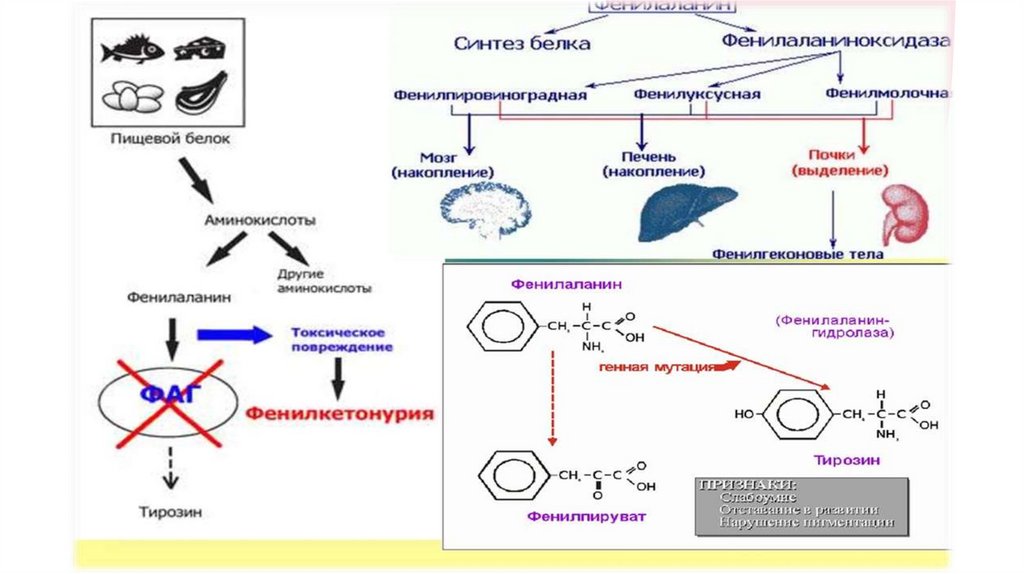
Фенилкетонурия (ФКУ, фенилпировиноградная олигофрения,болезнь Феллинга) – гиперфенилаланинемия (ГФА),
обусловленная недостаточностью активности
фенилаланингидроксилазы (ФАГ) и приводящая к накоплению в
организме ФА и продуктов его метаболизма.
Гиперфенилаланинемии – группа аутосомно-рецессивных
заболеваний, обусловленных нарушением обмена незаменимой
аминокислоты фенилаланина (ФА), поступающей в организм
человека с белковой пищей. Гиперфенилаланинемии (ГФА)
объединяют несколько генетически гетерогенных форм
нарушений обмена ФА, сходных по клиническим признакам:
фенилкетонурия и нарушения обмена тетрагидробиоптерина.
3.
Распространенность:- Частота ГФА среди населения планеты значительно варьирует в
зависимости от популяции: от 1:4370 в Турции до 1:80500 в Японии. Наибольшая распространенность заболевание отмечена среди
европеоидной расы.
- В России по данным неонатального скрининга частота ГФА
составляет 1:7000 и колеблется в различных регионах от 1:4735 в
Курской области до 1:18000 в Республике Тыва.
- ФАГ-дефицитная ГФА выявляется в 97-98% случаев.
- Аутосомно-доминантный тип наследования.
- Ген находится на длинном плече 12 хромосомы.
4.
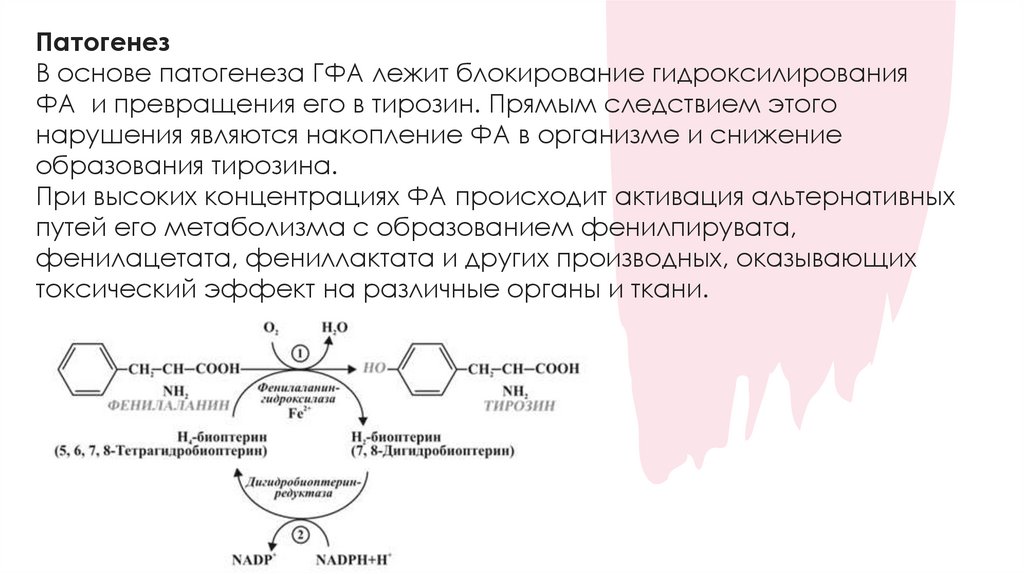
ПатогенезВ основе патогенеза ГФА лежит блокирование гидроксилирования
ФА и превращения его в тирозин. Прямым следствием этого
нарушения являются накопление ФА в организме и снижение
образования тирозина.
При высоких концентрациях ФА происходит активация альтернативных
путей его метаболизма с образованием фенилпирувата,
фенилацетата, фениллактата и других производных, оказывающих
токсический эффект на различные органы и ткани.
5.
6.
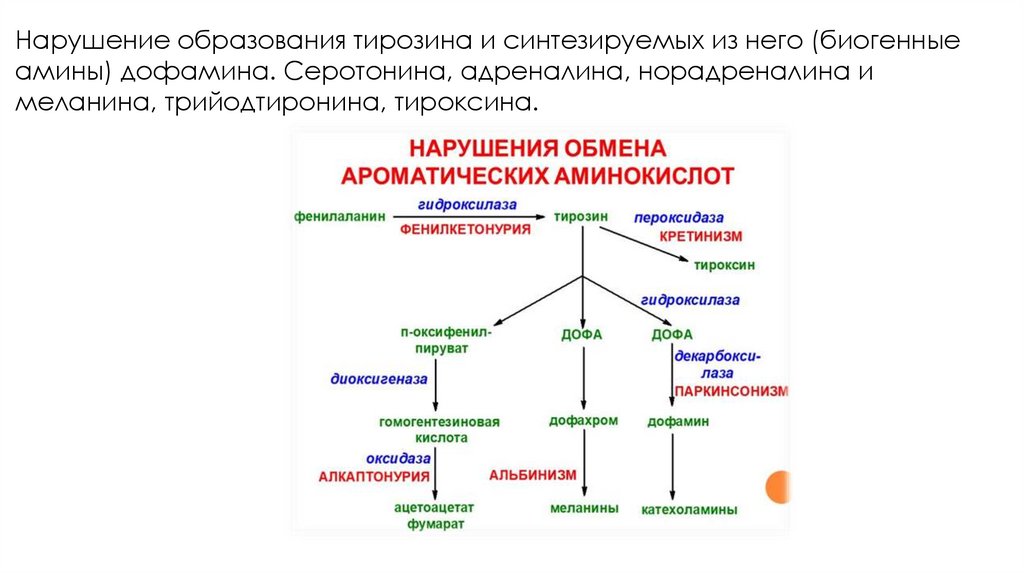
Нарушение образования тирозина и синтезируемых из него (биогенныеамины) дофамина. Серотонина, адреналина, норадреналина и
меланина, трийодтиронина, тироксина.
7.
Классическая фенилкетонурия (фенилкетонурия 1типа)-заболевание вызвано мутацией гена
фенилаланингидроксилазы, локализующегося на
длинном плече хромосомы 12, участке 12q22-q24.1.
Фенилкетонурия 2 типа-заболевание вызвано
мутацией структурного гена
цитозольной дигидроптеридинредуктазы. Ген QDPR
локализован на хромосоме 4p15.3.
Фенилкетонурия 3 типа вызвана мутацией
структурного гена цитозольной 6пирувоилтетрагидроптеринситазы PTS, что приводит к
ее недостаточности в печени и эритроцитах. Ген
PTSрасположен на длинном плече хромосомы 11 в
районе q22.3-23.3.
8.
Для всех указанныхметодов уровень
ФА в крови
человека выше 2,0
мг/дл (120
мкмоль/л)
квалифицируют
как ГФА. ГФА с
уровнем выше 10,0
мг/дл (600
мкмоль/л) относят
к различным
формам ФКУ.
До появления данных молекулярно-генетических
исследований ГФА считалось, что тяжесть заболевания и
степень поражения интеллекта зависят только от уровня
фенилаланина в крови, что тесно связано со степенью
активности фермента.
9.
Симптомы фенилкетонурии:- Новорожденные с фенилкетонурией не имеют клинических
признаков заболевания.
- Обычно манифестация фенилкетонурии у детей происходит в
возрасте 2-6 месяцев. С началом кормления в организм ребенка
начинает поступать белок грудного молока либо его заменителей,
что приводит к развитию первых, неспецифических симптомов –
вялости, иногда – беспокойства и гипервозбудимости, срыгивания,
мышечной дистонии, судорожного синдрома.
Одним из ранних патогномоничных признаков фенилкетонурии
служит упорная рвота, которая нередко ошибочно расценивается
как проявление пилоростеноза.
10.
Ко второму полугодиюстановится заметным
отставание ребенка в
психомоторном развитии.
Ребенок становится менее
активным, безучастным,
перестает узнавать близких,
не пытается садиться и
вставать на ножки.
Аномальный состав мочи и
пота обусловливают
характерный «мышиный»
запах (запах плесени),
исходящий от тела. Часто
наблюдается шелушение
кожи, дерматиты, экзема,
склеродермия.
11.
У детей с фенилкетонуриейне получающих лечение,
выявляется микроцефалия
(уменьшение размера
черепа и головного мозга),
прогнатия, позднее (после
1,5 лет) прорезывание зубов,
гипоплазия эмали.
Отмечается задержка
речевого развития, а к 3-4
годам выявляется глубокая
олигофрения и практически
полное отсуствие речи. Дети
часто белокурые, светлая
кожа, голубые глаза
(дефицит мелатонина)
12.
ЭПИЛЕПТИЧЕСКИЕ ПРИСТУПЫ ВСТРЕЧАЮТСЯ ПОЧТИ УПОЛОВИНЫ НЕЛЕЧЕНЫХ БОЛЬНЫХ И В НЕКОТОРЫХ СЛУЧАЯХ
МОГУТ СЛУЖИТЬ ПЕРВЫМ ПРИЗНАКОМ БОЛЕЗНИ. ОБЫЧНО
ОТМЕЧАЮТСЯ ГЕНЕРАЛИЗОВАННЫЕ ПАРОКСИЗМЫ ПО ТИПУ
ИНФАНТИЛЬНЫХ СПАЗМОВ В ВИДЕ «САЛААМОВЫХ СУДОРОГ»,
КИВКОВ; МОГУТ НАБЛЮДАТЬСЯ АБСАНСЫ. ПРИСТУПЫ НОСЯТ
УПОРНЫЙ ХАРАКТЕР И ПЛОХО ПОДДАЮТСЯ
АНТИКОНВУЛЬСАНТНОЙ ТЕРАПИИ.
13.
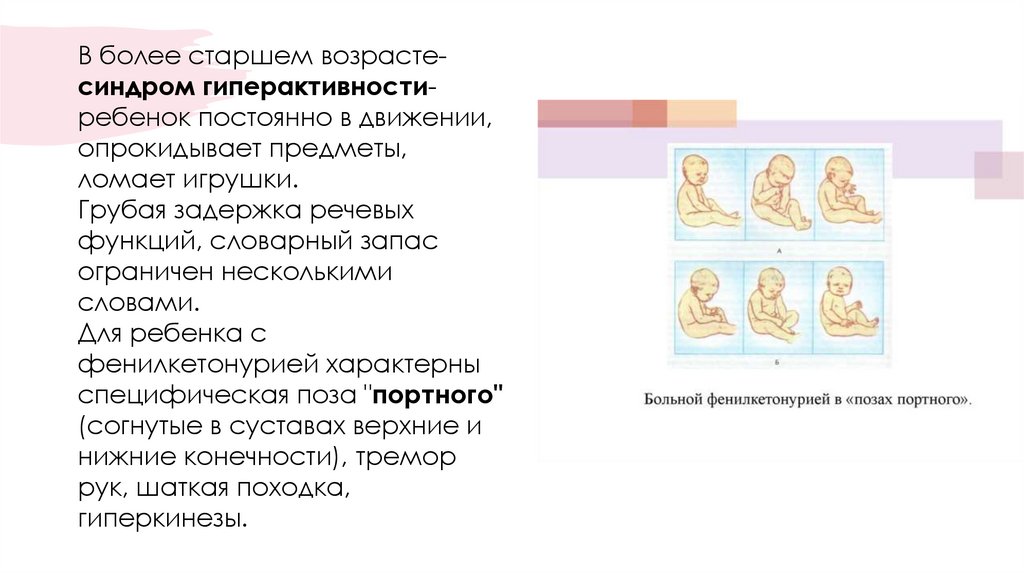
В более старшем возрастесиндром гиперактивностиребенок постоянно в движении,опрокидывает предметы,
ломает игрушки.
Грубая задержка речевых
функций, словарный запас
ограничен несколькими
словами.
Для ребенка с
фенилкетонурией характерны
специфическая поза "портного"
(согнутые в суставах верхние и
нижние конечности), тремор
рук, шаткая походка,
гиперкинезы.
14.
Грубые психические нарушения, принегрубых неврологических симптомах:
1. Повышение или понижение мышечного
тонуса.
2. Пирамидный синдром.
3. Мышечная дистония.
4. Легкая мозжечковая атаксия.
5. Затруднение движений- в большей
степени из-за корковых нарушений.
15.
Диагностика:1. Рекомендуется проведение неонатального скрининга
(определение концентрации фенилаланина в сухих
пятнах крови) для доклинической диагностики ГФА и
своевременного начала патогенетической терапии
2. Нагрузочный тест с фенилаланином (180 мг/кг)- не
увеличивается уровень биоптерина.
3. Нагрузочный тест с тетрагидробиоптерином (5-20 мг/кг)нормализация плазменного фенилаланина через 4-8
часов.
4. Нахождение кетокислот в моче (зеленое окрашивание
мочи при добавлении 5% раствора хлорида железа
(проба Фелинга)).
5. Хроматографическое исследование аминокислот в
плазме крови и в моче.
6. ДНК
16.
Диагностика:1. Рекомендовано проведение электроэнцефалографии для
выявления паттернов гипсаритмии (даже при отсутствии
клинических судорожных приступов.
2. Проведение магнитно-резонансной томографии с целью
выявления очагов перивентрикулярной лейкопатии, кортикальной
атрофии и других изменений у пациентов старше 12 лет.
3. Рекомендовано проведение ультразвукового исследования
брюшной полости и почек для диагностики дискинезии желчных
путей, диффузных изменений печени и поджелудочной железы,
мочекаменной болезни.
4. Проведение эзофагогастрофиброскопии для диагностики
поражения слизистой оболочки желудка (по показаниям).
5. Рекомендовано психолого-педагогическое консультирование и
логопедическое тестирование.
17.
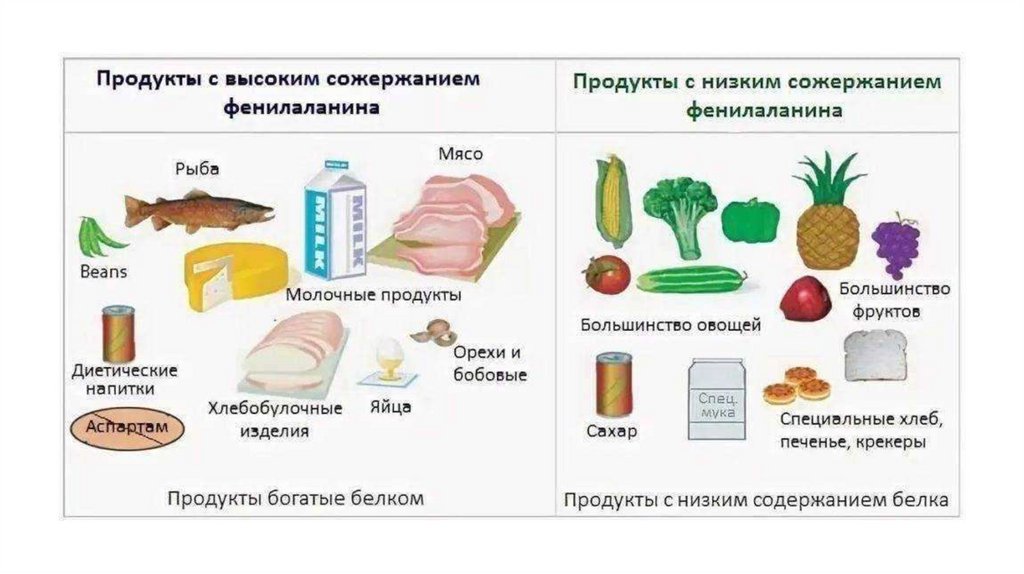
Лечение :Важно- не позже 3х недельного возраста!
Согласно современным данным, диетическое лечение назначают
при уровне фенилаланина на скрининге ≥360 мкмоль/л (≥6 мг/дл).
1. Диета с ограничением фенилаланина (до 250-500 мг/сут).
2. Исключить высокобелковые продукты ( мясо, рыба, печень, яйца,
творог, бобовые).
3. Можно употреблять овощи, фрукты, мед, подсолнечное масло.
4. Употребление специальных пищевых смесей без фенилаланина.
18.
В зависимости от переносимости пищевого фенилаланинадопустимое и безопасное его количество в сутки составляет от 90 до
35 мг/кг массы тела для детей первого года жизни. В питании детей
старше 1 года допустимое количество фенилаланина постепенно
снижается с 35 до 10 мг/кг массы тела ребенка.
19.
20.
1. Рекомендуется (по показаниям) медикаментозная терапиясапроптерином ФКУ, обусловленной дефектами обмена ФАГ (легкие и
умеренные формы), после проведения теста и подтверждения
чувствительности к сапроптерину.
(Начальная доза сапроптерина дигидрохлорида от 2 до 5 мг/кг массы тела
при приеме 1 раз в день. Доза может быть увеличена до 20 мг/кг массы тела
в день. Для достижения оптимального терапевтического эффекта суточная
доза препарата может быть разделена на 2 или 3 приема в течение дня).
2. В комплекс лечения также рекомендовано включать препараты леводопы
(10−15 мг/кг в сутки) в сочетании с карбидопой в дозировке 1−1,5 мг/кг в сутки,
5-гидрокситриптофан (10 мг/кг в сутки) (препарат в Российской Федерации в
настоящее время не зарегистрирован), 5- формилтетрагидрофолат
(кальция фолинат) в средней дозе 25 мг/сут, в некоторых случаях диета с
ограничением фенилаланина и фолиевой кислоты.
21.
Профилактика1. Пренатальная диагностика-определение активности
дигидроптерилинредкутазы в культуре амниоцитов.
2. Массовый скрининг новорожденных (доклиническая диагностика).
3. Уровень ФА в крови новорожденных в условиях регионального МГК.
1 этап. Уровень ФА выше 2 мг%- требуется повторный контроль уровня
ФА.
2 этап. Уровень 2-6мг%- гиперфенилаланинемия. Ограничение
потребления белка+динамический контроль.
3 этап. Уровень ФА выше 6 мг- ФКУ. Дополнительная диагностикаподтверждение диагноза. Жесткая диета, лечебные смеси, лечение
по стандарту ФКУ.