



")




")
")
")
")
")
")



 Медицина
МедицинаПохожие презентации:










Анализ особенностей наследственных нейрометаболических заболеваний у детей
1. Кафедра нервных болезней, нейрохирургии и медицинской генетики Уральский государственный медицинский университет
Анализ особенностейнаследственных
нейрометаболических
заболеваний у детей
Кабдуакитова А.Т
Куратор к.м.н., Овсова О.В.
2. Лейкодистрофии Наследственные лейкоэнцефалопатии
группанаследственных
нейродегенеративных
характеризующихся прогрессирующей дистрофией
головного и/или спинного мозга.
заболеваний,
белого вещества
Частота встречаемости наследственных болезней обмена
1:3000 живых новорожденных
Лейкодистрофии(ЛД):
Метохроматическая ЛД
ЛД Краббе,
Х-сцепленная адренолейкодистрофия,
болезнь Канаван,
болезнь Александера,
Лейкоэнцефалопатия с макроцефалией и субкортикальными кистами,
Лейкоэнцефалопатия с поражением ствола головного мозга, спинного
мозга и повышением лактата,
Лейкоэнцефалопатия с «исчезающим» белым веществом.
3.
Аутосомно- рецессивный тип наследованияМутациями в белковых (транспортных/ сигнальных) системах/
мутациями в генах, кодирующих ферменты.
Возраст манифестации зависит от степени нарушения
метаболического пути (МП):
1.
полное выключение
раннее проявление
тяжелое
клиники
течение
МП
2. частичный дефицит
субклиническая
МП
картина
Младенческая (0-1 год)
истощении
поздний
процессов
дебют
Детская (2-6 лет)*
Взрослая
(лейкоэнцефалопатия Кри)
Прогредиентное течение
Нарушения вскармливания
Туловищная мышечная гипотония
Гипертонус конечностей
Судороги
У ряда пациентов Катаракта,
гепатоспленомегалия, дисгенезия яичников
Мозжечковая атаксия
Спастический тетрапарез
Когнитивные нарушения
Атрофия зрительных нервов
Эпиприступы
Ухудшение состояния после
инфекционного процесса, травмы
или стресса
Медленнопрогрессирующее
Дебют с психиатрических
нарушений/ эпиприпадков
Интеллектуальные расстройства
Двигательные нарушения
Дисгенезия яичников у женщин
4. МРТ- диагностика
ДНК- диагностика:Энзимодиагностика:
Мутации в гене:
DARS 2
LBSL
GALK
Болезнь Краббе (БКр)
ASA
Метохроматическая ЛД
(МЛД)
GFAP Болезнь Александера (БА)
активности Арилсульфатазы А (МЛД)
активности N-ацетиласпартата (БК)
активности Галактоцереброзидазы (БКр)
МРТ- диагностика
ЛД
БКр
Демиелинизация подкорковых структур, мозжечка
БА
Демиелинизация преимущественно лобного отдела
БК
Диффузные изменения в белом веществе
Х-АЛД
Демиелинизация заднетеменных отделов
LBSL
Симметричные изменения белого вещества ствола,
мозжечка и спинного мозга
МЛД
Демиелинизация ПВ областей, «тигроидный» паттерн
БКр- Болезнь Краббе; БА- болезнь Александера; БК- Болезнь Канаван; Х-АЛД – х-сцепленная адренолейкодистрофия; LBSL
Лейкоэнцефалопатия с поражением ствола головного мозга, спинного мозга и повышением лактата; МЛД – Метохроматическая ЛД;
5. Нейрональный цероидный липофусциноз (НЦЛ)
Наследственное нейродегенеративное заболевание, характеризующееся эпилептическимиприступами, резистентными к АЭТ, прогрессирующими нарушениями интеллектуального,
двигательного развития, снижением зрения.
Частота 1:25 000 в мире
В России диагностировано 30 случаев НЦЛ: 1,2 и 3 типов.
Аутосомно-рецессивный тип наследования
1. Врожденная форма (CNCL)
CTSD
2. Младенческая форма (Сантавуори- Халтиа) (INCL)
PPT 1
3. Поздняя младенческая: (LINCL)
Классическая (Янского-Бильшовского) (cLINCL)
TTP1, PPT1, CLN 5,6,8
Финский (fvLINCL)
CLN 5
Цыганский/индийский (vLINCL)
CLN 6
Турецкий (tLINCL)
CLN 7
4. Юношеские формы (JNCL)
Болезнь Батена/Шпильмейера-Вогта
CLN 3, РРТ1, ТТР1
Вариант юношеской формы
CLN 9
5.Северная эпилепсия (NE,PEMR)
CLN 8
6. Взрослая форма (болезнь Куфса) (ANCL)
РРТ1,CLN 3,4
6.
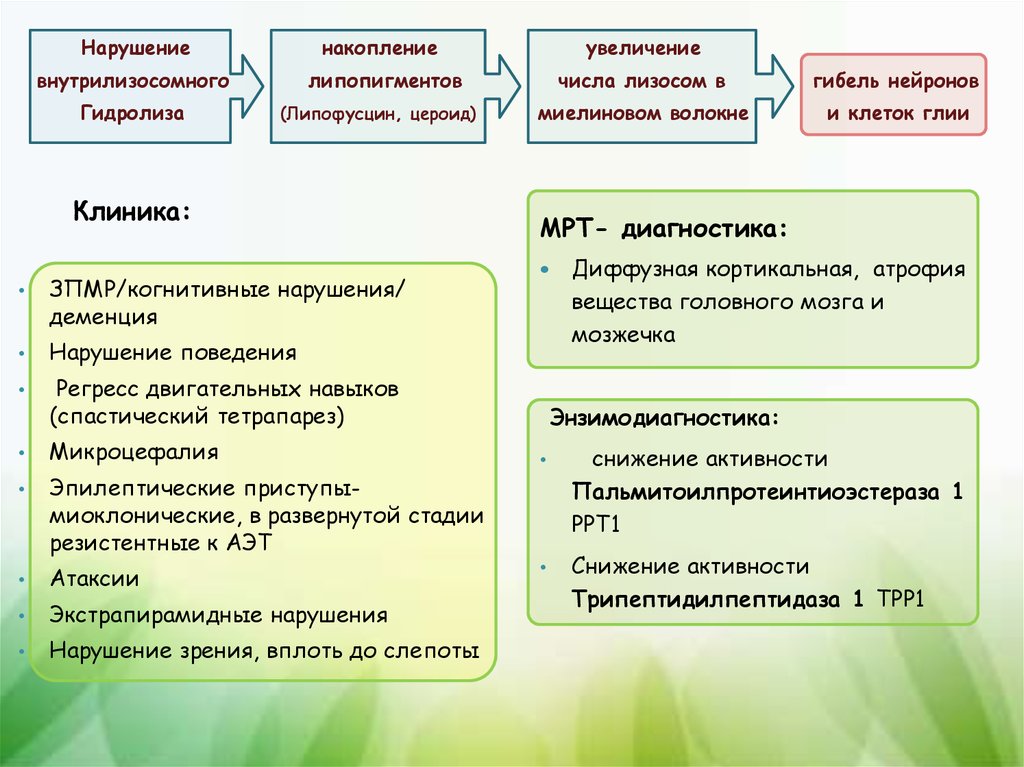
Нарушениенакопление
увеличение
внутрилизосомного
липопигментов
числа лизосом в
гибель нейронов
миелиновом волокне
и клеток глии
Гидролиза
(Липофусцин, цероид)
Клиника:
ЗПМР/когнитивные нарушения/
деменция
Нарушение поведения
Регресс двигательных навыков
(спастический тетрапарез)
Микроцефалия
Эпилептические приступымиоклонические, в развернутой стадии
резистентные к АЭТ
Атаксии
Экстрапирамидные нарушения
Нарушение зрения, вплоть до слепоты
МРТ- диагностика:
Диффузная кортикальная, атрофия
вещества головного мозга и
мозжечка
Энзимодиагностика:
снижение активности
Пальмитоилпротеинтиоэстераза 1
РРТ1
Снижение активности
Трипептидилпептидаза 1 ТРР1
7. Актуальность работы
Частота встречаемости наследственных болезней обмена 1:3000 живыхноворожденных.
Диагностика затруднительна ( манифестацией в раннем возрасте с
неспецифического поражения нервной системы, полиморфизмом клинических
проявлений, наличием атипичных форм)
Прогностически неблагоприятные, с быстрым прогрессирующим течением и
высоким летальным исходом.
Цель исследования - показать особенности клинической картины у
детей с нейрометаболическими заболеваниями.
Задачи:
• Изучить нозологическую структуру, клинические и
нейрорадиологические проявления по данным литературы и среди
пациентов психоневрологического профиля
• Исследовать клинические проявления заболевания
• Установить нейрорадиологические/ лабораторные данные
характерные для каждой группы НМЗ
• Предложить рекомендации по терапии и методам профилактики
8. 20%
По данным регистра наследственных заболеваний в Свердловской областизарегистрировано 15 пробандов с первичными Лейкодистрофиями (n=15) и 6
пробандов с Нейрональным цероидным липофусцинозом детского
возраста (n=6).
Лейкодистрофии
Болезнь Канаван
6,6%
6,6%
20%
НЦЛ
ЛД Краббе
6,6%
26,6%
20%
13,3%
LBSL
аутосомнорецессивные ЛД
Х-АЛД
НЦЛ 1
типа
50%
50%
МЛД
неуточненная ЛД
Наибоее часто встречающиеся формы первичных лейкодистрофии в
Свердловской области – Х-сцепленная адренолейкодистрофия 26,6% (n=4),
Лейкоэнцефалопатия с поражением ствола головно мозга, спинного мозга и
повышением лактата (LBSL) по 20%(n=3) и Болезнь Краббе по 20%(n=3).
НЦЛ 2
типа
9.
Анализ 6 случаев лейкодистрофий и 3 случаев нейрональногоцероидного липофусциноза у пациентов наблюдающихся в КДЦ
«Охрана здоровья матери и ребенка», отделение МГК (зав. к.м.н.
Никитина)
LBSL (n=3)
НЦЛ 1 тип (n=2)
Метохроматическая ЛД (n=1)
НЦЛ 2 тип (n=1)
ЛД Краббе (n=1)
Х-Адренолейкодистрофия (n=1)
Ранние клинические проявления:
Ранние клинические проявления:
ЗСМР, ЗРР - 83,3%
ЗСМР, ЗРР – 100%
Регресс навыков – 66,6%
Регресс навыков – 100%
Мышечный гипертонус - 50%
Нарушение походки – 60%
Нарушение походки – 83,3%
Тремор – 30%
Мозжечковая атаксия – 33,3%
Эпилепсия – 100%
Тремор - 50%
Эпилепсия – 16,6%
10.
ЛейкодистрофииНЦЛ
Средний возраст дебюта заболевания
6 лет 5 месяцев (6,51±5,92).
Средний возраст дебюта заболевания
1 год 8месяцев (1,88±1,11).
Средний возраст постановки диагноза
8 лет 4 месяца (8,35±6,03).
Средний возраст постановки диагноза
4 года 2 месяца (4,23±0,77).
Минимальный и максимальный возраст
дебюта 1 год и 15 лет.
Минимальный и максимальный возраст
дебюта 1 год и 5 лет.
В 66,6% (n=6) на фоне полного здоровья, с предшествующим
периодом нормального развития;
В 33,3% (n=3)
заболевание связывают с перенесенной инфекцией
(ветряная оспа) и вакцинацией (от гепатита В, АКДСМ);
11. Клинические симптомы НМЗ (n=9)
Для всех форм характерно прогрессирующее
течение, с разным по продолжительности
периодом нормального развития (от 1 до 14
лет)
Для 88,8% характерен регресс
психомоторных навыков, с развитием
когнитивного дефицита, развитием
психоорганического синдрома вплоть до
деменции
У 66,6% развился центральный
(спастический) парез из них тетрапрез(n=5),
нижний парапарез (n=1), гемипарез(n=1), с
сухожильной гиперрефлексией и
патологическими стопными знаками.
Эпелпсия у 55,5%, у большинства в виде
симптоматической парциальной
эпилепсии(n=3)
Мозжечковая атаксия в 22,2%
Экстрапирамидные нарушения в виде тремора
(рук, предплечий/туловища /головы /ног)
44,4% снижение остроты зрения, зрительная
агнозия за счет поражения затылочной доли
У одного пациента с НЦЛ 1 типа развилось
явление декортикационной рагидности
100,00%
80,00%
60,00%
40,00%
Декортикационная регидность
Тремор
Поведенческие расстройства
Атаксия
Эпилепсия
Снижение о.зрения
Псевдобульбарный синдром
Центральные парезы
ЗПР/ЗРР
Период N развития
0,00%
Прогредиентное течение
20,00%
12. Типы эпилептических приступов НМЗ (n=6)
Тип приступаКоличество
пациентов
Симптоматическая парциальная эпилепсия
3 (33,3%)
Эпилепсия по типу инфантильного спазма флексорного/
экстензорного характера
1 (11,1%)
Аксиальные тонические спазмы
1 (11,1% )
Прогрессирующая миоклоническая эпилепсия
1 (11,1%)
У всех пациентов с НЦЛ эпилепсия манифестировала в дебюте заболевания:
Эпилепсия по типу инфантильного спазма флексорного/ экстензорного характера
(наклоны головы) (n=1), симптоматическая парциальная эпилепсия (миоклоникоастетическая форма) (n=1), аксиальные тонические спазмы (n=1).
У пациентов с первичной лейкодистрофией эпилепсия развилась по мере
прогрессирования заболевания: Симптоматическая парциальная эпилепсия (височно
-лобнодолевая с соматовегетативными приступами в руках) (n=2), и
прогрессирующая миоклоническая эпилепсия (n=1).
13. Двигательные нарушения при НМЗ (n=8)
90,00%80,00%
Для 77,7% характерен регресс двигательных
навыков, с предшествующим нормальным
периодом развития
Двигательные нарушения в виде центральных
(спастических) парезов (n=7) из них
тетрапрез(n=5) , нижний парапарез (n=1),
гемипарез(n=1), с сухожильной
гиперрефлексией и патологическими
стопными знаками и в 66,6% случаев с потерей
способности к самостоятельной ходьбы
88,8% сухожильная гиперрефлексия может
говорить о нарастании двигательных
нарушений , с дальнейшим формированием
спастичности и снижением мышечной силы у
пациентов без двигательных расстройств.
Мозжечковая атаксия в 22,2% (n=2)
55,5% Экстрапирамидные нарушения в виде
тремора (рук , предплечий(n=3), туловища
(n=2), головы (n=2), ног (n=1)
70,00%
60,00%
50,00%
40,00%
30,00%
20,00%
10,00%
0,00%
Двигательные стереотипии в руках: (n=2)
22,2%
В руках «хлопанье в ладоши», потирания
В руках подергивание, перебирание
пальцев
14. Спектр нейрорадиологических изменений при НМЗ (n=9)
100%
90%
80%
70%
60%
50%
40%
30%
20%
10%
0%
Поражение сп/м
Гидроцефалия
СК
Тем/зат обл
Лобн. обл
Вис. обл
ПВ
Подкорк/стр.
Мозжечок
Ствол
Атрофия коры гм
НЦЛ 2
НЦЛ 1
Х-АЛД
МЛД
ЛДКр
LBSL
Наиболее обширные поражения при
LBSL (n=3): субкортикальные,
перивентрикулярные области;
Теменно-затылочные, лобные,
височные доли; поражение
подкорковых структур, мозжечка,
ствола гм и диффузные поражения
спинного мозга.
Болезнь Краббе: СК , ПВ области и
ствол; гидроцефалия.
Метохроматическая ЛД: Поражение
ПВ области и подкорковых структур.
Адренолейкодистрофия: Теменнозатылочные и височные области;
подкорковые структуры.
НЦЛ:
1 тип- Атрофия коры гм, поражение
СК и ПВ областей, мозжечка;
гидроцефалия.
2 тип – Атрофия коры гм;
15. Экстраневральные нарушения (n=5, 55,5%)
Среди первичных лейкодистрофии у 22,2%
наблюдалось спленомегаия (n=2) и также у
22,2% миокардодистрофия, что объяснимо
тем, что блокированные метаболические пути
(ферментные, белковые) являются общими
для многих органов и систем, а выраженность
клиники зависит от тканеспецифичности
ферментного блока.
У 11,1% (n=1) патология желчевыводящих
путей , возможно связанная с лекарственной
терапией
У пациента с Х-сцепленной
Адренолейкодистрофией, симптомокомплекс
экстраневральных нарушений укладывается в
клинический вариант Адреномиелопатию с
надпочечниковой недостаточность , ихтиозом,
аллопециями и дерматитом.
Среди пациентов НЦЛ (n=1) реактивные
изменения в поджелудочной железе и
увеличение объема почек
25,00%
20,00%
15,00%
10,00%
5,00%
0,00%
16. Поведенческие нарушения (n=8; 88,8%)
В 88,8% (n=8) случаев отмечается регресс
психомоторного развития
33,3% Гипердинамический синдром
22,2% (n=2) Органическое астеническое
расстройство и также 22,2% (n=2)
эмоциональное лабильное расстройство
44,4% грубый когнитивный дефицит со
снижением интеллекта, вплоть до деменции
(n=1)
Аустиподобное поведение 22,2% наблюдалось
только у пациентов с НЦЛ
В 22,2% (n=2) брадилалия, сенсоматорная
алалия, вплоть до мутизма(n=1), пациент НЦЛ
1 типа, с тяжелым прогрессирующим
течением
100,00%
80,00%
60,00%
40,00%
20,00%
0,00%
17. Молекулярно- генетическая диагностика
Лаборатория НБО МГНЦ РАМН (к.м.н. Захарова)LBSL Выявлены молекулярные дефекты в 3,5 экзонах гена DARS2.
3 экзоне мутация делеция 228-20_-21delTTinsC в гетерозиготном состоянии,
в 5 экзоне гена мутация с.492+2Т>C в гетерозиготном состоянии (n=1).
мутации CS072179 c.228-11C>G и СМ071698 с.455G>T (р.Су s152Phe) в гетерозиготном
состоянии (n=1).
Мутации CX072638 c/228-20_21delTTinsC в гетерозиготном состоянии и мутация,
CS072183 c/492+2T>c в гетерозиготном состоянии (n=1).
ЛД Краббе полный анализ гена GALС. Исследованы с 1 по 17 экзоны и
прилегающие интронные области, выявлены
полиморфные варианты в гетерозиготном состоянии c 1350C>T (p Ser450) и
варианты в гомозиготном состоянии с 1685T>C (p Ile562Thr) с 1620G>A (p
Thr540) c 1698A>T (p Val566), c 1765+5C>G c 1921A>G (p Thr641Ala) (n=1).
НЦЛ типа 1. Обнаружены мутации в гене PPT1 (пальмитоил протеин
тиоэстераза)
p.Arg122Trp, p.Arg151 Stop-мутация, C 169-170insA, p.Arg122Trp(n=2);
НЦЛ тип 2 в гене ТТР1(трипептидил пептидаза 1)
с.89+5G>C, p.Arg208Term (n=1).
Генетический риск для сибсов 25%
18. Лабораторная диагностика
Определение активности ферментов методомспектрофотометрии/ спектрофлюорометрии:
ЛД Краббе - фермент галактоцереброзидаза (0,1
мкМ/л/ч) референсные значения (0,70-10,00 мкМ/л/ч) (n=1);
МЛД - снижение активности фермента арилсульфатазы А
20,5 ЕД (референсные значения 120 ЕД)(n=1);
НЦЛ 1 типа - резкое снижение активности пальмитоил протеин
тиоэстеразы 1 до 0,1 до 9,6 нМ/мг/час (норма - 27-100 нМ/мг/час).
Определение уровня органических и жирных кислот с очень
длинной цепью методом
• АЛД - увеличена концентрация кислоты С26 3,9 мМ/мл (0,22- 2,20
мМ/мл), а также повышены соотношения С24/С22 (1,29 мМ/мл (0,88-0,66
мМ/мл) и С26/С22 0,08 мМ/мл (0,01-0,02 мМ/мл) (n=1).
19.
Выводы1.
Для всех выявленных нейрометаболических заболеваний характерно тяжелое,
прогредиентное течение, с регрессом двигательных функций, грубой задержкой
психического развития, мозжечковыми, экстрапирамидными нарушениями, в трети
случаев развитием эпилептических припадков, резистентных к противосудорожной
терапии.
2.
Нейрорадиологическое исследование продемонстрировало, что (МРТ головного
мозга) для каждой нозологической формы характерны определенные изменения
белого вещества мозга, что важно для дифференциальной диагностики НМЗ.
3.
Средний возраст дебюта заболевания в исследуемой группе составил 6 лет 5 месяцев
(6,51±5,92).
4.
Манифестация заболевания в 77,7% случаев произошла на фоне полного здоровья, с
предшествующим периодом нормального развития.
5.
Учитывая тяжесть заболевания и неблагоприятный прогноз, необходимо для
верификации диагноза, выполнить молекулярно- генетическое исследование, с
дальнейшим проведением диагностики носительства мутаций у родителей, для
расчета риска рождения больного ребенка при планировании повторного
деторождения и пренатальной ДНК-диагностики плода на ранних сроках
беременности (до 8 недель гестации).