

























")
")
")
")
")
")
")

")


")

")
")
")
")











































 Медицина
МедицинаПохожие презентации:

 дистрофии")







 Дистрофии. Патоморфология нарушений обмена белков и жиров")
Стромально-сосудистые дистрофии
1. Стромально-сосудистые дистрофии
2. Белковые сосудисто-стромальные дистрофии
Стромально-сосудистые(мезенхимальные)
дистрофии развиваются на территории
гистиона (отрезок микроциркуляторного русла
с
окружающими
его
элементами
соединительной ткани и нервными волокнами)
в
результате
нарушений
обмена
в
соединительной ткани и выявляются в строме
органов и стенках сосудов.
3. Белковые сосудисто-стромальные дистрофии
Мукоидное набуханиеФибриноидное набухание
Гиалиноз
Амилоидоз
4. Белковые сосудисто-стромальные дистрофии
Мукоидноенабухание
–
поверхностная
и
обратимая
дезорганизация соединительной
ткани
(син.
слизеподобное
набухание, хромотропный отек)
5. Белковые сосудисто-стромальные дистрофии
Мукоидноенабухание
–
диагностируется при получении
метахроматического
окрашивания с красителем –
толуидиновым синим
6. Мукоидное набухание
7. Мукоидное набухание
8. Белковые сосудисто-стромальные дистрофии
Причины мукоидного набухания:- гипоксия
- инфекция
- реакции гиперчувствительности
9. Белковые сосудисто-стромальные дистрофии
Мукоидноенабухание
развивается при повышении
сосудистой проницаемости и
высоком содержании жидкости в
основном
веществе
соединительной ткани
10. Белковые сосудисто-стромальные дистрофии
Звеньяморфогенеза
набухания:
мукоидного
Накопление и перераспределение
гликозаминокликанов в основном веществе
соединительной ткани за счёт увеличения
содержания гиалуроновой кислоты.
Повышение сосудисто-тканевой
проницаемости.
Гидратация и набухание основного вещества и
коллагеновых волокон соединительной ткани.
11. Белковые сосудисто-стромальные дистрофии
Исходы мукоидного набухания:- полное восстановление ткани
- переход
в
фибриноидное
набухание
12. Белковые сосудисто-стромальные дистрофии
Фибриноидноенабухание
–
накопление в основном веществе
соединительной ткани белковых
масс
(плазменных
белков,
содержащих
фибриноген)
фибриноида.
При
образовании
комплексов с коллагеном процесс
становится необратимым
13. Белковые сосудисто-стромальные дистрофии
Звенья морфогенеза фибриноидногонабухания:
Резкое повышение сосудисто-тканевой
проницаемости.
Плазморрагия.
Выход грубодисперсных плазменных белков, в
первую очередь фибриногена.
Превращение фибриногена в фибрин.
Деструкция коллагена и основного вещества
соединительной ткани.
Образование фибриноида.
14. Белковые сосудисто-стромальные дистрофии
Фибриноидное набухание можетперейти в фибриноидный некроз
Исходы – склероз, гиалиноз
15. Окраска азокармином
16. Окраска азокармином
Окраска наэластические
волокна и по
Ван Гизон
17. Фибриноидный некроз
18. Белковые сосудисто-стромальные дистрофии
Гиалиноз – отложение белковыхмасс,
которое
при
макроскопическом исследовании
напоминает гиалиновый хрящ
19. Белковые сосудисто-стромальные дистрофии
Взависимости
от
преимущественной локализации
выделяют:
– гиалиноз сосудов
– гиалиноз стромы (собственно
соединительной ткани)
20. Белковые сосудисто-стромальные дистрофии
По составу выделяют:– простой гиалин
– сложный гиалин
– липогиалин
21. Белковые сосудисто-стромальные дистрофии
Простой гиалин возникает врезультате пропитывания стенки
сосуда белками плазмы при
гипертонической
болезни
и
атеросклерозе
22. Белковые сосудисто-стромальные дистрофии
Сложный гиалин состоит изиммунных комплексов, фибрина
и
компонентов
сосудистой
стенки,
образуется
при
иммунопатологических
процессах
23. Белковые сосудисто-стромальные дистрофии
Исходы гиалиноза:ослизнение ткани;
липоидоз;
обызвествление;
рассасывание гиалиновых масс.
24. Гиалиноз селезенки – «глазурная селезенка»
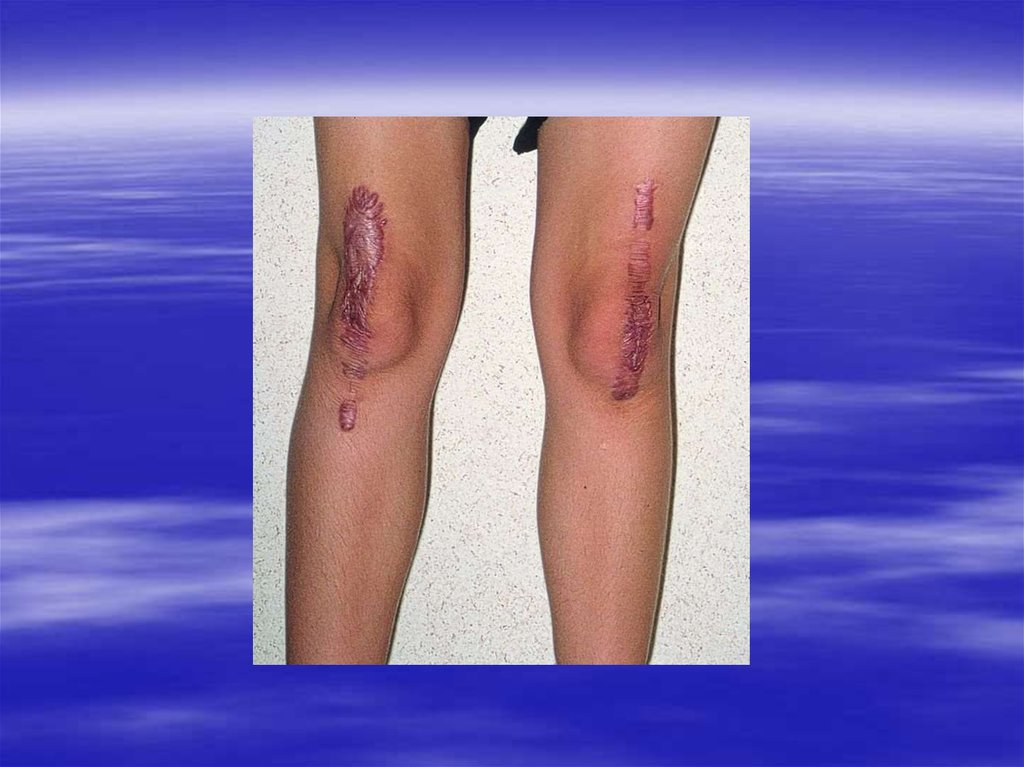
25. Склероз – рубцовая ткань
26. Окраска трихром по Массону
27.
28.
29.
30. Жировые сосудисто-стромальные дистрофии (ожирение)
Нарушения обмена нейтральныхжиров
проявляются
в
увеличении или уменьшении их
запасов в жировой ткани, могут
иметь общий или местный
характер.
31. Жировые сосудисто-стромальные дистрофии (ожирение)
Ожирение (тучность) –увеличение количества
нейтральных жиров в жировых
депо, имеющее общий характер
32. Жировые сосудисто-стромальные дистрофии (ожирение)
Локализация отложения жира:подкожная жировая клетчатка
сальник
брыжейка
средостение
эпикард
33. Жировые сосудисто-стромальные дистрофии (ожирение)
Классификацияожирения
учитывает следующие признаки:
этиология
внешние проявления
степень превышения массы тела
морфологические изменения
жировой ткани
34. Жировые сосудисто-стромальные дистрофии (ожирение)
По механизму:- Наследственное
- Алиментарное
- Эндокринное
- Церебральное
- Смешанное
35. Жировые сосудисто-стромальные дистрофии (ожирение)
По внешним проявлениям:- симметричное
- верхнее
- среднее
- нижнее
36. Жировые сосудисто-стромальные дистрофии (ожирение)
По превышению массы тела:- 1 степени (избыточная масса
составляет 20-29%)
- 2 степени (30-49%)
- 3 степени (50-99%)
- 4 степени (100% и более)
37.
38. Жировые сосудисто-стромальные дистрофии (ожирение)
По локализации:- общее
- местное
39. Ожирение миокарда
40. Ожирение лимфатического узла
41. Жировые сосудисто-стромальные дистрофии (ожирение)
По морфологии:- Гипертрофическое – увеличение
размеров
жировых
клеток
(липоцитов)
- Гиперпластическое – увеличение
количества клеток
42. Гиперплазия и гипертрофия липоцитов
43. Жировые сосудисто-стромальные дистрофии (ожирение)
Липоматоз – диффузное илимножественное опухолевидное
разрастание жировой ткани в
организме.
44. Жировые сосудисто-стромальные дистрофии (ожирение)
Вакатное ожирение – местноеувеличение количества жировой
ткани при атрофии органа
(жировое замещение).
45. Жировые сосудисто-стромальные дистрофии (ожирение)
Истощение – снижение веса,сопровождающееся сокращением
количества нейтральных жиров в
жировых депо, уменьшением объема
и массы органов с утратой
способности к выполнению
физиологических функций.
Кахексия – резко выраженное
истощение организма.
46. Жировые сосудисто-стромальные дистрофии (ожирение)
Регионарные липодистрофиихарактеризуются очаговой
деструкцией жировой ткани и
распадом жиров, часто с
воспалительной реакцией и
образованием липогранулём.
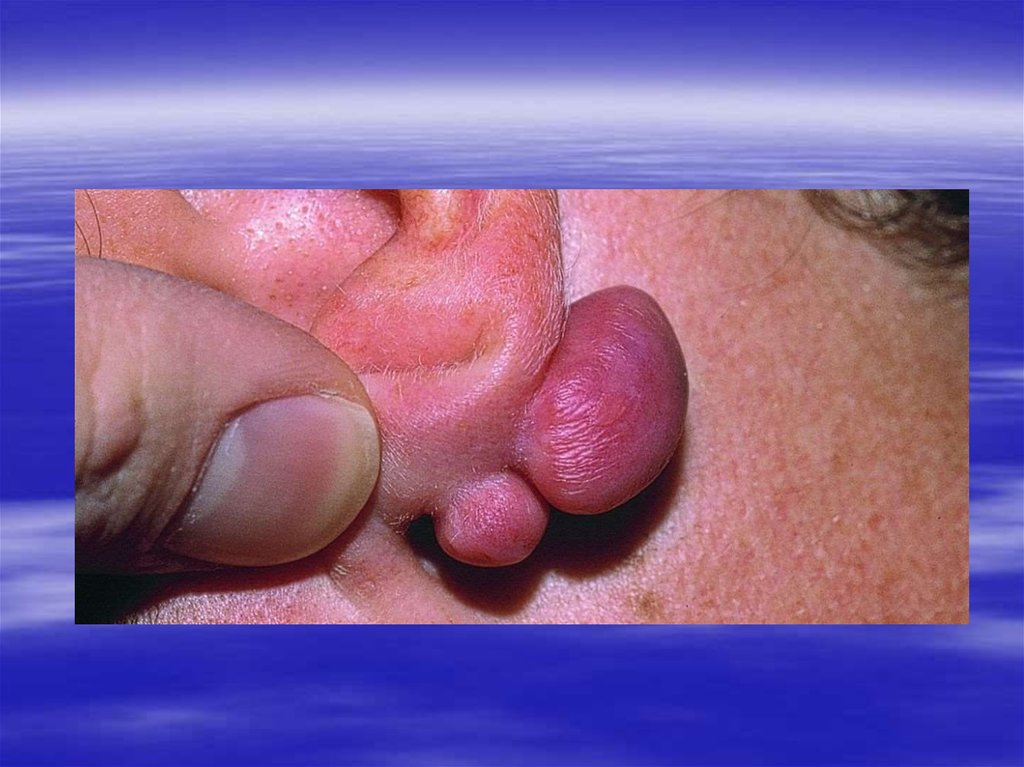
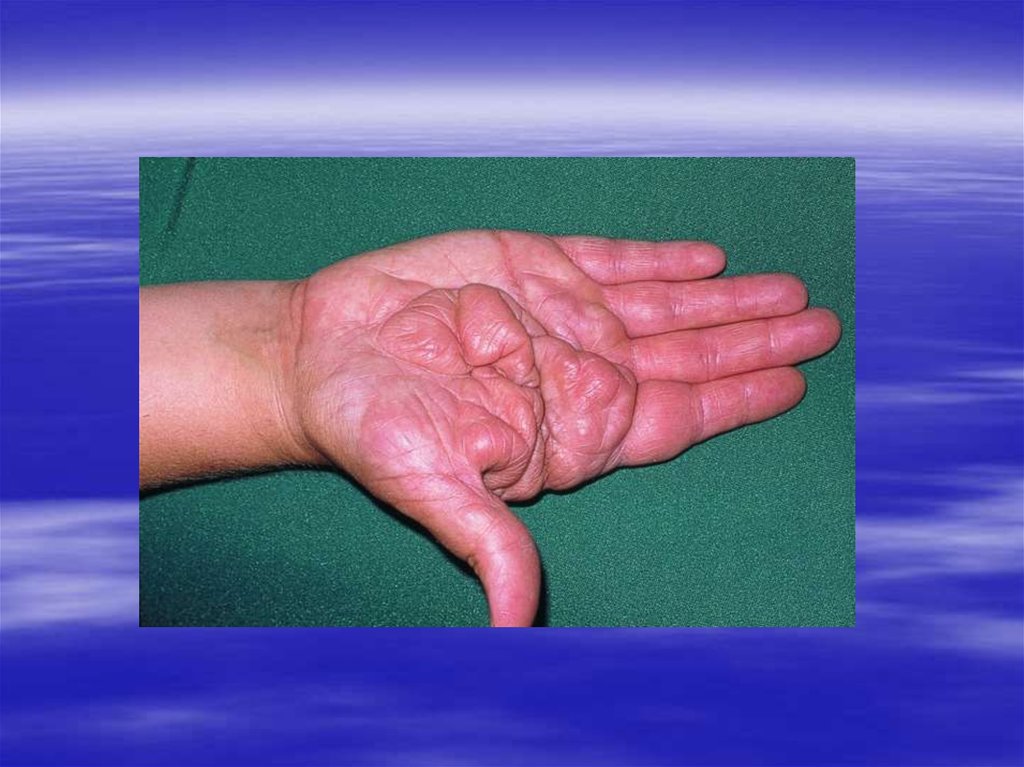
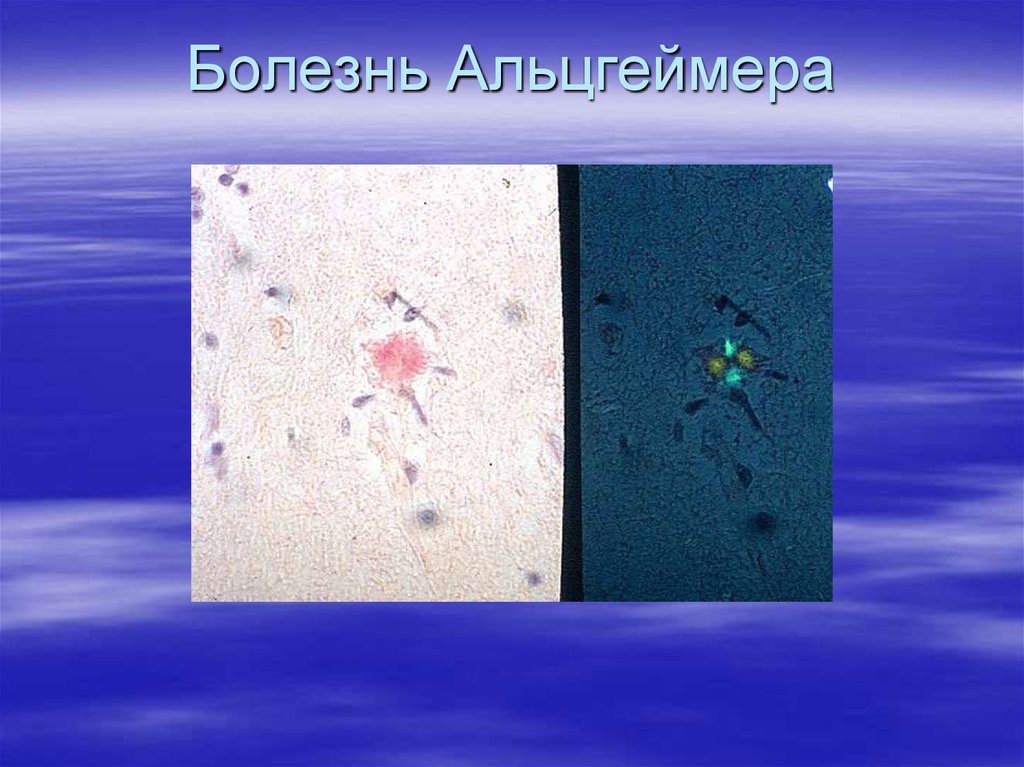
47. Амилоидоз
Группа заболеваний,характеризующихся отложением
гомогенных эозинофильных
нерастворимых белковых масс –
амилоида.
Амилоид окрашивается красителем
Конго рот в красный цвет и дает
двойное лучепреломление и
зеленое свечение в
поляризационном свете.
48. Амилоидоз
49. Амилоидоз
50.
51. Амилоидоз
Впервые описалКарл Рокитанский –
«сальная болезнь»
Название предложил
Рудольф Вирхов –
«амилоид» – подобный
крахмалу
52. Амилоидоз
53. Амилоидоз
54. Амилоидоз
«Большая сальная почка» отложение амилоидаобнаруживается в клубочках, стенках
сосудов и строме
«Сальная селезенка» («ветчинная
селезенка») – диффузно в строме и
капсуле
«Саговая селезенка» преимущественно в проекции
лимфоидных фолликулов
55. Амилоидоз
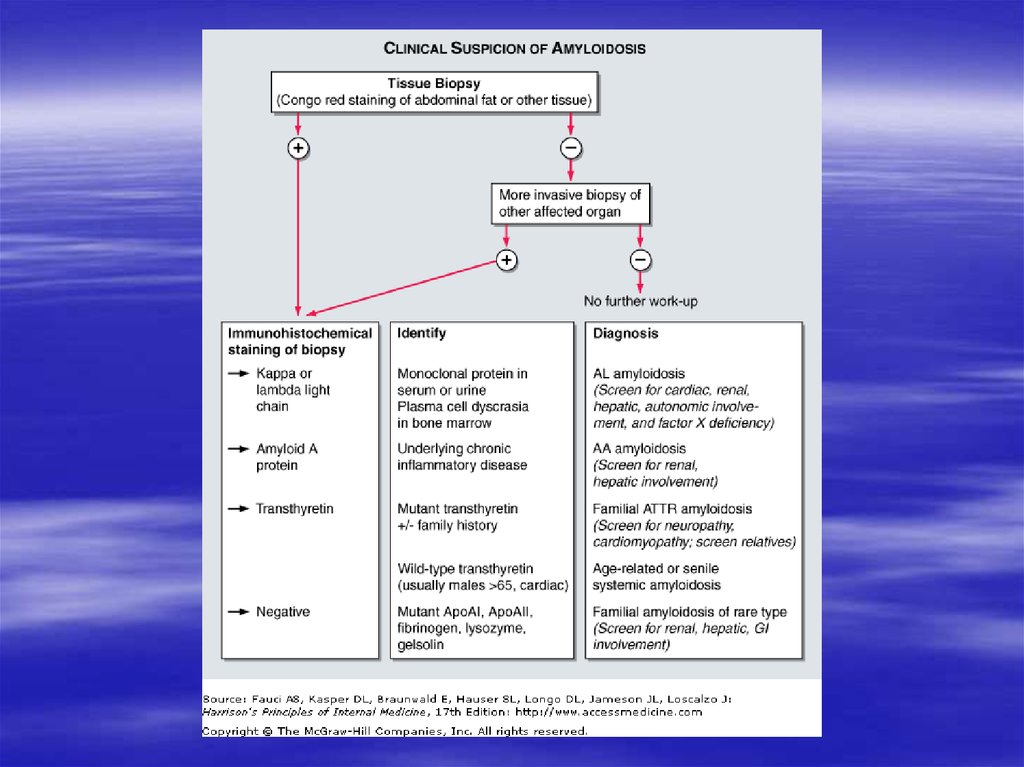
Диагноз морфологическийДля постановки диагноза исследуют
биопсии:
- Подкожно-жировой клетчатки
- Слизистой оболочки прямой кишки
- Десны
- Синовиальной оболочки суставов
- Печени
- Почки
56. Амилоидоз
57. Амилоидоз
58. Амилоидоз
Актуальность:- Журнал «Amyloid. The Journal of
Protein Folding Disoders» выходит с
1994 года
- Amyloidosis Support Network http://
www.amyloidosis.org
- Международные конгрессы и
симпозиумы по амилоидозу
59. Амилоидоз
Патогенез:- Стимул
- Растворимый белок предшественник
- Образование нерастворимых
фибрилл амилоида
60. Амилоидоз
Причины отложения амилоида:- Нарушения в структурной
организации белков
- Замены (мутации) в аминокислотной
последовательности белков
61. ПРИНЦИПЫ КЛАССИФИКАЦИИ АМИЛОИДОЗА:
• ПО ВХОДЯЩЕМУ В СОСТАВ АМИЛОИДА БЕЛКУ– на сегодняшний день выделено более 25 белков
• ПО РАСПРОСТРАНЕННОСТИ
– системный
– местный
• ПО ПРОИСХОЖДЕНИЮ
– наследственный
– приобретенный
• ПО КЛИНИЧЕСКИМ ПРОЯВЛЕНИЯМ
– с преимущественным поражением тех или иных
органов и систем (почек, печени, ЖКТ и т.д.)
62.
Название белкаАмилоида
БелокСистемный (S)
предшественник или локальный
(L)
Синдром или
пораженные ткани
и органы
AL
Immunoglobulin light
chain
S, L
AH
Immunoglobulin heavy
chain
S, L
ATTR
Transthyretin
S
L?
Primary
Myeloma-associated
Primary
Myeloma-associated
Familial
Senile systemic
Tenosynovium
S
L?
S
Hemodialysis
Joints
Secondary, reactive
Familial
Aortic
Familial
A 2M
2-microglobulin
AA
(Apo)serum AA
AApoAI
Apolipoprotein AI
AApoAII
Apolipoprotein AII
S
L
S
AGel
Gelsolin
S
Familial
ALys
Lysozyme
S
Familial
AFib
Fibrinogen -chain
S
Familial
ACys
Cystatin C
S
Familial
ABria
ABriPP
L, S?
Familial dementia, British
ADana
ADanPP
L
Familial dementia, Danish
A
A protein precursor (A PP) L
Alzheimer's disease, aging
APrP
Prion protein
L
Spongiform encephalopathies
ACal
(Pro)calcitonin
L
C-cell thyroid tumors
AIAPP
Islet amyloid polypeptide
L
AANF
Atrial natriuretic factor
L
Islets of Langerhans
Insulinomas
Cardiac atria
APro
Prolactin
L
Alns
Insulin
L
Aging pituitary
Prolactinomas
Iatrogenic
AMed
Lactadherin
L
Senile aortic, media
AKer
Kerato-epithelin
L
Cornea; familial
A(tbn)b
tbnb
L
Pindborg tumors
ALac
Lactoferrin
L
Cornea; familial
63. Амилоидоз
Наиболее клинически значимыесистемные амилоидозы:
- Первичный
- Вторичный
- Наследственный
- Диализ-ассоциированный
64. Амилоидоз
Первичныйамилоидоз
AL
амилоидоз (в состав амилоида
входят
легкие
цепи
иммуноглобулинов)
Патогенез:
- Неизвестный стимул (канцероген?)
- Моноклональная пролиферация Влимфоцитов
- Секреция
плазматическими
клетками моноклональных L-цепей
65. Амилоидоз
Первичный амилоидоз (частота встречаемости8 на 1000000 населения в год, остается стабильной в
течение 40 лет):
- Наблюдается при миеломной болезни
- При заболеваниях, сопровождающихся
избыточной продукцией моноклональных
иммуноглобулинов
- Идиопатический
66. Первичный амилоидоз
67. Амилоидоз
Вторичный (реактивный) амилоидоз - AАамилоидоз (в состав амилоида входит
SAA-белок – сывороточный белок
предшственник)
Патогенез:
- Хроническое воспаление
- Активация макрофагов
- Продукция интерлейкинов ИЛ1 и ИЛ6
- Продукция гепатоцитами белка SAA
- Образование АА амилоида
68. Амилоидоз
Вторичный (реактивный) амилоидознаблюдается при:
- Туберкулез, лепра (до 17% больных)
- Анкилозирующий спондилит,
ревматоидный артрит, болезнь Крона
(0.5-13%)
- Хронический остеомиелит, хронический
абсцесс легкого, бронхоэктазы
- Периодическая болезнь
69. Амилоидоз
Наследственный амилоидоз:- ATTR амилоидоз (транстиретин,
преальбумин – плазменный белок
связывающий и переносящий
тироксин и ретинол) обнаруживается
при семейных амилоидных
полинейропатиях и старческом
семейном амилоидозе
70. Амилоидоз
Диализ-ассоциированныйамилоидоз:
- Aβm амилоидоз (β2-микроглобулин
входит в состав главного комплекса
гистосовместимости 1 класса) и
определяется при длительном
гемодиализе
71.
72. Амилоидоз
73. Амилоидоз
74. Амилоидоз
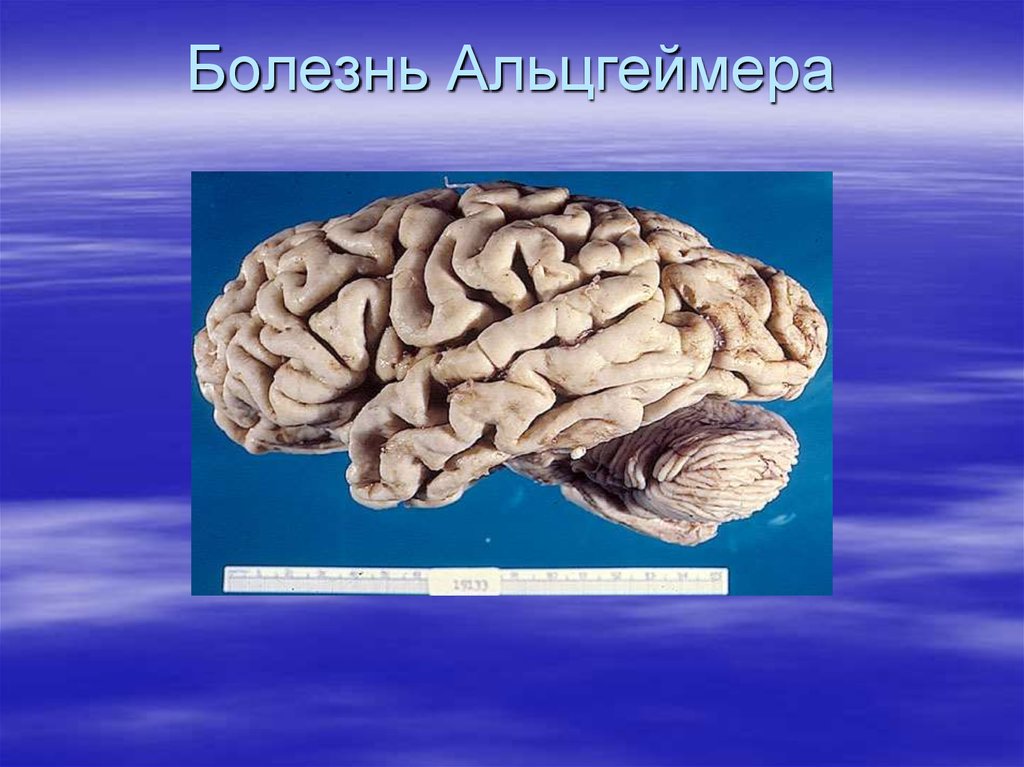
Наиболее клинически значимыелокальные амилоидозы:
- Болезнь Альцгеймера
- Сердечный
- Инсулярный
- Опухолевый
75. Амилоидоз
Этиологическиеформы
церебральный
амилоидоз
изолированный
амилоидоз
предсердий
амилоидоз островков
поджелудочной
железы
при медуллярной
карциноме
щитовидной железы
Предшественник
белка амилоида
трансмембранный
гликопротеид (АРР)
предсердный
натрийуретический
фактор (ANF)
Белок амилоида
A-β-2-протеин
AANF
амилин
IAPP
кальцитонин
ACal
76.
Болезнь Альцгеймера77.
Болезнь Альцгеймера78.
Болезнь Альцгеймера79. Сердечный Амилоидоз
80. Эндокринный Амилоидоз
81. Опухолевый Амилоидоз
82. Опухолевый Амилоидоз
83. СОСТАВ СОЕДИНИТЕЛЬНОЙ ТКАНИ:
• КЛЕТКИ–
–
–
–
фибробласты
жировые клетки (адипозоциты)
недифференцированные мезенхимальные клетки
клетки гемопоэтичесой ткани
• ВОЛОКНА ВНЕКЛЕТОЧНОГО МАТРИКСА
– коллагеновые волокна
– ретикулиновые волокна
– эластические волокна
• ОСНОВНОЕ ВЕЩЕСТВО
– протеогликаны
– гиалуроновая кислота
• ТКАНЕВАЯ ЖИДКОСТЬ
84. НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ СОЕДИНИТЕЛЬНОЙ ТКАНИ:
Синдром Элерса-Данлоса(Ehlers-Danlos syndrome (EDS))
• Хондродисплазия
(chondrodysplasias (CDs))
• Синдром Марфана (Marfan
syndrome (MFS))
85. ВРОЖДЕННЫЕ НАРУШЕНИЯ МЕТАБОЛИЗМА СОЕДИНИТЕЛЬНОЙ ТКАНИ:
• Мукополисахаридозы–
группа
заболеваний, обусловленных генетическим
дефектом
ферментного
расщепления
углеводной
части
молекулы
мукополисахаридов (гликозоаминогликанов),
при этом в тканях (преимущественно в
фибробластах и мезенхимальных клетках)
накапливаются
хондроитинсульфат
В
и/или гепаранмоносульфат
86. МУКОПОЛИСАХАРИДОЗЫ:
• Мукополисахаридоз I типа – обусловлендефицитом альфа-L-идуронидазы, которая
является лизосомальной гидролазой – главным
ферментом катаболизма мукополисахаридов.
Дефицит идуронидазы приводит к аккумуляции
гепарансульфата и дерматансульфата.
Выделяют три фенотипа болезни:
синдром Гурлера (мукополисахаридоз I H –
Hurler)
синдром Шейе (мукополисахаридоз I S –
Scheie)
синдром Гурлера-Шейе (мукополисахаридоз I
H/S – Hurler-Scheie).
87. МУКОПОЛИСАХАРИДОЗЫ:
Мукополисахаридоз II типа (синдромХантера) cвязан с дефицитом фермента,
альфа-L-идуроносульфатсульфатазы .
Синдром описан в 1917 году Hunter. Он
встречается реже, чем мукополисахаридоз I
типа, составляя приблизительно 14—15 %
от всех форм мукополисахаридозов.
88. МУКОПОЛИСАХАРИДОЗЫ:
• Мукополисахаридоз III типа (синдромСанфилиппо) обусловлен дефицитом трех
разных ферментов, но во всех случаях в
лизосомах накапливается один тип
гликозаминогликанов – гепарансульфат:
болезнь Санфилиппо А (дефицит фермента
гепарансульфатазы),
болезнь Санфилиппо В (дефицит фермента Nацетил-а-D-глюкозаминидазы),
болезнь
Санфилиппо
С
(дефицит
аглюкозаминидазы).
Синдром описан в 1963 г. S. Sanfilippo с соавт.
89. Углеводные сосудисто-стромальные дистрофии
Углеводные сосудистостромальные дистрофииСтромально-сосудистые
углеводные дистрофии могут
быть связаны с нарушением
баланса
гликопротеидов
и
гликозаминогликанов
90. Углеводные сосудисто-стромальные дистрофии
Углеводные сосудистостромальные дистрофииОслизнение тканей – стромальнососудистая дистрофия, связанная с
нарушением обмена гликопротеидов,
при которой хромотропные вещества
высвобождаются из связей с белками и
накапливаются в межуточном веществе
соединительной ткани.
91. Углеводные сосудисто-стромальные дистрофии
Углеводные сосудистостромальные дистрофииОсновные причины ослизнения
тканей:
истощение, кахексия любого
генеза;
дисфункция эндокринных желез.
92. Углеводные сосудисто-стромальные дистрофии
Углеводные сосудистостромальные дистрофииНаследственные нарушения обмена
гликозаминогликанов
(мукополисахаридов) представлены
большой группой болезней накопления
– мукополисахаридозами.