



















 Медицина
МедицинаПохожие презентации:






")



Хронічні лейкемії
1.
Хронічні лейкеміїВиконала: студентка 1А групи, 5 курсу
Січко Лідія Сергіївна
2.
• Злоякісні клональні захворювання кісткового мозку, що розвиваються внаслідоквиникнення мутацій в стовбурових клітинах кісткового мозку та полягають в
акумуляції лейкемічних клітин з різних ступенем диференціювання та дозрівання в
кістковому мозку та екстрамедулярно.
• В залежності від ступеня дозрівання лейкемічних клітин всі лейкемії поділяють на
гострі та хронічні.
• Гострі лейкемії – лейкемічні клітини здатні диференціюватись до IV класу клітин
кісткового мозку (бластів).
• Хронічні лейкемії – лейкемічні клітини здатні диференціюватись до V та VI класу
клітин кісткового мозку (дозріваючі та зрілі клітини).Лейкемічний провал відсутній.
Хронічні мієлоїдні лейкози
Мієлоцитарний Еритробластний
Хронічні лімфоїдні лейкози
В-лімфоцитарний
Моноцитарний
Еозинофільний
Т-лімфоцитарний
Мегакаріоцитарний
3.
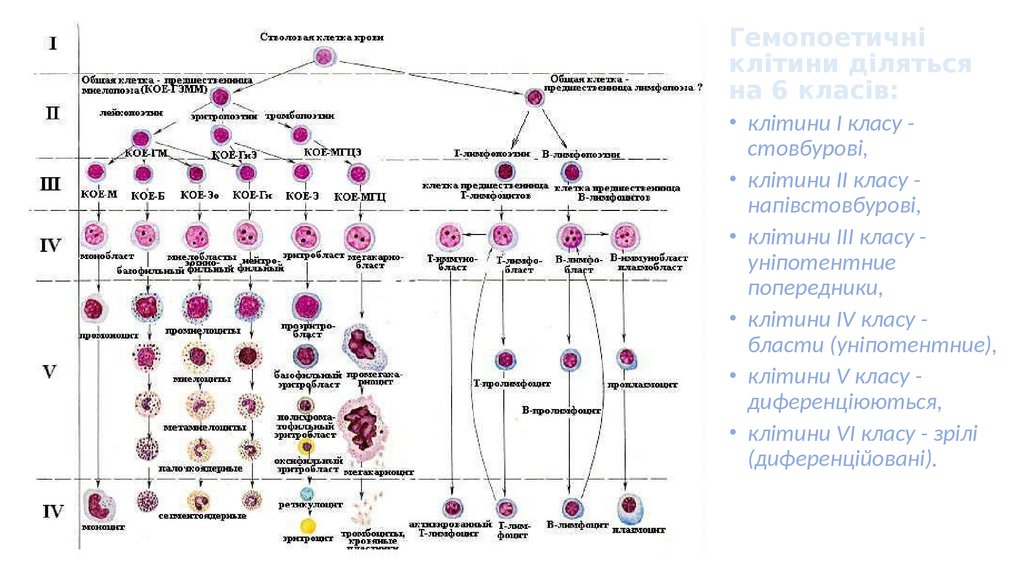
Гемопоетичніклітини діляться
на 6 класів:
• клітини I класу стовбурові,
• клітини II класу напівстовбурові,
• клітини III класу уніпотентние
попередники,
• клітини IV класу бласти (уніпотентние),
• клітини V класу диференціюються,
• клітини VI класу - зрілі
(диференційовані).
4.
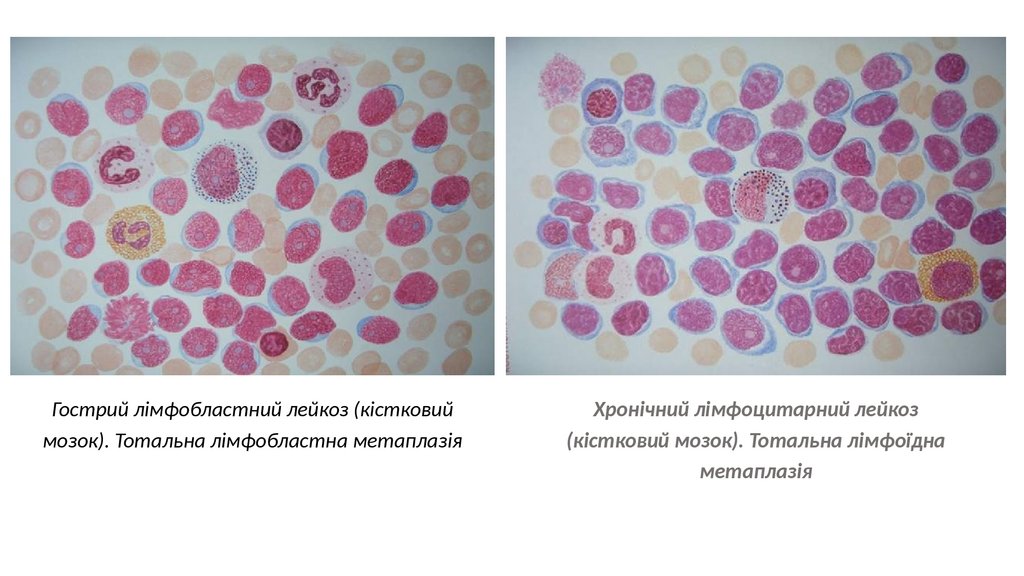
Гострий лімфобластний лейкоз (кістковиймозок). Тотальна лімфобластна метаплазія
Хронічний лімфоцитарний лейкоз
(кістковий мозок). Тотальна лімфоїдна
метаплазія
5.
Класифікація мієлоїдних неоплазій,ВООЗ, 2016 р.
1. Мієлопроліферативні неоплазії(MPN) 2. Мієлоїдні, лімфоїдні неоплазії з еозинофілією
1.1. Хронічна мієлоїдна лейкемія,BCR-ABL1–
позитивна (ХМЛ)
1.2. Справжня поліцитемія (СП)
1.3. Есенціальна тромбоцитемія (ET)
1.4. Первинний мієлофіброз (ПМФ)
1.5. Хронічна нейтрофільна лейкемія (ХНЛ)
1.6. Хронічна еозинофільна лейкемія(ХЕЛ)
1.7. Хвороба з тучних клітин (ТКХ)
1.8. Мієлопроліферативні неоплазії,
некласифіковані.
та аномаліями of PDGFRA, PDGFRB, and FGFR1.
3. MDS/MPN
3.1. Хронічна мієломоноцитарна лейкемія (ХММЛ)
3.2. Ювенільна мієломоноцитарна лейкемія (ЮММЛ)
3.3. Атипова хронічна мієлоїдна лейкемія, BCR-ABL–
негативна (aХМЛ).
3.4. МДС/МПН, некласифіковані.
4. Мієлодиспластичні синдроми (МДС)
5. Гостра мієлоїдна лейкемія (ГМЛ).
6.
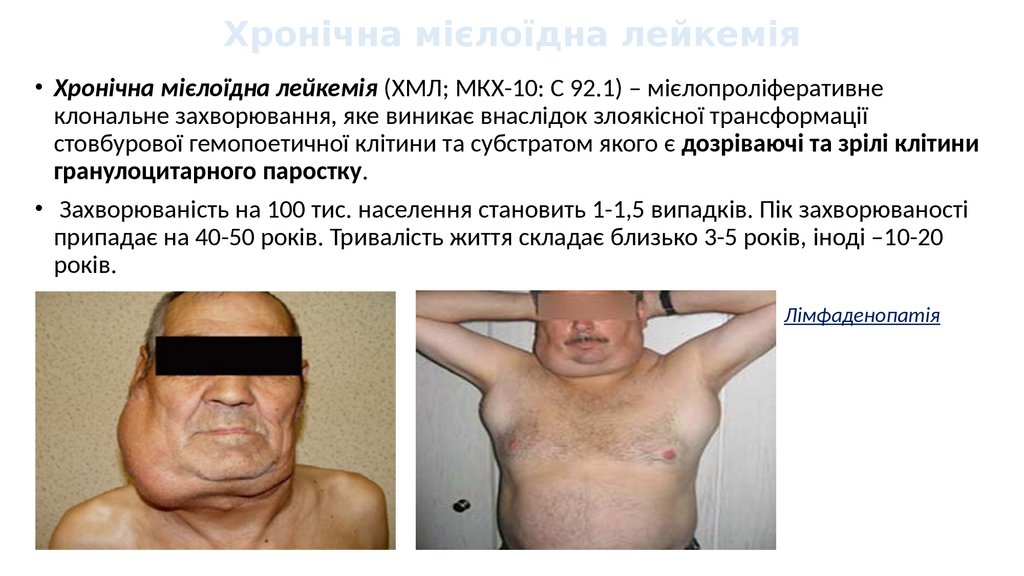
Хронічна мієлоїдна лейкемія• Хронічна мієлоїдна лейкемія (ХМЛ; МКХ-10: С 92.1) – мієлопроліферативне
клональне захворювання, яке виникає внаслідок злоякісної трансформації
стовбурової гемопоетичної клітини та субстратом якого є дозріваючі та зрілі клітини
гранулоцитарного паростку.
• Захворюваність на 100 тис. населення становить 1-1,5 випадків. Пік захворюваності
припадає на 40-50 років. Тривалість життя складає близько 3-5 років, іноді –10-20
років.
Лімфаденопатія
7.
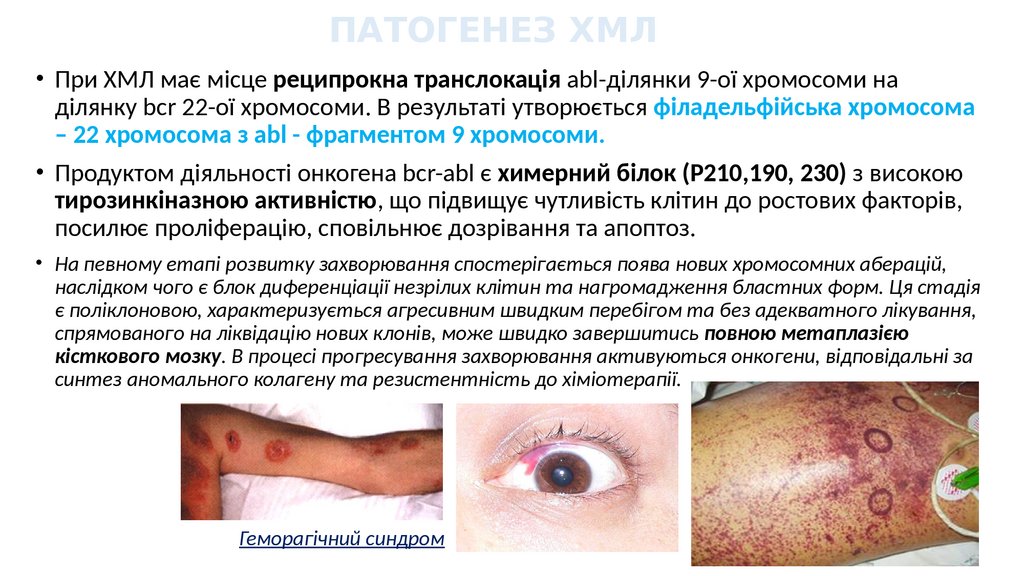
ПАТОГЕНЕЗ ХМЛ• При ХМЛ має місце реципрокна транслокація abl-ділянки 9-ої хромосоми на
ділянку bcr 22-ої хромосоми. В результаті утворюється філадельфійська хромосома
– 22 хромосома з abl - фрагментом 9 хромосоми.
• Продуктом діяльності онкогена bcr-abl є химерний білок (Р210,190, 230) з високою
тирозинкіназною активністю, що підвищує чутливість клітин до ростових факторів,
посилює проліферацію, сповільнює дозрівання та апоптоз.
• На певному етапі розвитку захворювання спостерігається поява нових хромосомних аберацій,
наслідком чого є блок диференціації незрілих клітин та нагромадження бластних форм. Ця стадія
є поліклоновою, характеризується агресивним швидким перебігом та без адекватного лікування,
спрямованого на ліквідацію нових клонів, може швидко завершитись повною метаплазією
кісткового мозку. В процесі прогресування захворювання активуються онкогени, відповідальні за
синтез аномального колагену та резистентність до хіміотерапії.
Геморагічний синдром
8.
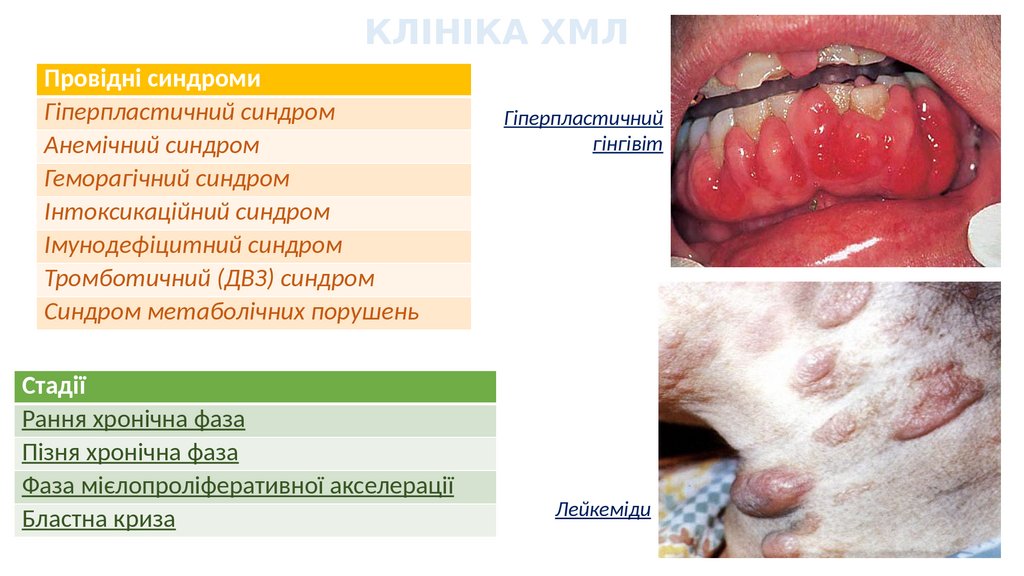
КЛІНІКА ХМЛПровідні синдроми
Гіперпластичний синдром
Анемічний синдром
Геморагічний синдром
Інтоксикаційний синдром
Імунодефіцитний синдром
Тромботичний (ДВЗ) синдром
Синдром метаболічних порушень
Стадії
Рання хронічна фаза
Пізня хронічна фаза
Фаза мієлопроліферативної акселерації
Бластна криза
Гіперпластичний
гінгівіт
Лейкеміди
9.
КЛІНІКА ХМЛІнтоксикаційний синдром
•підвищення температури до тіла до субфебрильних цифр, а в фазу акселерації до 38,0 С (протягом 2
тижнів),
•профузна пітливість,
•втрата апетиту та зниження маси тіла на 10 % за 6 міс.
Тромбо-геморагічний синдром (метапластична тромбоцитопенія)
•Хронічний синдром ДВЗ (порушення в системі коагулянтів, антикоагулянтів та фібринолізу)
•Характерні артеріальні та венозні тромбози, з утворенням тромбоцитарних, лейкоцитарних,
фібринових тромбів.
•Геморагічні прояви (петехії, екхімози, синці, кровотечі зі слизових) виникають внаслідок
тромбоцитопенії та ДВЗ.
Анемічний синдром.
Імунодефіцитний синдром.
• рецидивуючі бактеріальні захворювання з боку оргаів;
• рецидивуючі вірусні інфекції;
10.
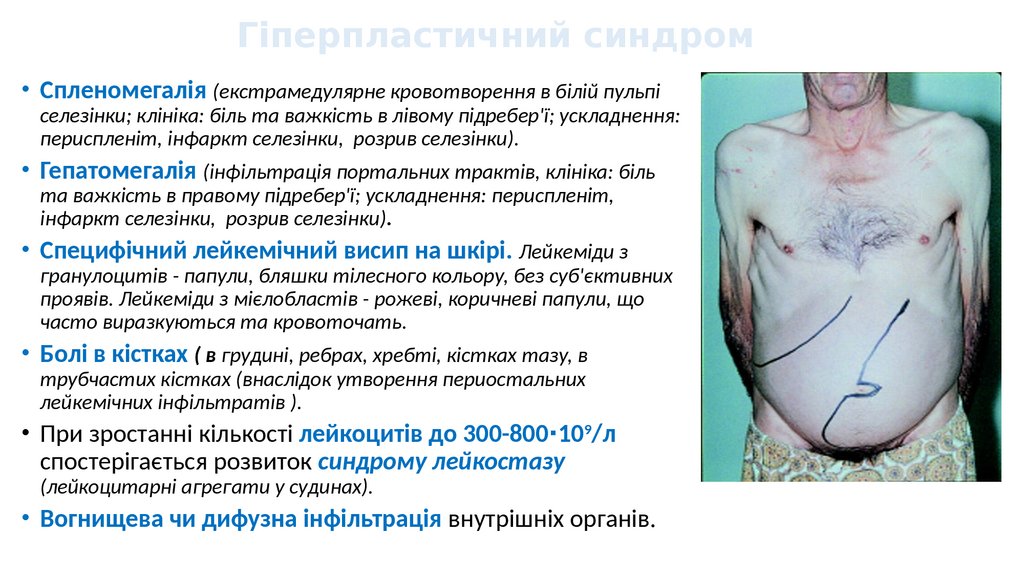
Гіперпластичний синдром• Спленомегалія (екстрамедулярне кровотворення в білій пульпі
селезінки; клініка: біль та важкість в лівому підребер'ї; ускладнення:
периспленіт, інфаркт селезінки, розрив селезінки).
• Гепатомегалія (інфільтрація портальних трактів, клініка: біль
та важкість в правому підребер'ї; ускладнення: периспленіт,
інфаркт селезінки, розрив селезінки).
• Специфічний лейкемічний висип на шкірі. Лейкеміди з
гранулоцитів - папули, бляшки тілесного кольору, без суб'єктивних
проявів. Лейкеміди з мієлобластів - рожеві, коричневі папули, що
часто виразкуються та кровоточать.
• Болі в кістках ( в грудині, ребрах, хребті, кістках тазу, в
трубчастих кістках (внаслідок утворення периостальних
лейкемічних інфільтратів ).
• При зростанні кількості лейкоцитів до 300-800∙109/л
спостерігається розвиток синдрому лейкостазу
(лейкоцитарні агрегати у судинах).
• Вогнищева чи дифузна інфільтрація внутрішніх органів.
11.
Гіперпластичний синдромНервова система: Нейролейкемія – дисемінація лейкемічних клітин в оболонках та речовині
головного та спинного мозку, нервових стовбурах.
Клінічні форми: менінгеальна, енцефалітична, діенцефальна, мієлітична, полірадикулоневритична.
Нирки: специфічна нефропатія; нефромегалія; сечовий синдром з гематуричним компонентом,
хронічна ниркова недостатність; гостра ниркова недостатність (під час ХТ внаслідок гіперурикемії та
обтурації ниркових канальців уратами).
Легені:
•лейкемічний пульмоніт (вогнищеві або дифузні інфільтрати) (задишка, кашель, кровохаркання,
крепітація та вологі хрипи), розпад інфільтратів з формуванням каверн; сухий чи ексудативний плеврит,
• компресії бронхів збільшеними лімфовузлами, ателектази легень.
Міокард та перикард: специфічний міокардит, перикардит; порушення ритму, провідності,
прогресування серцевої недостатності.
Клініка:
- некардіогенний набряк легень (РДС): задишка, дист. хрипи, ціаноз;
- енцефалопатія, набряк головного мозку (галюцинації, порушення мови, головний біль, блювання, судоми);
- зміни на очному дні (набряк диску зорового нерву, крововиливи),
- враження статевих органів (пріапізм).
12.
Діагностичний протокол для ХМЛ1. Загальний аналіз крові.
2. Стернальна пункція та цитологічне дослідження КМ.
гіперклітинність: розширення гранулоцитарного та мегакаріоцитарного але, звуження
еритроїдного паростків; збільшення кількості дозріваючих гранулоцитів та мієлоблостів
3. Трепанбіопсія та гістологічне дослідження кісткового мозку.
збільшення вмісту колагену ІІІ типу (ретикуліновий фіброз)
4. Цитогенетичне дослідження периферичної крові та кісткового
мозку методами каріотипування чи FISH (для виявлення
Філадельфійської хромосоми).
5. ПЛР для виявлення транскриптів гена BCR-ABL.
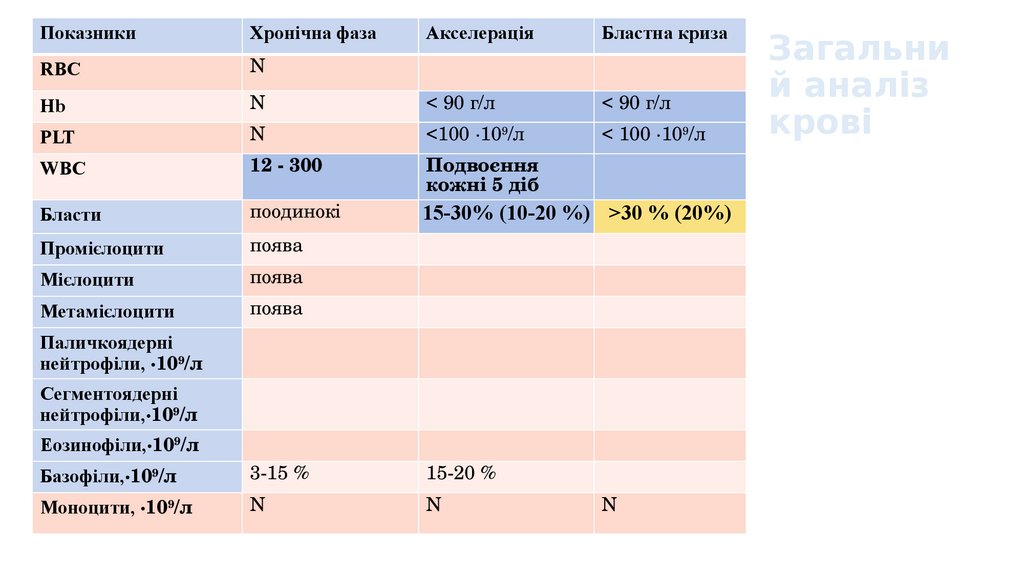
13.
ПоказникиХронічна фаза
Акселерація
Бластна криза
RBC
N
Hb
N
< 90 г/л
< 90 г/л
PLT
N
<100 ∙109/л
< 100 ∙109/л
WBC
12 - 300
Подвоєння
кожні 5 діб
Бласти
поодинокі
15-30% (10-20 %) >30 % (20%)
Промієлоцити
поява
Мієлоцити
поява
Метамієлоцити
поява
Паличкоядерні
нейтрофіли, ∙109/л
Сегментоядерні
нейтрофіли,∙109/л
Еозинофіли,∙109/л
Базофіли,∙109/л
3-15 %
15-20 %
Моноцити, ∙109/л
N
N
N
Загальни
й аналіз
крові
14.
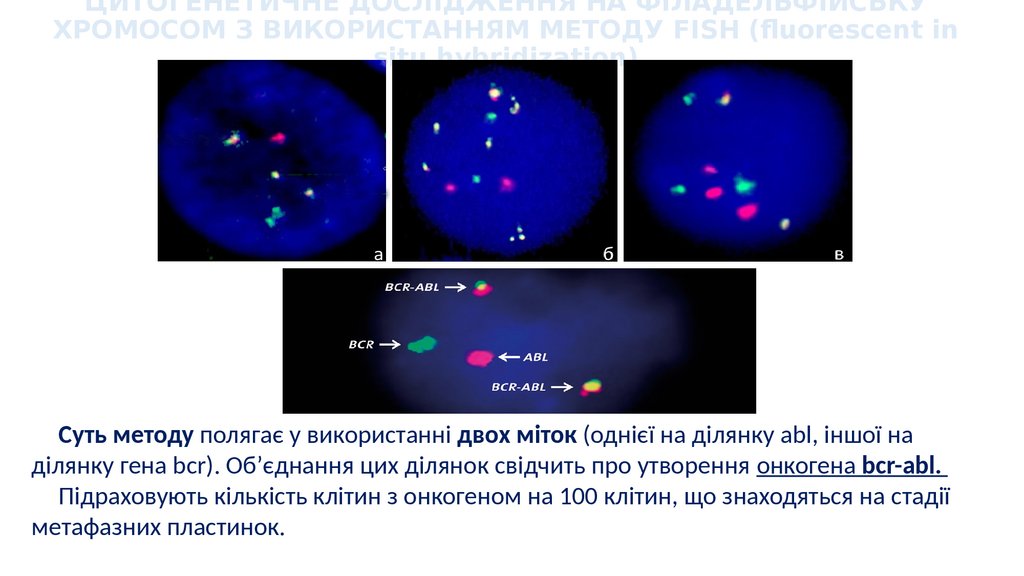
ЦИТОГЕНЕТИЧНЕ ДОСЛІДЖЕННЯ НА ФІЛАДЕЛЬФІЙСЬКУХРОМОСОМ З ВИКОРИСТАННЯМ МЕТОДУ FISH (fluorescent in
situ hybridization)
Суть методу полягає у використанні двох міток (однієї на ділянку abl, іншої на
ділянку гена bcr). Об’єднання цих ділянок свідчить про утворення онкогена bcr-abl.
Підраховують кількість клітин з онкогеном на 100 клітин, що знаходяться на стадії
метафазних пластинок.
15.
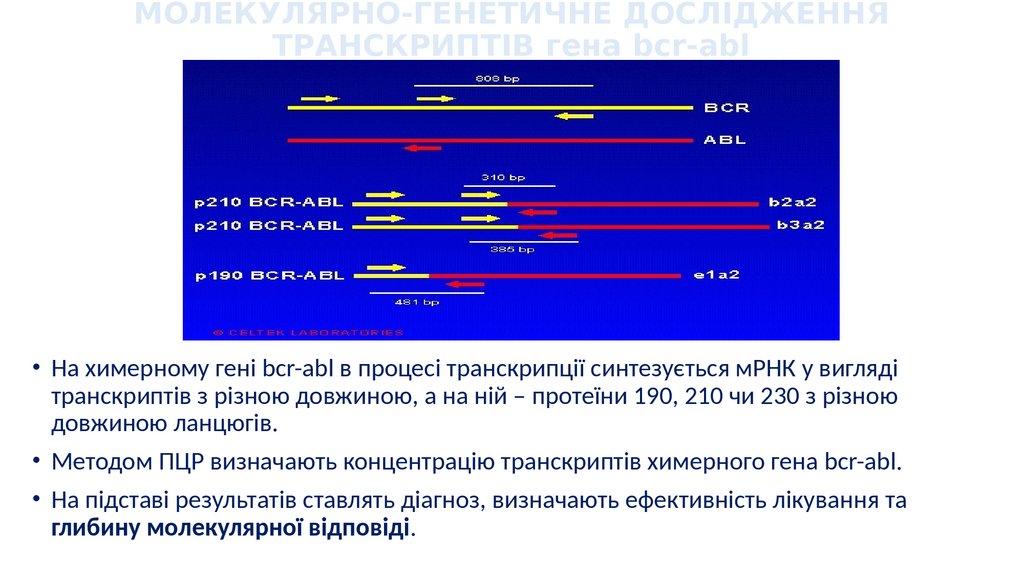
МОЛЕКУЛЯРНО-ГЕНЕТИЧНЕ ДОСЛІДЖЕННЯТРАНСКРИПТІВ гена bcr-abl
• На химерному гені bcr-abl в процесі транскрипції синтезується мРНК у вигляді
транскриптів з різною довжиною, а на ній – протеїни 190, 210 чи 230 з різною
довжиною ланцюгів.
• Методом ПЦР визначають концентрацію транскриптів химерного гена bcr-abl.
• На підставі результатів ставлять діагноз, визначають ефективність лікування та
глибину молекулярної відповіді.
16.
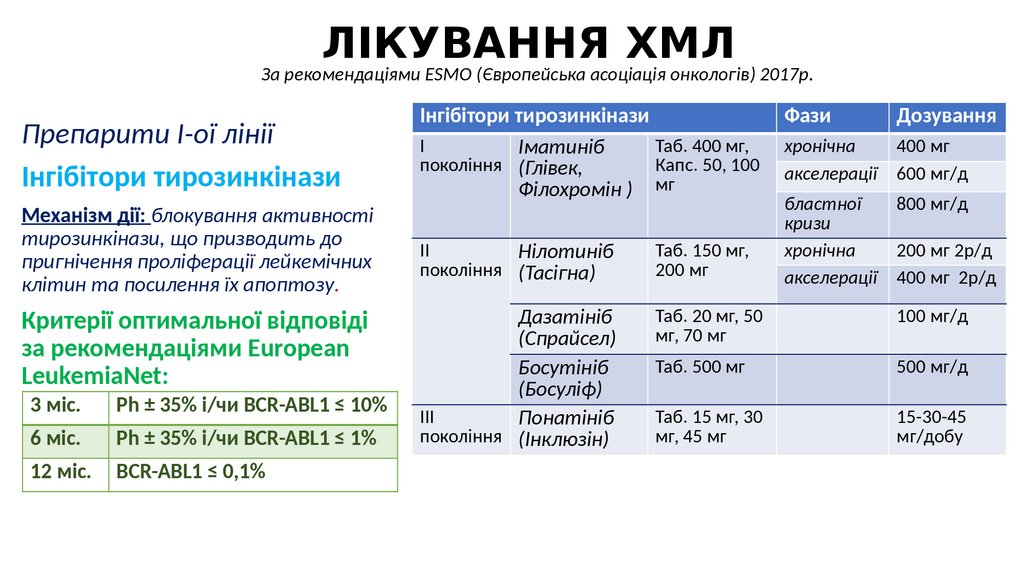
ЛІКУВАННЯ ХМЛЗа рекомендаціями ESMO (Європейська асоціація онкологів) 2017р.
Препарити І-ої лінії
Інгібітори тирозинкінази
Механізм дії: блокування активності
тирозинкінази, що призводить до
пригнічення проліферації лейкемічних
клітин та посилення їх апоптозу.
Критерії оптимальної відповіді
за рекомендаціями European
LeukemiaNet:
3 міс.
Ph ± 35% і/чи BCR-ABL1 ≤ 10%
6 міс.
Ph ± 35% і/чи BCR-ABL1 ≤ 1%
12 міс.
BCR-ABL1 ≤ 0,1%
Інгібітори тирозинкінази
І
Таб. 400 мг,
Іматиніб
покоління (Глівек,
Капс. 50, 100
Філохромін ) мг
Фази
Дозування
хронічна
акселерації
400 мг
600 мг/д
бластної
кризи
хронічна
акселерації
800 мг/д
ІІ
Нілотиніб
покоління (Тасігна)
Таб. 150 мг,
200 мг
Дазатініб
(Спрайсел)
Босутініб
(Босуліф)
ІІІ
Понатініб
покоління (Інклюзін)
Таб. 20 мг, 50
мг, 70 мг
100 мг/д
Таб. 500 мг
500 мг/д
Таб. 15 мг, 30
мг, 45 мг
15-30-45
мг/добу
200 мг 2р/д
400 мг 2р/д
17.
ЛІКУВАННЯ ХМЛ• У випадках неможливості забезпечення пацієнта препаратами та,
коли ІТК не можуть бути використані, призначають
гідроксисечовину та/або ІФН-α.
Гідроксисечовина
Інтерферон-α Цитарабін
Механізм дії: блокування
рибонуклеотидредуктазного
комплексу, що пригнічує синтез ДНК)
(роферон, інтрон,
лаферон)
Антинеопластичні
засоби. Структурні
аналоги піримідину
40 мг/кг при лейкоцитозі 40-100
∙109/л,
30 мг/кг при лейкоцитозі 20-40∙109/л,
20 мг/кг при лейкоцитозі 5-20 ∙109/л .
5 млн МО/м2; в
монотерапії чи в
комбінації з
гідроксисечовино
ю.
6-меркаптопурин
(ПУРИ-НЕТОЛ)
Антинеопластичні
засоби.
Структурні
аналоги пурину.
10мг/м2/добу - 10 діб 50-100 мг/добу.
щомісяця, в хронічну
фазу,
20 мг/м2/добу - 14
діб щомісяця в фазу
акселерації.
18.
Справжня поліцитеміяЕритремія – хронічне мієлопроліферативне
Провідні синдроми
клональне захворювання, що розвивається
Плеторичний синдром
внаслідок ураження клітини-попередниці
мієлопоезу та характеризується проліферацією Синдром мікроциркуляторних порушень
клітин еритроїдного, мегакаріоцитарного та
Тромбо-геморагічний синдром
гранулоцитарного паростків.
Інтоксикаційний синдром
Етіологія та патогенез
•Мутація гену рецептора до еритопоетину
jak-2 призводить до гіперчутливості
еритроїдних клітин патологічного клону до
еритропоетину.
•Клітини цієї популяції швидко проліферують,
а здатність до апоптозу різко знижується.
Патологічний клон витісняє нормальний клон з
кісткового мозку.
Імунодефіцитний синдром
Гіперпластичний синдром
(гепатоспленомегалія)
Синдром метаболічних порушень
(гіперурикемія, вторинна подагра)
19.
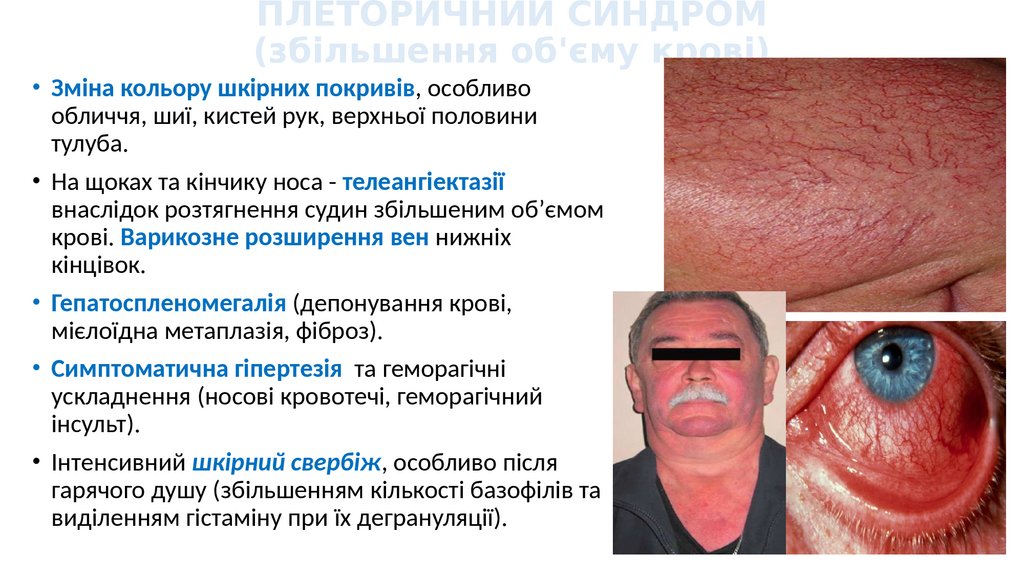
ПЛЕТОРИЧНИЙ СИНДРОМ(збільшення об'єму крові)
• Зміна кольору шкірних покривів, особливо
обличчя, шиї, кистей рук, верхньої половини
тулуба.
• На щоках та кінчику носа - телеангіектазії
внаслідок розтягнення судин збільшеним об’ємом
крові. Варикозне розширення вен нижніх
кінцівок.
• Гепатоспленомегалія (депонування крові,
мієлоїдна метаплазія, фіброз).
• Симптоматична гіпертезія та геморагічні
ускладнення (носові кровотечі, геморагічний
інсульт).
• Інтенсивний шкірний свербіж, особливо після
гарячого душу (збільшенням кількості базофілів та
виділенням гістаміну при їх дегрануляції).
20.
Тромбо-геморагічний синдромЗростання кількості еритроцитів та тромбоцитів, їх адгезивно-агрегаційних
властивостей призводить до вповільнення кровотоку, формування
тромбоцитарно-еритроцитарних агрегатів, гіпоксії тканин.
• Еритромелалгії – сильні пекучі болі в кінчиках пальців рук та ніг (ішемія кінчиків
пальців).
• Синдром Рейно.
• Стенокардія, дифузний кардіосклероз та серцева недостатність (ішемія міокарду).
• Хронічна енцефалопатія.
• Дифузний пневмосклероз, емфізема, ЛН.
• Виразки шлунка (ішемія слизової);
• Ішемічний коліт, абдомінальний ішемічний синдром.
21.
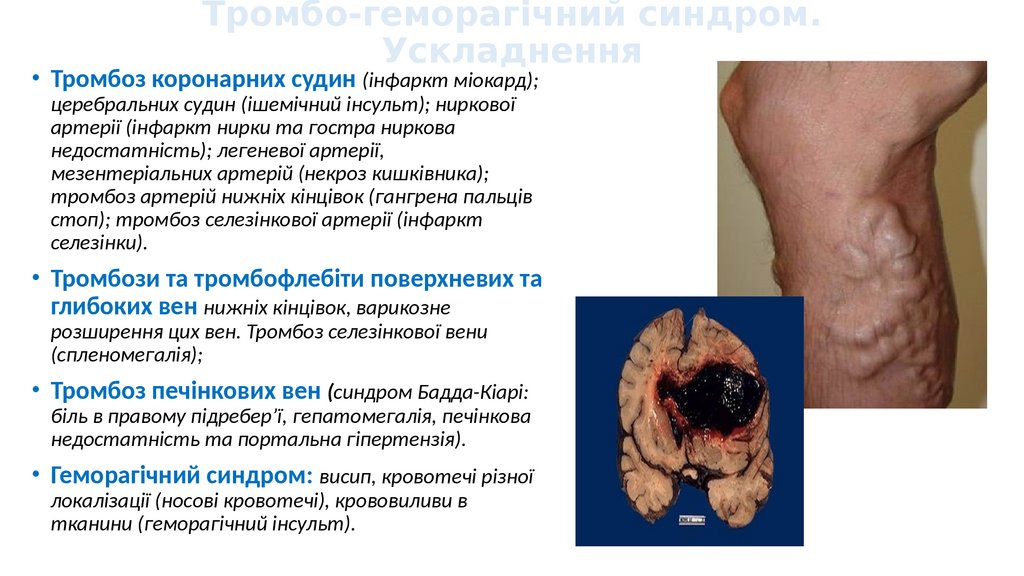
Тромбо-геморагічний синдром.Ускладнення
• Тромбоз коронарних судин (інфаркт міокард);
церебральних судин (ішемічний інсульт); ниркової
артерії (інфаркт нирки та гостра ниркова
недостатність); легеневої артерії,
мезентеріальних артерій (некроз кишківника);
тромбоз артерій нижніх кінцівок (гангрена пальців
стоп); тромбоз селезінкової артерії (інфаркт
селезінки).
• Тромбози та тромбофлебіти поверхневих та
глибоких вен нижніх кінцівок, варикозне
розширення цих вен. Тромбоз селезінкової вени
(спленомегалія);
• Тромбоз печінкових вен (синдром Бадда-Кіарі:
біль в правому підребер’ї, гепатомегалія, печінкова
недостатність та портальна гіпертензія).
• Геморагічний синдром: висип, кровотечі різної
локалізації (носові кровотечі), крововиливи в
тканини (геморагічний інсульт).
22.
Діагностичний протокол1. Загальний аналіз крові.
2. Стернальна пункція та цитологічне дослідження КМ.
гіперплазія гранулоцитарного, мегакаріоцитарного та еритроїдного паростків
3. Трепанбіопсія та гістологічне дослідження кісткового мозку.
панмієлоз, зменшення вмісту жирової тканини, порушення диференціації еритробластів; в
термінальній стадії- картина мієлофіброзу
4. Цитогенетичне дослідження
мутація jak 2-V617F
5. Вміст еритропоетину знижений.
23.
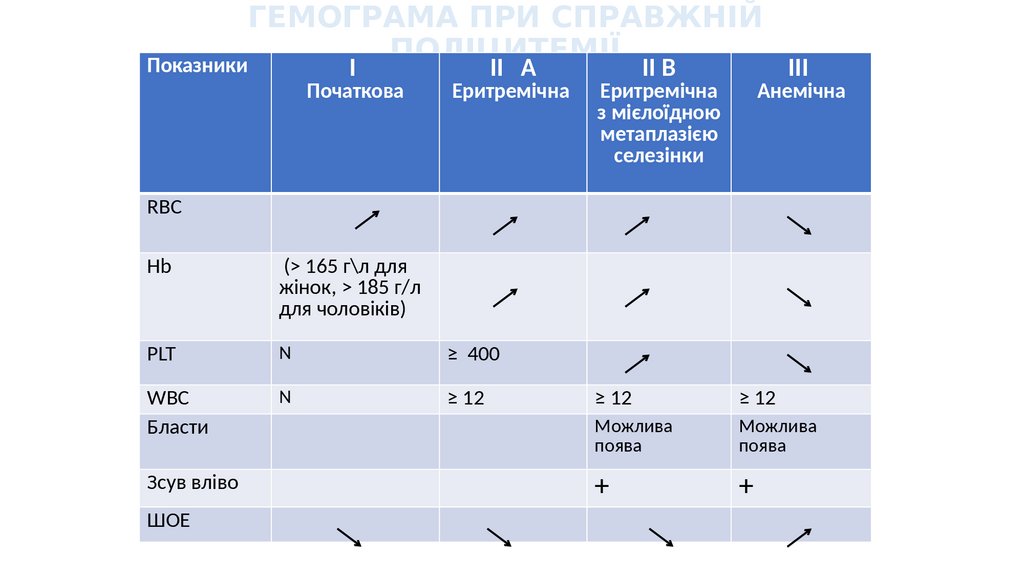
ГЕМОГРАМА ПРИ СПРАВЖНІЙПОЛІЦИТЕМІЇ
Показники
I
Початкова
II А
Еритремічна
II В
Еритремічна
з мієлоїдною
метаплазією
селезінки
III
Анемічна
RBC
Hb
(> 165 г\л для
жінок, > 185 г/л
для чоловіків)
PLT
N
≥ 400
WBC
Бласти
N
≥ 12
Зсув вліво
ШОЕ
≥ 12
≥ 12
Можлива
поява
Можлива
поява
+
+
24.
Діагностичні критерії (ВООЗ, 2008рік)
Великі критерії:
1. Підвищення вмісту гемоглобіну: у чоловіків >185, у жінок> 165 г/л.
2. Наявність jak 2-V617F або подібних мутацій.
Малі критерії:
1. Трьохлінійна проліферація кісткового мозку.
2. Зниження вмісту еритропоетину в сироватці крові.
3. Збільшення числа колоній в культурі тканини.
Якщо пацієнт має 2 великих критерії та щонайменше 1 малий або якщо пацієнт
має 1 великий критерій і щонайменше 2 малих, то діагноз справжньої поліцитемії є
вірогідний.
25.
НИЗЬКИЙ РИЗИК ТРОМБОЗІВЛІКУВАННЯ
British Committee for Standards in
Haematology, 2014
ВИСОКИЙ РИЗИК ТРОМБОЗІВ
Кровопускання
1. Інгібітор Jak-кіназ (руксолітініб). По 10 мг 2/д.
• Призначають при І та ІІА стадії, пацієнтам до 50
років
• 1 курс на 6 місяців для підтримання
гематокриту ≤ 45%
2. Препарати ІФН-α-2β
Кровопускання виконують в об’ємі 500 мл через
1-2 дні; у пацієнтів старечого віку, хворих на
серцево-судинні захворювання та тих, що
перенесли порушення мозкового кровообігу,
ексфузії крові здійснюють в об’ємі 350 мл та
перерви подовжують.
Напередодні рекомендується розпочати
антиагрегантну терапію та закінчити її через 2
тижні після останньої ексфузії.
Безпосередньо перед кровопусканням вводять
в/в плазмозамінник (400 мл реополіглюкіну) та
гепарин (5тис. ОД).
Протипокази: погана переносимість
кровопускань, низька ефективність, ознаки
прогресування хвороби (панцитоз, прогресуючий
ріст селезінки).
При недостатній ефективності ексфузій крові в молодому віці, коли є
протипокази до призначення цитостатиків. Дозволяє контролювати
еритроцитоз, тромбоцитоз, зменшує розміри селезінки. Призначають по 3
млн. ОД щодня до нормалізації гематокриту, після чого редукують дозу до
підтримуючої (3 млн. ОД 2-3 рази на тиждень).
3. Інгібітор фосфодіестерази-ІІІ (анагрелід).
При еритремії зі значним тромбоцитозом 800∙109/л і більше.
Порушує дозрівання мегакаріоцитів та призводить до зменшення к-ості
тромбоцитів; у високих дозах - виявляє антиангрегантну дію. Призначається
в дозі 0,5 мг 4 рази на добу (або 1 мг 2 рази на добу) протягом 1 тижня, далі
знижують дозу до мінімально ефективної для підтримуючого лікування.
Початковий терапевтичний ефект спостерігається через 14-21 день,
повний - через 4-12 тижнів.
4. Цитостатична терапія (гідроксисечовина, бусульфан,
піпеброман, цитозин-арабінозид).
Покази: 1. Перебіг еритремії з лейкоцитозом та тромбоцитозом. 2. Неможливість
проведення кровопускань. 3. II Б стадія захворювання.
Протипокази: 1. Дитячий та юнацький вік. 2. Анемічна стадія захворювання. 3.
Рефрактерніть до цитостатиків.
26.
Цитостатична терапіяОсновні групи цитостатиків, що застосовують в лікуванні еритремії:
1. Алкілюючі агенти: мієлосан, мієлобромол, алкеран.
2. Антиметаболіти: гідроксисечовина, тіогуанін, 6-меркаптопурин,
цитозінарабінозид.
• На сьогодні в лікуванні еритремії серед цитостатиків надається перевага
гідроксисечовині. Призначають препарат в дозі 15-30 мг/кг щоденно, з
корекцією дози за рівнем лейкоцитів. Показники крові моніторують 1 раз в 7-10
діб протягом перших 3-х тижнів, далі 1 раз на 5 діб. При кількості лейкоцитів
3∙109/л і кількості тромбоцитів 150∙109/л лікування переривають.
• В подальшому показники перевіряють 1 раз на 2 тижні протягом 3-х місяців.
Гідроксисечовина добре контролює лейкоцитоз та тромбоцитоз, дещо слабше
впливає на еритроцитоз та розміри селезінки.
27.
Симптоматичне лікуванняДля зменшення свербіжу рекомендують використовувати антигістамінні препарати тавегіл,
фенкарол, лоратадин, цетиризин та Н2-гістаміноблокатори (циметидин, фамотидин). Однак, їх
ефективність є сумнівною.
Еритромелалгії лікують антиагрегантами (клопідогрел, тиклопідин, пентоксифілін, дипірідамол,
ацетилсаліцилова кислота), нестероїдними протизапальними засобами; а при неефективності гепарином.
При тромбозах будь-якої локалізації необхідне призначення антиагрегантів (ацетилсаліцилова
кислота, трентал, курантил, тиклопідин), прямих антикоагулянтів (гепарин 5-10 тис. ОД 4 рази на
добу, фраксипарин, еноксипарин, далтепарин), а через 7 діб – непрямих антикоагулянтів
(варфарин).
У випадку тромбофлебіту поверхневих вен рекомендують застосовувати міхур з льодом,
гепаринову мазь та мазь Вишневського.
При артеріальній гіпертензії застосовують β-адреноблокатори, інгібітори
ангіотензинконвертуючого ферменту та інші гіпотензивні.
Порушення метаболізму сечової кислоти (гіперурикемія, сечокислий діатез) корегують
призначенням урикодепресантів – аллопуринол 200-1000 мг/добу; уриколітичних засобів
(блемарен, магурліт, уродан) та лужного пиття.
28.
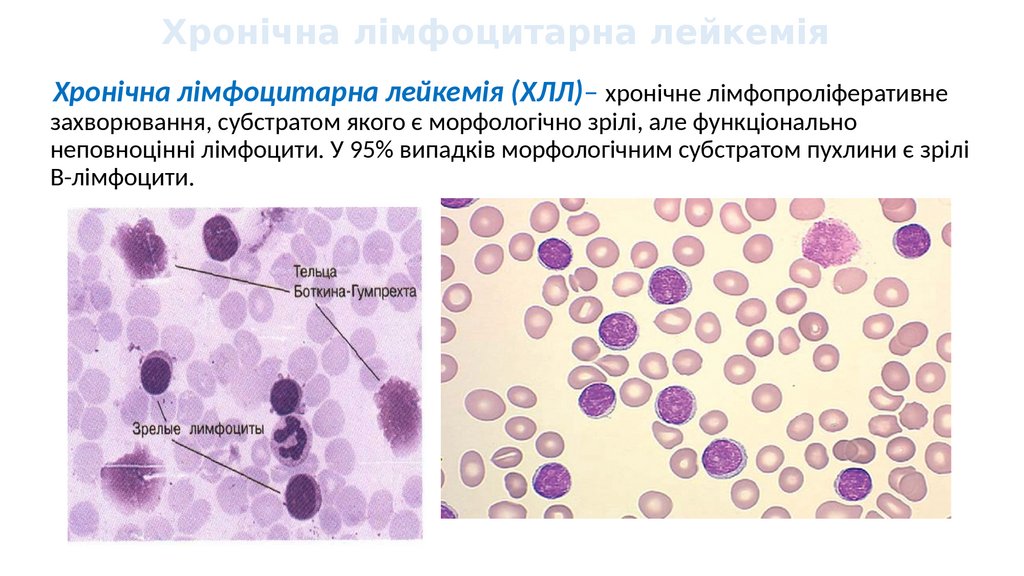
Хронічна лімфоцитарна лейкеміяХронічна лімфоцитарна лейкемія (ХЛЛ)– хронічне лімфопроліферативне
захворювання, субстратом якого є морфологічно зрілі, але функціонально
неповноцінні лімфоцити. У 95% випадків морфологічним субстратом пухлини є зрілі
В-лімфоцити.
29.
ЕТІОЛОГІЯ ХЛЛ• Поліетіологічне захворювання. Однак, найбільш очевидними чинниками на сьогодні вважається
вірусна інфекція та електромагнітне випромінювання.
• У хворих на ХЛЛ, приблизно у 50 % випадків виявляють хромосомні аномалії переважно 12, 13, 14,
рідше 17, 18 хромосом. Деякі генетичні аномалії мають прогностичне значення та важливі для
підбору тактики лікування. Найчастіше зустрічається трисомія 12 хромосоми. На 14 хромосомі
розміщується ген, що кодує синтез важких ланцюгів імуноглобулінів, транслокація частини 11
хромосоми на 14 призводить до порушення експресії цього гену.
• Клінічно важливим є індукція експресії гену мультилікарської резистентності на 7 хромосомі у
приблизно 40% хворих на ХЛЛ, що зумовлює стійкість до ряду цитостатиків (доксорубіцину,
вінкрістину).
• Важливим є виявлення мутації гену р53 на 17 хромосомі, що призводить до значного пригнічення
фізіологічного апоптозу лімфоцитів та зумовлює резистентність до флударабіну. У хворих з цією
аберацією спостерігається важкий перебіг захворювання та вкорочення тривалості життя в
порівнянні з тими, хто не має даної мутації.
• У 5% хворих можна виявити аберацію 18 хромосоми, що обумовлює порушення експресії легких
ланцюгів імуноглобулінів.
30.
ПАТОГЕНЕЗ• Патологічні В-лімфоцити при ХЛЛ не диференціюються до плазмоцитів, які повинні синтезувати
антитіла, що призводить до частих інфекційно-запальних процесів. Функціонально неповноцінні Тлімфоцити не здатні забезпечити достатній противірусний захист. Функціональна неповноцінність
лімфоцитів є причиною розвитку частих аутоімунних конфліктів.
• Клон патологічних лімфоцитів має продовжену тривалість життя, що обумовлює накопичення їх в
кістковому мозку та різних органах. Акумуляція лімфоцитів у внутрішніх органах призводить як до
органомегалії, так і до дистрофічно-запальних змін в них.
• В процесі прогресування захворювання пухлинна маса поступово витісняє нормальні паростки
кровотворення з кісткового мозку, обумовлюючи розвиток метапластичної анемії, тромбоцитопенії.
Важливим фактором пригнічення кровотворення є розвиток фіброзу кісткового мозку під впливом
цитокінів.
Провідні синдроми
Гіперпластичний синдром
Анемічний синдром
Геморагічний синдром
Інтоксикаційний синдром
Імунодефіцитний синдром
31.
ГІПЕРПЛАСТИЧНИЙ СИНДРОМ• Системна лімфаденопатія. Лімфатичні вузли -тістуваті,
безболісні чи мало болісні, рухомі, не спаяні між
собою та зі шкірою, розміри можуть коливатись від
1,5 до 5-6 см в діаметрі. Лімфатичні вузли можуть
спричиняти компресію судин, нервів, порожнистих
органів – жовчно-вивідних шляхів, кишківника,
сечоводів.
• Гепатоспленомегалія.
• Лейкеміди - інфільтрація шкіри лейкемічними
клітинами - макуло-папулольозні, пухлиноподібні чи
еритродермічні утворення, тілесного, рожевого чи
червоного кольору, іноді зі свербежем.
• Інфільтрація щільно інервованого периосту кісток
спричиняє інтенсивний біль в трубчастих та плоских
кістках (оссалгії), в першу чергу в грудині (стерналгія).
32.
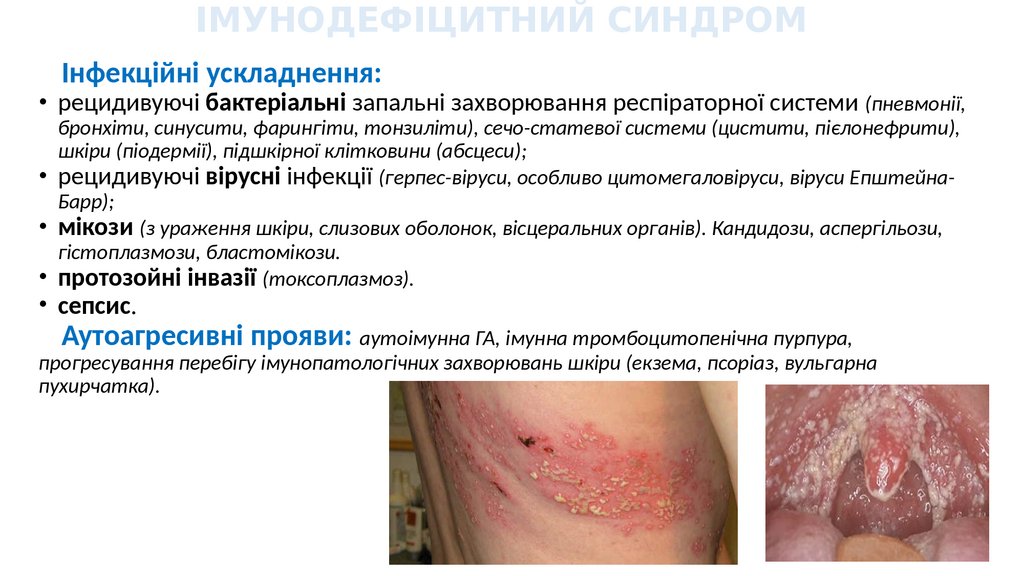
ІМУНОДЕФІЦИТНИЙ СИНДРОМІнфекційні ускладнення:
• рецидивуючі бактеріальні запальні захворювання респіраторної системи (пневмонії,
бронхіти, синусити, фарингіти, тонзиліти), сечо-статевої системи (цистити, пієлонефрити),
шкіри (піодермії), підшкірної клітковини (абсцеси);
• рецидивуючі вірусні інфекції (герпес-віруси, особливо цитомегаловіруси, віруси ЕпштейнаБарр);
• мікози (з ураження шкіри, слизових оболонок, вісцеральних органів). Кандидози, аспергільози,
гістоплазмози, бластомікози.
• протозойні інвазії (токсоплазмоз).
• сепсис.
Аутоагресивні прояви: аутоімунна ГА, імунна тромбоцитопенічна пурпура,
прогресування перебігу імунопатологічних захворювань шкіри (екзема, псоріаз, вульгарна
пухирчатка).
33.
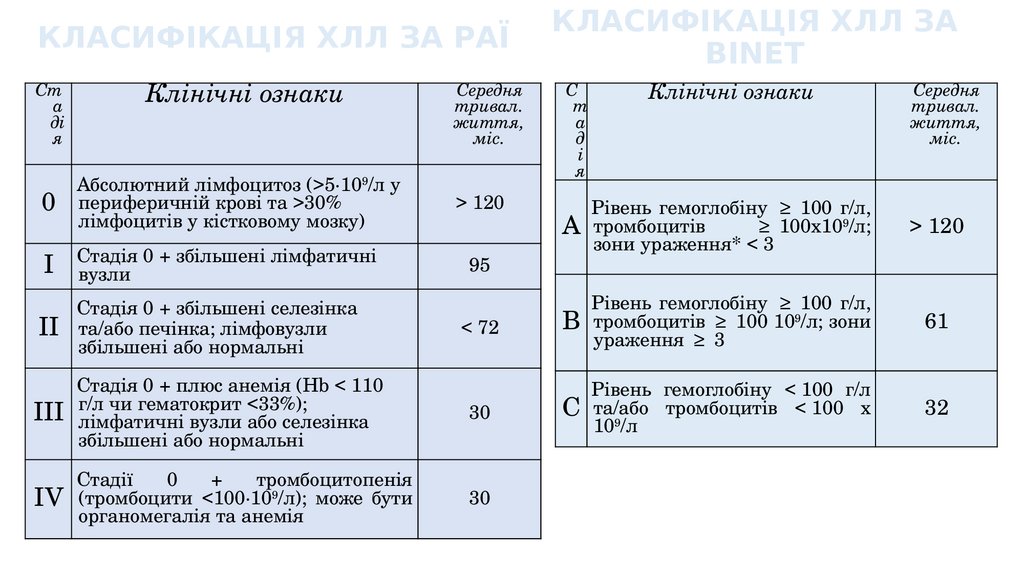
КЛАСИФІКАЦІЯ ХЛЛ ЗА РАЇСт
а
ді
я
Клінічні ознаки
КЛАСИФІКАЦІЯ ХЛЛ ЗА
BINET
Клінічні ознаки
Середня
тривал.
життя,
міс.
0
Абсолютний лімфоцитоз (>5∙109/л у
периферичній крові та >30%
лімфоцитів у кістковому мозку)
С
т
а
д
і
я
Середня
тривал.
життя,
міс.
> 120
> 120
I
Стадія 0 + збільшені лімфатичні
вузли
95
Рівень гемоглобіну ≥ 100 г/л,
≥ 100х109/л;
A тромбоцитів
зони ураження* < 3
II
Стадія 0 + збільшені селезінка
та/або печінка; лімфовузли
збільшені або нормальні
< 72
Рівень гемоглобіну ≥ 100 г/л,
B тромбоцитів ≥ 100 109/л; зони
ураження ≥ 3
61
Стадія 0 + плюс анемія (Hb < 110
чи гематокрит <33%);
III г/л
лімфатичні вузли або селезінка
збільшені або нормальні
30
Рівень гемоглобіну < 100 г/л
тромбоцитів < 100 х
C та/або
9
10 /л
32
Стадії
0
+
тромбоцитопенія
IV (тромбоцити <100∙109/л); може бути
органомегалія та анемія
30
34.
Загальний аналізкрові
Показники
RBC
Hb
PLT
WBC
Паличкоядерні
нейтрофіли
Сегментоядерні
нейтрофіли
Еозинофіли
Базофіли
Лімфоцити
Моноцити
Тіні БоткінаГумпрехта
Зміни показників
N чи
N чи
N чи
N
N чи
N чи
N чи
> 5 ∙ 109/л
N
напівзруйновані ядра
лімфоцитів
Мієлограма
• Лімфоцитарна інфільтрація
(>30%);
• Редукція інших паростків
кровотворення.
35.
ДІАГНОСТИЧНІ КРИТЕРІЇ ХЛЛ(ESMO, 2015)
1. Абсолютний лімфоцитоз в периферичній
крові >5∙109/л.
2. В кістковому мозку лімфоцитів > 30%.
3. Імунофенотипові характеристики
лімфоцитів СD5+, 10-, 19+, 23+, 43+/FMC7-. Низький рівень експресії СD20+,
22+, 79b+, низька щільність поверхневих
імуноглобулінів (sIgG, D).
36.
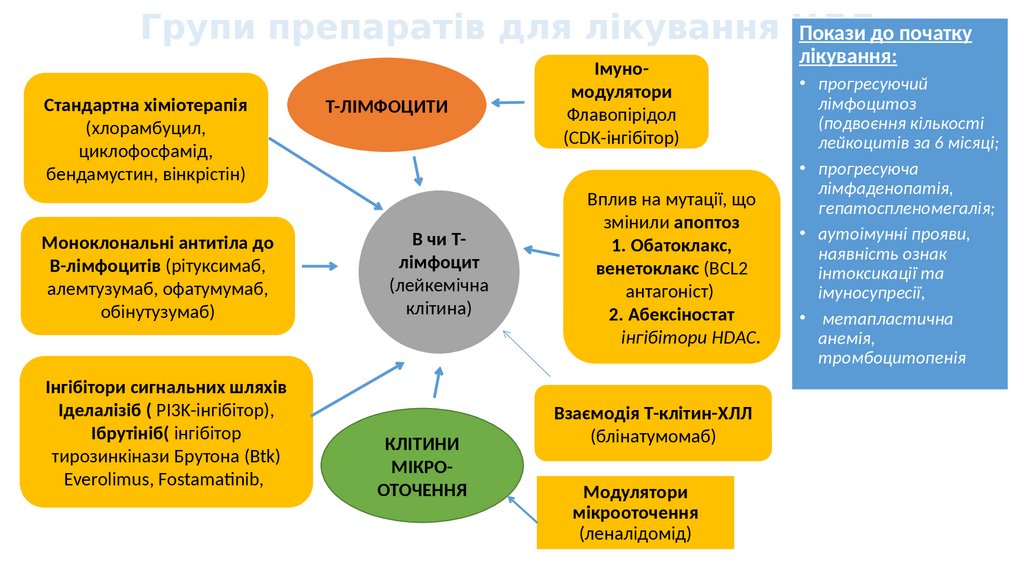
Групи препаратів для лікування ХЛЛПокази до початку
Стандартна хіміотерапія
(хлорамбуцил,
циклофосфамід,
бендамустин, вінкрістін)
Моноклональні антитіла до
В-лімфоцитів (рітуксимаб,
алемтузумаб, офатумумаб,
обінутузумаб)
Інгібітори сигнальних шляхів
Іделалізіб ( PI3K-інгібітор),
Ібрутініб( інгібітор
тирозинкінази Брутона (Btk)
Everolimus, Fostamatinib,
Т-ЛІМФОЦИТИ
В чи Тлімфоцит
(лейкемічна
клітина)
КЛІТИНИ
МІКРООТОЧЕННЯ
Імуномодулятори
Флавопірідол
(CDK-інгібітор)
Вплив на мутації, що
змінили апоптоз
1. Обатоклакс,
венетоклакс (BCL2
антагоніст)
2. Абексіностат
інгібітори HDAC.
Взаємодія Т-клітин-ХЛЛ
(блінатумомаб)
Модулятори
мікрооточення
(леналідомід)
лікування:
• прогресуючий
лімфоцитоз
(подвоєння кількості
лейкоцитів за 6 місяці;
• прогресуюча
лімфаденопатія,
гепатоспленомегалія;
• аутоімунні прояви,
наявність ознак
інтоксикації та
імуносупресії,
• метапластична
анемія,
тромбоцитопенія
37.
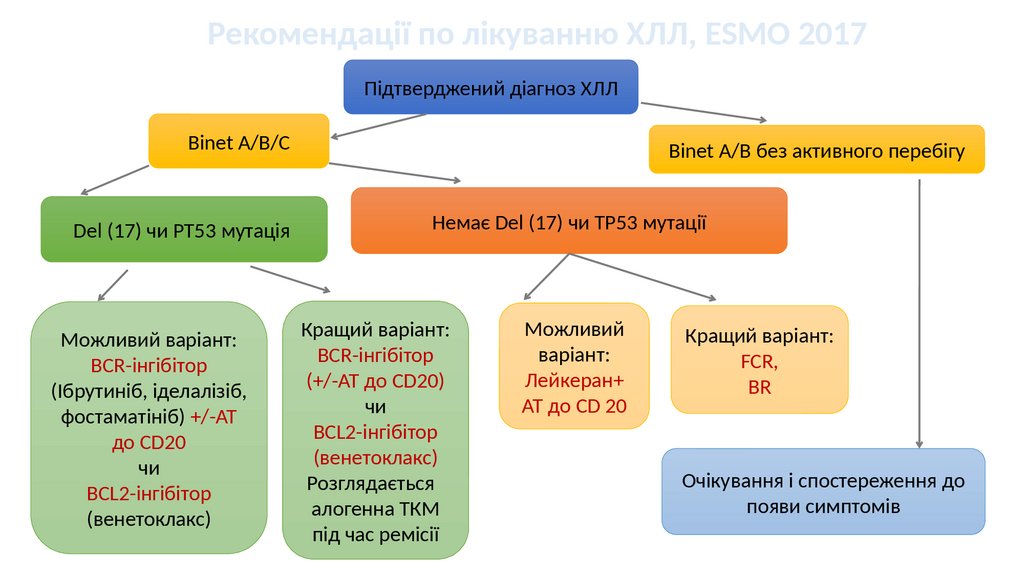
Рекомендації по лікуванню ХЛЛ, ESMO 2017Підтверджений діагноз ХЛЛ
Binet A/B/C
Del (17) чи PT53 мутація
Можливий варіант:
BCR-інгібітор
(Ібрутиніб, іделалізіб,
фостаматініб) +/-АТ
до CD20
чи
BCL2-інгібітор
(венетоклакс)
Binet A/B без активного перебігу
Немає Del (17) чи ТР53 мутації
Кращий варіант:
BCR-інгібітор
(+/-АТ до CD20)
чи
BCL2-інгібітор
(венетоклакс)
Розглядається
алогенна ТКМ
під час ремісії
Можливий
варіант:
Лейкеран+
АТ до CD 20
Кращий варіант:
FCR,
BR
Очікування і спостереження до
появи симптомів
38.
ХІМІОТЕРАПЕВТИЧНЕ ЛІКУВАННЯЛікування I-ої лінії.
Хворі до 70 років.
• FCR (флударабін, циклофосфамід, рітуксимаб)
• FC (флударібін, циклофосфамід)
• FR (флударабін, рітуксимаб)
Хворі після 70 років.
• COP (циклофосфамід, вінкрістін, преднізолон)
• LP (лейкеран, преднізолон)
• FR (флударабін, рітуксимаб)
Лікування II-ої лінії.
• FCR (флударабін, циклофосфамід, рітуксимаб)
• CHOP-R (циклофосфамід, доксорубіцин, вінкрістін, преднізолон)
• AF (алемтузумаб, флударабін), CFAR (циклофосфамід, флударабін, алемтузумаб, ритуксимаб) ,
OFAR (оксаліплатин, флударабін, цитарабін, ритуксимаб), високі дози метилпреднізолону з
ритуксимабом.
Алогенна трансплантація кісткового мозку для молодших 65 років, хто не відповів на терапія , має
прогресію хвороби в межах 1 року чи 2–х років на флударабін-вмісних режимах, хто має del (17 p).
39.
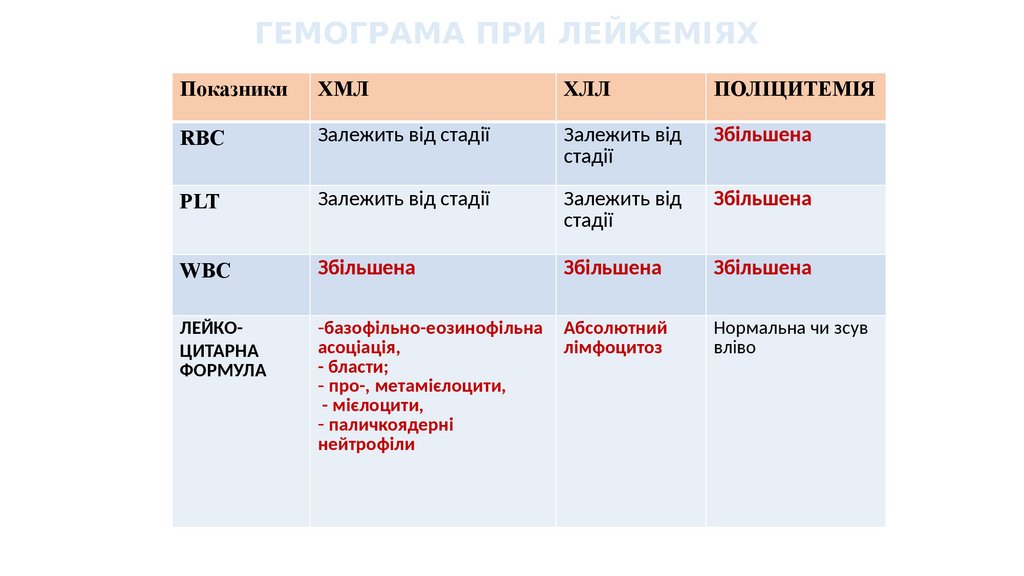
ГЕМОГРАМА ПРИ ЛЕЙКЕМІЯХПоказники
ХМЛ
ХЛЛ
ПОЛІЦИТЕМІЯ
RBC
Залежить від стадії
Залежить від
стадії
Збільшена
PLT
Залежить від стадії
Залежить від
стадії
Збільшена
WBC
Збільшена
Збільшена
Збільшена
ЛЕЙКОЦИТАРНА
ФОРМУЛА
-базофільно-еозинофільна
асоціація,
- бласти;
- про-, метамієлоцити,
- мієлоцити,
- паличкоядерні
нейтрофіли
Абсолютний
лімфоцитоз
Нормальна чи зсув
вліво
40.
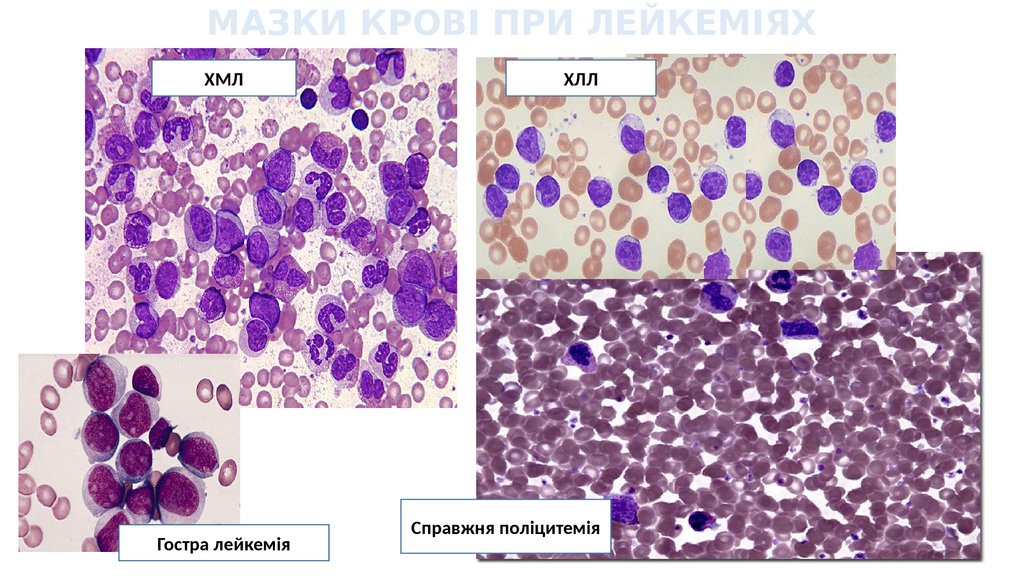
МАЗКИ КРОВІ ПРИ ЛЕЙКЕМІЯХХМЛ
Гостра лейкемія
ХЛЛ
Справжня поліцитемія