



















 Медицина
МедицинаПохожие презентации:
")

")




")
")

Болезнь Вильсона - Коновалова, болезнь Марфана, Факоматозы
1. Болезнь Вильсона-Коновалова, болезнь Марфана, Факоматозы: этиология, патогенез, классификация, клиническая картина,
диагностика, диф.диагностика, медикаментозныеи немедикаментозные методы терапии.
Выполнила: студентка 4 курса 3 группы
Пшенова М.А.
Преподаватель: Шевченко В.В.
2. Болезнь Вильсона-Коновалова
Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация)относится к наследственным заболеваниям центральной нервной
системы и внутренних органов, но неврологические проявления —
одно из характерных проявлений этой патологии.
Заболевание характеризуется
нарушением обмена меди, из-за чего
избыточные её количества
накапливаются в печени, головном
мозге, роговице и других тканях и
органах, приводя к нарушению их
функций.
3. Этиология
Заболевание впервые описано в 1912 году английским неврологом Семюэлем Вильсоном. В своём печатном труде онописал симптомы и особенности изменения внутренних органов при этом заболевании. Обычно первые проявления
становились заметными в молодом возрасте, позже прогрессировала ригидность, сложности при глотании, снижение
двигательной способности мягкого нёба, языка, губ (причина расстройства речи — дизартрии). Развиваются
непроизвольные движения, расстройства психики — пониженное или необоснованно повышенное настроение,
немотивированная агрессия, которая на поздних стадиях сменяется безучастностью, бредовые идеи, галлюцинации.
Российский невролог Николай Васильевич Коновалов изучал болезнь Вильсона на протяжении многих лет. Это
позволило ему создать оригинальную полноценную классификацию различных форм заболевания. За огромный вклад
Коновалова в исследование проблемы фамилия учёного навсегда дополнила название болезни.
Частота встречаемости — от 1 до 9 случаев на 100000 населения.
Основная причина возникновения болезни — мутация гена с названием ATP7B, который ответственен за встраивание
ионов меди в белок церулоплазмин. Всего описано более 300 мутаций этого гена. База данных мутаций постоянно
расширяется новыми вариантами. По международным оценкам, носителем аномального гена является примерно 1
человек из 100. При данном виде наследования заболевание проявляется симптоматикой только в том случае, если
патологический ген был унаследован от обоих родителей. Мальчики и девочки болеют с одинаковой частотой.
4. Патогенез
Основной механизм развития болезни Вильсона — Коновалова — это нарушениеобменных процессов в организме, в результате которых медь накапливается в
различных органах и тканях с прогрессирующим нарушением их функций. Кроме
того, за различные симптомы отвечает разнообразие мутаций гена ATP7B,
определяющего, как именно проявится заболевание.
Ген ATP7B кодирует белки, ускоряющие химические реакции меди у человека.
Накопление меди в организме вызывает хроническое отравление (интоксикацию).
Накапливаясь в печени и мозге, медь способствует гибели клеток этих органов. Это
вызывает воспалительную реакцию и разрастание соединительной ткани в печени
— фиброз печеночных протоков с нарушением их функций, из-за чего позже
формируется цирроз. Гибель нервных клеток и их растворение (лизис) в головном
мозге образует полости (кисты). Изменения других органов и тканей, как правило,
незначительны.
При выбросе меди из разрушенных клеток в кровь под воздействием внешних
факторов (инфекции, интоксикации, реакции на медикаменты) концентрация меди в
плазме крови может повыситься в несколько раз. Это вызывает массированный
распад эритроцитов, что приводит к тяжелому, чаще смертельному осложнению —
фульминантной печеночной несостоятельности.
Всё многообразие симптомов болезни Вильсона — Коновалова вызывает не только
накопление меди, но и отравление продуктами распада собственных клеток
(аутоинтоксикация)
5. Классификация Клиническая картина
В России чаще всего применяется классификация,которая построена на клинических особенностях
болезни, сочетания поражения печени и
центральной нервной системы. Течение болезни
Вилсона — Коновалова подразделяют на:
• Неврологические проявления обычно возникают на
более поздних стадиях. Некоторые из них:
1. бессимптомную форму;
2. печеночную форму;
• тремор конечностей и головы;
• мышечная дистония — непроизвольное сокращение
мышц с изменением нормального положения тела;
• ригидность мышц;
• проблемы с речью и глотанием (дизартрия, дисфагия);
3. церебральную форму;
• повышенное слюноотделение.
4. смешанную форму.
• У детей ранние неврологические признаки:
поведенческие нарушения, отставание в учёбе,
проблемы при выполнении заданий, требующих
сочетанной координации работы рук и глаз,
ухудшение мелкой моторики, изменение почерка.
По течению заболевание можно разделить на две
сменяющиеся стадии:
латентная — характеризуется отсутствием
внешних проявлений болезни, характерные
изменения определяются только при лабораторном
исследовании;
стадия клинических проявлений — появляются
специфические симптомы болезни гепатоцеребеллярной дегенерации.
• Характерный симптом: кольцо Кайзера-Флейшера —
зеленовато-коричневое кольцо по краю роговицы,
которое видно при обследовании глаз с помощью
щелевой лампы.
6. Также применяется классификация Коновалова, которая включает пять форм гепато-церебральной дистрофии:
Также применяется классификация Коновалова, которая включает пять форм гепатоцеребральной дистрофии:• Брюшная (абдоминальная) форма — тяжёлое поражение печени, которое проявляется гепатопатией, вильсоновским
гепатитом, циррозом печени и фульминарной печёночной несостоятельностью. Может привести к смерти до появления
симптомов со стороны нервной системы. Продолжительность от нескольких месяцев до 3-5 лет;.
• Ригидно-аритмогиперкинетическая (ранняя) форма — характеризуется быстрым течением и начинается в детском
возрасте. Среди симптомов преобладает мышечная скованность, приводящая к изменениям суставов и их тугоподвижности.
Движения замедляются, руки и ноги могут непроизвольно двигаться спирально и червеобразно в сочетании с быстрыми
непроизвольными сокращениями мышц. Характерны нарушения речи (дизартрия) и глотания (дисфагия),
насильственный, непроизвольный смех и плач, нарушения эмоционального состояния, умеренное снижение интеллекта.
Заболевание продолжается 2-3 года, заканчивается смертельным исходом.
• Дрожательно-ригидная форма встречается чаще остальных; начинается в юношеском возрасте, протекает медленно, иногда
с периодами полного или неполного восстановления и внезапными ухудшениями, сопровождающиеся повышением
температуры тела до 37–38 °C; характерно одновременное развитие тяжёлой скованности мышц и ритмичного дрожания
частотой 2-8 подёргиваний в секунду. Эти симптомы резко усиливаются при движениях и волнении, но исчезают в покое и
во сне. Иногда наблюдаются дисфагия и дизартрия. Средняя продолжительность жизни около 6 лет.
• Дрожательная форма проявляется с возраста 20-30 лет, течёт относительно медленно (10-15 лет и более); преобладает
дрожание, ригидность появляется лишь в конце болезни, иногда наблюдается пониженный тонус мышц; отмечается
отсутствие мимики, медленная монотонная речь, тяжёлые изменения психики, частые эмоциональные вспышки,
судорожные припадки.
• Экстрапирамидно-корковая форма встречается реже других. Типичные для гепато-церебральной дистрофии нарушения в
дальнейшем осложняются внезапно развивающимися двигательными расстройствами по типу параличей (пирамидными
парезами), судорожными (эпилептиформными) припадками и слабоумием тяжёлой степени. Длится 6-8 лет, заканчивается
летально.
7. Диагностика Диф. диагностика
• Клиническое обследование: анализ симптомов, таких какжелтуха, неврологические нарушения, психические
расстройства.
Дифференциальная диагностика болезни Вильсона включает в себя
несколько заболеваний и состояний, которые могут иметь схожие
клинические проявления. Вот некоторые из них:
• Лабораторные тесты:
– Уровень меди в сыворотке крови и моче.
– Уровень церулоплазмина.
– Печеночные пробы (АЛТ, АСТ, щелочная фосфатаза).
• Генетическое тестирование: для подтверждения мутаций в гене
ATP7B.
• Ультразвуковое исследование печени: для оценки состояния
печени и наличия фиброза или цирроза.
• Биопсия печени: для оценки накопления меди в тканях.
1. Гепатиты (вирусные, токсические, аутоиммунные):
– Вирусные гепатиты (A, B, C)
– Алкогольный гепатит
2. Цирроз печени:
– Первичный билиарный цирроз
– Первичный склерозирующий холангит
3. Психические расстройства:
– Другие неврологические заболевания (например, болезнь
Паркинсона)
– Шизофрения или другие психозы
4. Неврологические расстройства:
– Деменция
– Синдромы, связанные с дефицитом витаминов (например, B12)
8. Лечение
Основой патогенетического лечения является назначение тиоловых препаратов, в первую очередь — Dпеницилламина либо унитиола. Главное преимущество купренила — низкая токсичность и возможность длительногоприема при отсутствии побочных эффектов. Его назначают по 0,15 г (1 капсула) в сутки (только после еды), в
дальнейшем, в течение 2,5-3 месяцев дозу увеличивают до 6-10 капсул/сутки (оптимальная доза). Лечение Dпеницилламином проводится годами и даже пожизненно с небольшими перерывами (на 2-3 недели) в случае
появления побочных эффектов (тромбоцитопения, лейкопения, обострения язвенной болезни желудка и т. д.).
Унитиол назначают в случае непереносимости (плохой переносимости) D-пеницилламина. Длительность одного
курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца. В большинстве случаев наступает
улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов). В
случае доминирования гиперкинезов рекомендовано назначение небольших курсов нейролептиков, при ригидности
— леводопы, карбидопы, тригексифенидила.
В случае тяжелого течения болезни Вильсона, при неэффективности консервативного лечения за рубежом прибегают
к трансплантации печени. При положительном исходе операции состояние пациента улучшается, восстанавливается
обмен меди в организме. В дальнейшем лечение пациента составляет иммуносупрессивная терапия. В России на
сегодня постепенно внедряется в клиническую практику метод биогемоперфузии с изолированными живыми
клетками селезенки и печени (т. н. аппарат «вспомогательная печень). Немедикаментозное лечение состоит в
назначении диеты (стол №5) в целях исключения продуктов богатых медью (кофе, шоколад, бобовые, орехи и т. д.).
9. Болезнь Марфана
Синдром Марфана — генетическое заболевание соединительной ткани, которое проявляетсямножественными нарушениями в скелетной, сердечно-сосудистой системах и органах
зрения. Затрагивает опорно-двигательный аппарат, глаза, сердечно-сосудистую систему и
другие органы.
10. Этиология
Синдром Марфана относится к врожденным аномалиям, наследуемым поаутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей
экспрессивностью и высокой пенетрантностью. В основе синдрома Марфана
лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего
структурного белка межклеточного матрикса, придающего эластичность и
сократимость соединительной ткани. Аномалия и дефицит фибриллина при
синдроме Марфана приводят к нарушению формирования волокнистых
структур, потере прочности и упругости соединительной ткани,
невозможности выдерживать физиологические нагрузки. Гистологическим
изменениям в большей степени подвержены стенки сосудов эластического
типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза,
содержащие наибольшее количество фибриллина).
11. Патогенез
При болезни Марфана происходит замена нуклеотидов вгене, содержащем информацию о структуре пептида
фибриллина-1. Этот белок относится к гликопротеидам,
принимает участие в микрофибриллярном комплексе, он
обеспечивает
основу
эластических
фибрилл
соединительной ткани.
При синдроме Марфана значительно поражается
трансформирующий фактор роста бета (TGF-β),
нарушается связывание его неактивной формы, что
приводит к повышению биоактивности данного фактора,
с чем связано появление многих проявлений болезни.
Патология
фибриллина
приводит
к
патологии
формирования волокон, что вызывает утерю прочности и
эластичности кожи и других соединительнотканных
структур.
Изменение структуры коллагеновых волокон приводит к
нарушению первичного звена гемостаза у пациентов с
синдромом Марфана.
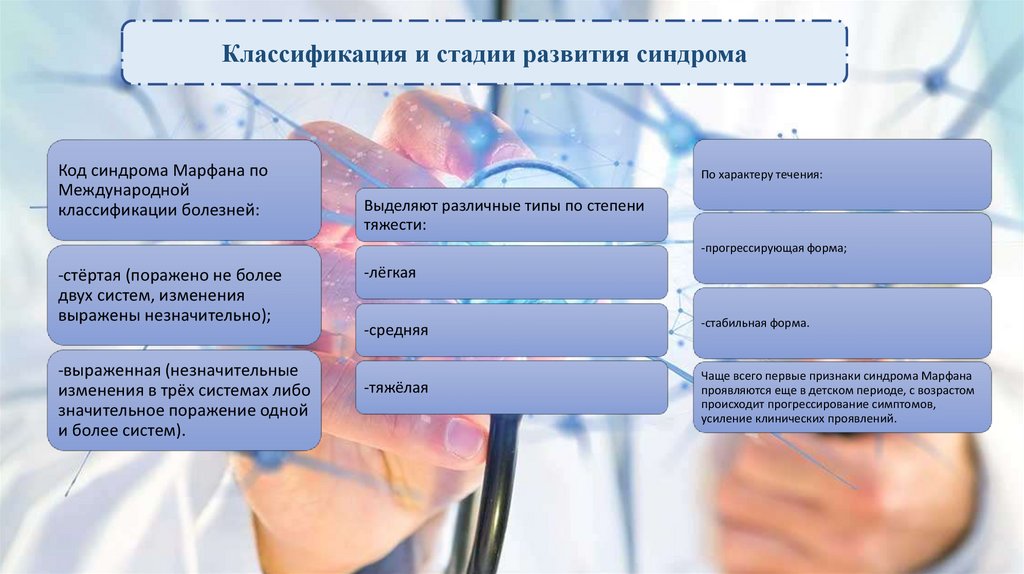
12.
Классификация и стадии развития синдромаКод синдрома Марфана по
Международной
классификации болезней:
По характеру течения:
Выделяют различные типы по степени
тяжести:
-прогрессирующая форма;
-стёртая (поражено не более
двух систем, изменения
выражены незначительно);
-выраженная (незначительные
изменения в трёх системах либо
значительное поражение одной
и более систем).
-лёгкая
-средняя
-тяжёлая
-стабильная форма.
Чаще всего первые признаки синдрома Марфана
проявляются еще в детском периоде, с возрастом
происходит прогрессирование симптомов,
усиление клинических проявлений.
13.
Клиническая картина-Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая
близорукость, вывих/подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы,
гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки.
-При синдроме Марфана отмечаются нарушение функции суставов (гипермобильность); деформация
грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз,
кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия
вертлужной впадины.
-Больные синдромом Марфана, как правило, отличаются высоким ростом, относительно коротким
туловищем с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными
паукообразными пальцами (арахнодактилией); астеническим телосложением со слаборазвитой
подкожной клетчаткой и мышечной гипотонией; длинным и узким лицевым скелетом (долихоцефалией);
наличием высокого аркообразного неба и нарушения прикуса (прогнатии).
14.
ДиагностикаДиагностика Синдрома Марфана основывается на клинических данных, выявлении изменений в
гене FBN1.
Способы обнаружения арахнодактилии:
-Симптом Steinberg (признак первого пальца). Первый палец виден из-под hypothenar при
напряжённом кулаке.
-Симптом Walker-Murdoch (признак запястья). При обхватывании кистью в области лучезапястного
сочленения контралатеральной верхней конечности первый палец заходит за пятый.
-Определение пястного индекса. Определяется при помощи рентгенографии. Средняя длина пясти,
делённая на усреднённую ширину отрезка от второй до четвертой пястной кости. При нормальном
соотношении этот показатель соответствует 5,4-7,9, в то время, как при синдроме Марфана —
больше 8,4.
Дифференциальный диагноз проводится с заболеваниями, сопровождающимися марфаноподобным
фенотипом. К таким заболеваниям относятся гомоцистинурия (сбой обменных процессов
метионина), арахнодактилия контрактурная врождённая (аномально тонкие и длинные пальцы),
артроофтальмопатия наследственная прогрессирующая (изменение лица, патология суставов,
поражение глаз потеря слуха) и другие дисплазии соединительной ткани.
15. Выявление болезни Марфана
16.
ЛечениеЛечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться
группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком,
терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования
заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При
диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или
ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца,
пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и
расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют
высокий процент послеоперационной 5-ти и 10-ти летней выживаемости. При необходимости
выполняют протезирование митрального клапана. У беременных с синдромом Марфана и
выраженной
сердечно-сосудистой
патологией
проводят
досрочное
оперативное
родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита
и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
17. Факаматозы
18.
Факоматоз — это генетическое заболевание, обусловленное нарушениями в процессах дифференцировки иразвития клеток в раннем эмбриональном периоде. С развитием генетики и методов ДНК-анализа для
некоторых заболеваний были установлены гены, аберрации в которых детерминируют данный факоматоз.
Зачастую мутации приводят к снижению синтеза факторов, блокирующих онкогенез, что считают вероятной
причиной множественного опухолевого роста, в большинстве случаев характеризующего факоматоз.
Исследования показали, что факоматозы имеют в основном аутосомно-доминантное наследование с неполной
пенетрантностью, благодаря которой болезнь проявляется не в каждом поколении.
Нарушения касаются преимущественно эктодермального зародышевого листка, который дает
начало всей нервной ткани, наружным слоям кожных покровов, придаткам кожи (ногтям,
волосам), сетчатке, эпителию слизистой рта и полости носа. Клетки, которые остались в фазе
перманентной эмбрионизации, т. е. не продолжили свое развитие, образуют врожденные
опухолевые образования — гамартомы. Эти эмбриональные опухоли различной локализации
часто сопровождают любой факоматоз.
Типичной чертой факоматоза выступает сочетанное полиморфное поражение кожи, нервной
системы и соматических органов. Причем одни клинические синдромы, чаще всего
неврологические и дерматологические, являются врожденными или манифестируют в раннем
детском возрасте, а другие — намного позже. В отдельных случаях факоматоз сочетается с
врожденным иммунодефицитом, преждевременным старением и/или риском развития
злокачественных образований.
19.
Диагностика-Неврологическая диагностика. Электроэнцефалография позволяет установить характер
эпиактивности головного мозга. Эхо-ЭГ выявляет признаки гидроцефалии. При помощи МРТ и
КТ головного мозга визуализируются морфологические изменения церебральных тканей, при
помощи ангиографии головного мозга или МРА — пороки церебральных сосудов.
-Офтальмологическая диагностика. Офтальмоскопия проводится в обязательном порядке,
позволяет диагностировать поражение органа зрения даже в случае его субклинического
течения.
-Исследование внутренних органов. Кардиологические исследования включают ЭКГ и УЗИ
сердца, гастроэнтерологические — УЗИ брюшной полости, при необходимости
рентгенографию желудка, рентгенконтрастное обследование тонкого и толстого кишечника.
Исследование почек проводится при помощи УЗИ, урографии и КТ.
20. Лечение
Медикаментозная терапия. На сегодняшний день ни один факоматоз не имеетспецифического лечения. Проводится симптоматическая терапия. По показаниям
применяется антиконвульсантное (вальпроаты, леветирацетам, карбамазепин,
топирамат), дегидратирующее (ацетазоламид), нейрометаболическое (витамины группы
В, глицин) лечение. Зачастую эпиприступы оказываются резистентными к проводимой
противосудорожной терапии, в связи с чем приходится менять препарат или переходить
на комбинированные схемы, включающие 2 антиконвульсанта. При наличии
эписиндрома противопоказаны нейрометаболиты стимулирующего действия (к-та гаммааминомасляная, пирацетам, пиритинол).
Хирургическое лечение. По показаниям проводится хирургическое лечение, целью которого
является удаление возникшего новообразования. Вмешательства проводятся при
подозрении на злокачественность опухоли, при нарастании обусловленных ею клинических
проявлений, быстром росте образования, развитии компрессионного синдрома. Если речь
идет о внутримозговых опухолях, то операцию проводят нейрохирурги. При опухолях
соматических органов оперируют соответствующие специалисты.
Психологическая коррекция. Наряду с фармакотерапией в лечении факоматоза большую
роль играет психокоррекция. Она направлена на развитие умственных и психических
способностей ребенка, индивидуальную коррекцию имеющихся отклонений, обучение
ребенка в доступном для него формате и его социальную адаптацию. В зависимости от вида
и степени психических нарушений рекомендованы занятия с психологом, детская
психотерапия, игровая терапия, АВА терапия, нейропсихологическая коррекция.
Осуществляется психологическое консультирование родителей. Возможно комплексное
психологическое сопровождение ребенка.