![Болезнь Вестфаля—Вильсона—Коновалова [Коновалов Н. В., 1948; Westphal С., 1883; Wilson К., 1912].](https://cf.ppt-online.org/files/slide/v/VO9ao41skzU3y7ZlKBn08PACFNmDgLw2HfTXtM/slide-0.jpg "Болезнь Вестфаля—Вильсона—Коновалова [Коновалов Н. В., 1948; Westphal С., 1883; Wilson К., 1912].")
")







")





























 Медицина
МедицинаПохожие презентации:
")
")


")





Гепатолентикулярная дегенерация (Болезнь Вильсона-Коновалова)
1. Болезнь Вестфаля—Вильсона—Коновалова [Коновалов Н. В., 1948; Westphal С., 1883; Wilson К., 1912].
Гепатолентикулярная дегенерация(Болезнь Вильсона-Коновалова)
Болезнь Вестфаля—Вильсона—Коновалова
[Коновалов Н. В., 1948; Westphal С., 1883;
Wilson К., 1912].
2. Болезнь Вильсона-Коновалова (ВК)
• - наследственное заболевание воснове которого лежит нарушение
метаболизма меди и накопление ее в
печени и других внутренних органах.
3. Накопление меди во внутренних органах
Распространенность в различных регионах мира в среднем 1:300004. ВК - Аутосомно-рецессивный тип наследования
• Нарушается синтез медь-транспортирующегобелка (АТР7В, ответственного за
внутриклеточный транспорт ионов меди ) на 13
хромосоме;
• Болезнь поражает 25% братьев и сестер в семье
при клинически здоровых родителях, которые
являются носителями аномального гена
(гетерозиготы);
• Заболевают только те индивидуумы, которые
унаследовали два мутантных гена (гомозиготные
носители мутации).
5. Патогенез ВК
6. Патогенез ВК:
• Отложение меди в печени;• Связывание с эритроцитами –
гемолитическая анемия;
• Почках;
• Роговой оболочке глаз;
• Подкорковых ядрах – чечевидное, хвостатое
ядро, таламус, кора (тремор, насильственные
движения, психические расстройства,
нарушение интеллекта, и пр.)
• Коже, суставах
7. Накопление меди в гепатоцитах
8. КОЛЬЦА КАЙЗЕРА-ФЛЕЙШЕРА - располагаются в месте соединения склеры и роговицы, желтовато-зелёное или зелено-коричневое окрашивание краёв р
КОЛЬЦА КАЙЗЕРА-ФЛЕЙШЕРА - располагаются вместе соединения склеры и роговицы, желтоватозелёное или зелено-коричневое окрашивание краёв
роговицы (цвета бутылочного стекла), которое
появляется в первую очередь на верхнем крае.
После лечения кольца могут исчезать.
9. Классификация
• В соответствии с клинической картинойотчетливо выделяются 3 формы
заболевания:
• протекающие с поражением
1) печени;
2) центральной нервной системы;
3) смешанная форма.
10. Формы гепатолентикулярной дегенерации (ГД)
Смешаннаяформа
Брюшная
форма:
-цирроз
-гепатит
-острая печеночная
недостаточность
Церебральная
Форма
-экстрапирамидные
-церебеллярные
-псевдобульбарные
-судорожные
-когнитивные
-психотические
Поражение:
-почек
-глаз
-суставов
-сердца
-эндокринной системы
-кожи..
нарушения
Н. В. Коновалов (1948)
11. Начало заболевания:
• Может дебютировать в детском, подростковом,юношеском, зрелом возрасте;
• Очень редко – дебют ВК в 50–60 лет;
• Чем раньше начинается заболевание, тем тяжелее
оно протекает (при отсутствии лечения).
12. Классификация поражений печений при ВК
• I - латентная с неспецифическимиморфологическими изменениями
печени или мелко- или
среднекапельный жировой дистрофией;
• II - ХПГ;
• III - цирротическая стадия.
13. Клиническая картина
• Дебют - в типичных случаях (у 42% больных) представлен одним извариантов поражения печени: острый или хронический гепатит, цирроз,
реже – острая печеночная недостаточность;
• может манифестировать нейропсихическими симптомами: мышечная
дистония, дизартрия, тремор, изменения личности, реже (в 6% случаев)
эпилептические припадки;
• Развитие нейропсихической симптоматики наблюдается чаще во 2-3
десятилетии жизни и обычно ассоциируется с появлением колец
Кайзера-Флейшера;
• У 15% больных заболевание проявляется картиной острой
гемолитической анемии;
• Артропатии у 25-50% больных старше 20 лет;
• Поражение почек (глюкозурия, аминоацидурия, гиперфосфатурия,
гиперкальциурия); сердца (аритмии, изменения сегмента ST и зубца Т
на ЭКГ); эндокринной системы (гинекомастия, нарушение
толерантности к глюкозе) и кожи (гиперпигментация, голубые лунки у
14. Диагностические критерии
При латентном течении болезни ВильсонаКоновалова ДЗ подтверждает:-снижение уровня церулоплазмина плазмы (< 0,2 г/л);
-снижение концентрации связанной меди крови(< 13,424,4 мкмоль/л)
-увеличение содержания не связанной с
церулоплазмином меди в сыворотке крови (300 мкг/л и
>);
-повышение (> 100 мкг/сут.) экскреции меди с мочой.
Следует помнить, что любой экстрапирамидный
синдром, впервые проявившийся в возрасте до 50 лет,
служит основанием для исключения
гепатолентикулярной дегенерации **
Celia H Chang, 2006 **Штульман Д. Р., Левин О. С., 2004; Левин О.С. 2005
15. Диагноз
• Анамнез и физикальное обследование• Диагностические тесты
– Развернутый анализ крови
– Сывороточный церулоплазмин
– Осмотр в щелевой лампе кольца Кайзера-Флейшера
(Kayser-Fleischer rings)
– Базальная 24-часовая экскреция мочи
– Пробное лечение пеницилламином у детей
– Содержание меди в паренхиме печени
– Печеночная биопсия (обсуждается, но специально не
рекомендуется)
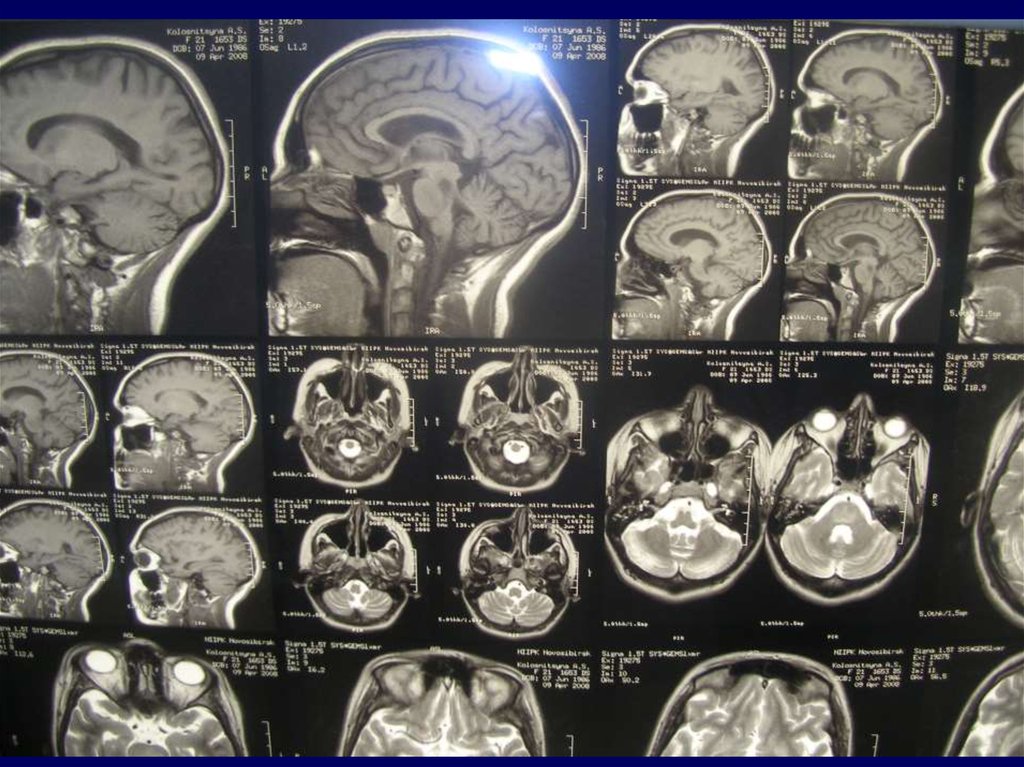
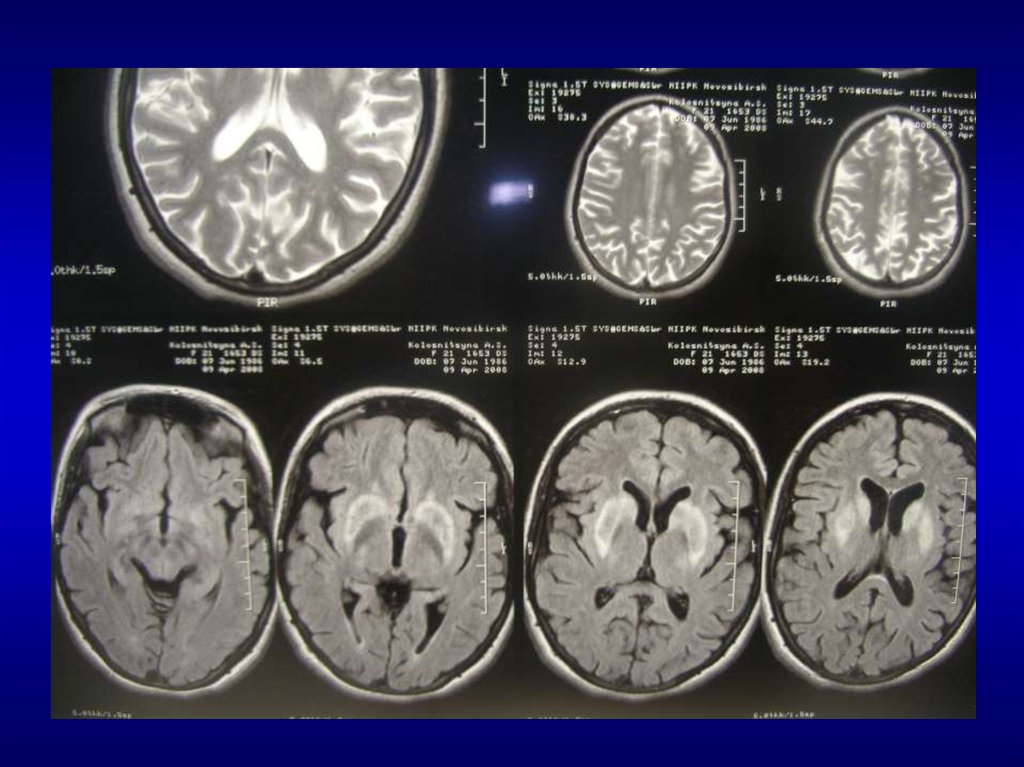
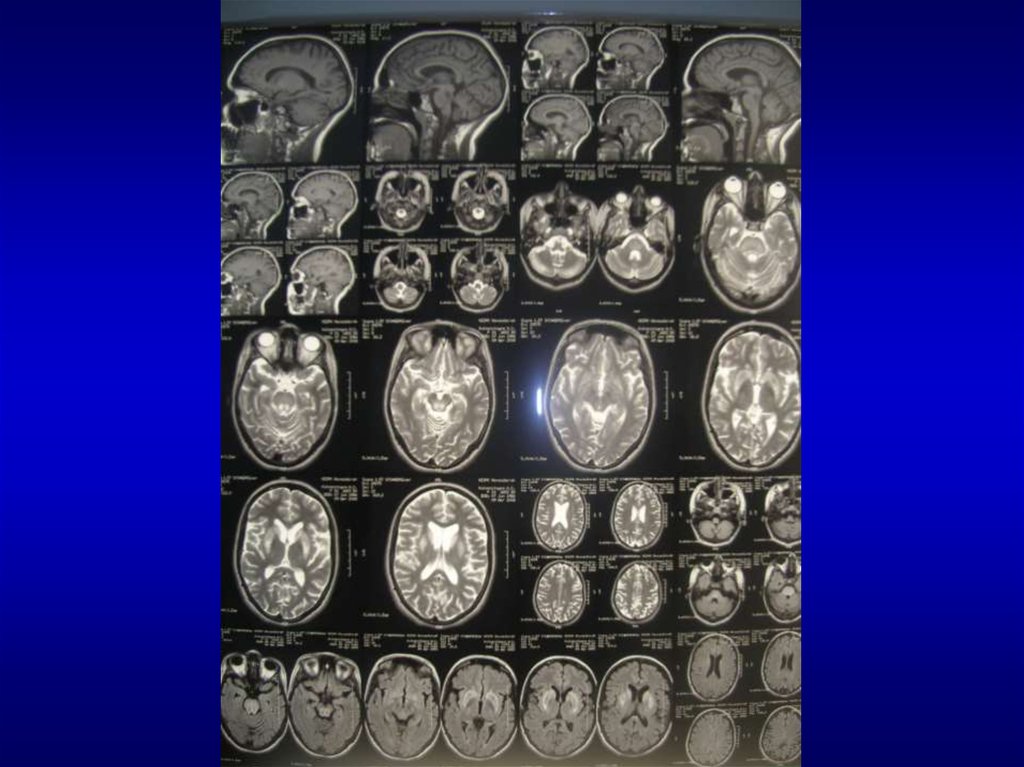
– Неврологическое и радиологическое визуализирующее
магниторезонансное исследование мозга (magnetic
resonance imaging - MRI)
– Генетическое исследование родственников первого
уровня родства на основании анализа гаплотипа
16. Диагноз
• У больных с клинически латентным течением диагнозставят при выявлении характерных изменений
показателей метаболизма меди:
• снижения уровня церулоплазмина плазмы (< 0,2 г/л),
• часто в сочетании со снижением концентрации
свободной меди крови (норма 13,4-24,4 мкмоль/л)
вследствие снижения фракции меди, связанной с
церулоплазмином
• повышения (> 100 мкг/сут.) экскреции меди с мочой.
17. Церулоплазмин
• Сывороточный церулоплазмин должен рутинноопределяться при обследовании больных с
неустановленной печеночной, неврологической патологией
и психиатрическим нарушениями у детей и пациентов
средних лет;
• Очень низкий уровень церулоплазмина сыворотки (<50 мг/Л
и <5 мг/dL) - сильное доказательство в пользу диагноза БВ;
• Случаи умеренных субнормальных показателей
церулоплазмина требуют дальнейшего обследования;
• Нормальные показатели сывороточного церулоплазмина не
исключают диагноз.
18. Экскреция меди с мочой
• Для диагностики - базальное 24-часовоеисследование мочи. Суточная экскреция мочи при БВ
обычно выше 100 мкг (1.6 ммолей), однако
показатели выше 40 мкг (>0.6 ммолей и >600
наномолей) могут свидетельствовать о БВ и требуют
дальнейших исследований.
• У детей проба с пенициллинамином может
подтвердиться диагноз БВ в том случае, если
выделяется более 1,600 мкг меди за 24 часа (>25
микромолей /24 часа) после назначения 500 мг Dпеницилламина в начале и через 12 часов после
начала 24 часового сбора мочи. Диагностическая
ценность этого теста у взрослых неизвестна.
19. Заподозрить раннюю стадию ВК можно на основании следующих признаков:
• Перенесенная желтуха• Повторные кровотечения из носа, десен либо
множественные кровоподтеки;
• Сосудистые “звездочки” на коже груди и спины;
• Своеобразные “полосы” (белых, меняющих
периодически окраску на красновато-синюшную) на
бедрах и в подмышечных областях;
• Гормональные нарушения в виде аменореи или
дисменореи у девушек, гинекомастии у юношей, а
также акромегалии;
• Снижение интеллекта и изменение психики в виде
чередования дурашливости и пониженного
настроения, трудности усвоения нового материала,
проблемы с успеваемостью в школе.
20.
К чему стремиться:постановка диагноза ВК в детской
сети
21. Дифференциальный диагноз
В зависимости от особенностей клинической картины БВ
дифференциальный диагноз следует проводить:
1) с острыми и хроническими заболеваниями печени
другой этиологии -вирусной, аутоиммунной,
метаболической (стеатогепатит, гемохроматоз);
2) с аутоиммунной гемолитической анемией;
3) с ревматическими заболеваниями (ревматоидный
артрит, полимиозит, склеродермия);
3) с болезнями нервной системы (рассеянный склероз),
экстрапирамидными и психическими заболеваниями.
22.
• Если своевременно не начать лечение,направленное на выведение токсичных
избытков меди из организма, то через
5–7 лет больной обречен на смерть.
23. Лечение
• Идеально начинать лечение в«досимптомной стадии» ВК;
• Необходимо тщательное обследование
всех братьев и сестер, если в семье
есть хоть один ребенок, страдающий
ВК.
24. Лечение:
• 1. Строгое соблюдение “печеночной” диеты (стол 5а):исключение богатых медью продуктов (шоколад, кофе,
орехи, бобовые и др.);
• 2. Постоянный прием препаратов, выводящих медь из
организма.
Основной препарат - D-пеницилламин. Триентин
• 3. Комбинированное лечение комплексообразователями и
препаратами цинка (оксид, сульфат);
• 4. Гепатопротекторная терапия;
• 5. Антиоксиданты?
• 6. Тетратиомолибдат?
• 7. Пересадка печени.
25. Лечение
Начальная доза D-пеницилламина - 0,25-0,5г/день с
постепенным повышением (каждые 7 дней на 0,25 г)
до 1-2 г/день.
• Суточная доза разделяется на 3-4 приема, разовая
доза принимается внутрь за полчаса до приема
пищи.
• Характерным побочным эффектом D-пеницилламина
является появление или ухудшение неврологической
симптоматики в начале лечения, что связано с
мобилизацией меди из печени и повышением ее
концентрации в ЦНС.
• Для купирования ухудшения необходимо временно
снизить дозу D-пеницилламина.
26.
• если к этому времени экскреция меди непревышает 150мкг/сут, после достижения
клинического улучшения, которое обычно
наступает через 6 месяцев от начала
лечения, переходят на поддерживающую
дозу (0,75-1,25 г/день).
• D-пеницилламин обладает
антипиридоксиновым эффектом, поэтому к
лечению добавляют пиридоксин (витамин В6)
в дозе 25 мг в день внутрь.
27.
• Оценку эффективности лечения следуетпроизводить не ранее, чем через 2 года от
начала терапии;
• Недопустимы перерывы в лечении Dпеницилламином, превышающие несколько
недель, так как рецидив заболевания может
протекать в виде острого поражения печени с
развитием острой печеночной недостаточности.
28. Триентин
• Дозы:Внутрь натощак
• Дети < 12 лет: 500—750 мг/сут в 2—
4 приема, максимальная доза 1,5 г/сут.
• Дети > 12 лет и взрослые: 750—
1250 мг/сут в 2—4 приема;
максимальная доза 2 г/сут.
29. Цинк - содержащие препараты*
• 25 мг – 3 - 4 раза в день*Sinha S; Taly AB, Neurol Sci. 2008; 264(1-2):129-32
30.
31.
32.
33. Мультисистемная атрофия
• спорадическая мультисистемная дегенерация• преимущественно вовлекает базальные
ганглии, оливы, мост, мозжечок, боковые рога
спинного мозга, ядро Онуфа в крестцовом
отделе спинного мозга
• сочетание паркинсонизма с вегетативной
недостаточностью, мозжечковым и пирамидным
синдромами.
• 2-6% случаев паркинсонизма и чаще всего
проявляется на шестом десятилетии жизни
34. Мультисистемная атрофия
• Критерии диагностики:- Вегетативная/тазовая дисфункция (ортостатическая
гипотензия со снижение систолического АД не менее чем
на 30 мм рт. ст., а диастолического - не менее чем на
15 мм рт. ст. в течение 3 мин стояния и/или недержание
мочи с перманентным непроизвольным частичным или
полным опорожнением мочевого пузыря и нарушением
эрекции у мужчин).
• Паркинсонизм (гипокинезия в сочетании с не менее чем
одним другим паркинсоническим симптомом: ригидностью,
тремором покоя или постуральной неустойчивостью, не
связанной с другими причинами).
35.
• Мозжечковая атаксия (статолокомоторнаяатаксия в сочетании с не менее чем
одним другим мозжечковым симптомом дизартрией, нистагмом, интенционным
тремором или дисметрией в конечностях).
36. Критерии, исключающие диагноз:
- Начало в возрасте до 30 лет.- Положительный семейный анамнез.
- Наличие анамнестических, клинических или
параклинических признаков иного заболевания,
способного вызвать аналогичные симптомы.
- Галлюцинации, не связанные с приемом
лекарственных средств.
- Наличие деменции или признаков нарушения
корковых функций (афазия, апраксия и др.).
- Резкое замедление вертикальных саккад или паралич
вертикального взора.
37. Болезнь Гентингтона
• наследственное нейродегенеративное заболевание свыраженной психопатологической симптоматикой имеет аутосомно-доминантный тип наследования со
100%-й пенетрантностью.
• Ген, ответственный за заболевание, расположен на
коротком плече хромосомы 4 и, как установлено,
отличается увеличением количества повторов
тринуклеотидов
• Патофизиология - дегенерация клеток в хвостатых
ядрах, гибель клеток в некоторых других структурах
головного мозга, включая кору и мозжечок
38. Болезнь Гентингтона
39.
Эпидемиология: 5-8/100.000 населенияКлиника:
• Обычно заболевание начинается в возрасте 3040 лет;
• типичная продолжительность - 17 лет
• клинический синдром - триада симптомов:
двигательные расстройства, деменция и другие
психические нарушения
40. Двигательные расстройства
• начинаются с нарушений координации,стереотипных мелких движений пальцев,
непроизвольного гримасничанья,
хаотического разбрасывания рук и ног, а
также патологических движений глаз;
• дистония и атетоз;
• хореические гиперкинезы, постепенно
охватывают и туловище;
• походка становится атактической;
• в конце заболевания развиваются
брадикинезия, дизартрия, дисфагия,
спастичность и недержание мочи.
41.
Деменция;
Депрессия;
Суициды;
Психозы.