")




















































 Маркетинг
Маркетинг Право
ПравоПохожие презентации:







")


Фармацевтическая разработка ICH Q8 (R2)
1. ФАРМАЦЕВТИЧЕСКАЯ РАЗРАБОТКА ICH Q8 (R2)
А.П. МешковскийПМГМУ им. Сеченова
meshkvskijj@mail.ru
2.
Кратко об истории вопроса• В странах ЕС существовал раздел
регистрационных требований
«фармацевтические аспекты разработки»
(Development pharmaceutics)
• В США также требовалось представить
пояснения о ходе создания продукта
(лекформы)
• Требование о наличии документа в составе
досье - пояснительной записки к прописи и
технологии
3.
В отличие от этого теперь:• Соответствующие регистрационные
требования изложены в другом документе:
раздел 3.2.P.2 «Общего технического
документа» (ICH M4)
• А фармацевтическая разработка – скорее
методические указания относительно
проведения определенных исследований
4.
Американская инициативав сфере GMP - 2002 г.
• Сближение регистрации, производства по GMP и
инспектирования по GMP
• Качество через дизайн, пространство дизайна, РАТ
• Управление рисками
• Системы качества
• Совершенствование практики инспектирования по
GMP
• Международная гармонизация требований GMP
• Международное
сотрудничество
по
всем
направлениям
4
5.
Ответ ЕС и Японии: 2003-2007новые документы ICH Q8, Q9 и Q10
Компоненты Американской
инициативы
Ответ международных
организаций
Качество через дизайн
Фармацевтическая разработка
(ICH Q8)
Управление рисками качества
(ICH Q9)
Управление рисками
Системы качества
Системы качества (ICH Q10)
Пространство дизайна
Элемент Фармацевтической
разработки (ICH Q8)
РАТ (Технология анализа процессов)
Рабочие группы в ЕС
Совершенствование практики
инспектирования по GMP
Вступление США в PIC/S
Все основные компоненты
Обсуждение
в
рамках
FIP
(Нордвайк, Каир, Лондон, Пекин)
6. Гармонизированное трехстороннее руководство
• МЕЖДУНАРОДНАЯ КОНФЕРЕНЦИЯ ПОГАРМОНИЗАЦИИ ТЕХНИЧЕСКИХ
ТРЕБОВАНИЙ ДЛЯ РЕГИСТРАЦИИ
лекарственных продуктов
медицинского назначения (ICH )
• Рекомендовано Руководящим комитетом ICH
для принятия на этапе 4 Процесса ICH
10 ноября 2005
7.
Статус документа Q8• Текст, утвержденный в ноябре 2005 г., рассматривался
как первая часть будущего документа.
• В 2008 г. принято дополнение Q8(R1)
• В 2009 г. принято дополнение Q8(R2)
• Одновременно пересматривается обзор раздела
«Качество» (Quality Overall Summary - QOS) ОТД.
• Возможен также пересмотр других документов ICH
8.
Дополнения к Q8-
Приняты в 2008 (R1) и в 2009 (R2) гг.
дальнейшие разъяснения новых понятий:
“качество через дизайн”
“пространство дизайна”
“целевой профиль продукта”
“критические свойства материалов”
“критические параметры процессов”
“стратегия контроля качества”
9.
Рекомендации ВОЗ• Документ ВОЗ “Фармацевтическая разработка для
дженериков” 2008 г.
• Рассматриваются вопросы:
- стратегии разработки воспроизведенных препаратов
- перичная оценка рисков качества
- выбор компараторов для испытания
биоэквивалентности
- те же разделы, что и для инновационных препаратов
- практические подходы (если не использовать Q8)
10.
Фармацевтическая разработка• Это действия, выполнение которых необходимо
для подтверждения того, что выбранная
лекформа отвечает своему назначению
• Позволяет выявить аспекты прописи и
процесса, критические для воспроизводимости
серий (внутрипроизводственный контроль
-ВПК)
• Помогает лучше понять дизайн продукта
• Является его научной базой
11. Цели руководства
Описывает содержание раздела 3.2.P.2 заявки на регистрацию в
формате ICH M4 «Общий технический документ» (CTD).
Дает возможность представить информацию, полученную путем
применения научных подходов и управления рисками качества
(Q9) в разработке продукта и производственного процесса.
Первоначально эти данные готовятся для включения в
регистрационное досье;
Они могут обновляться с тем, чтобы отражать новые знания,
полученные в течение жизненного цикла продукта.
Данные предназначены для обеспечения всестороннего
понимания продукта и производственного процесса теми, кто
рассматривает материалы заявки на регистрацию и инспекторами.
12.
Стратегические цели• Создать продукт высокого качества и соответствующий
производственный процесс,
• Обеспечивать клиническое действие продукта, предусмотренное
его дизайном.
• Информация и знания, полученные в результате исследований в
рамках фармацевтической разработки, а также опыт производства
позволяют достичь научного понимания, достаточного для
обоснования
установленного
пространства
дизайна,
спецификаций, и методов производственного контроля.
• Информация, полученная в результате исследований по
фармацевтической разработке, может быть основанием для
управления рисками качества.
13.
Связь с GMPНациональный конгресс в Харькове:
“Настоящее и будущее фармации”, апрель 2008 г.
В числе выводов: GMP без фармацевтической
разработки – евроремонт (не более того)
13
14.
Q8: содержание• Первая часть документа основана на Руководстве ЕС
по фармацевтическим исследованиям (Development
Pharmaceutics).
• Напоминает о тезисе «Качество не может быть
вложено в продукт путем его тестирования после
завершения производственного цикла. Оно должно
быть «встроено» (built in) в него, начиная с концепции
проекта и на протяжении всех этапов производства».
• Тезис сформулирован в США в 60-х годах прошлого
века, в период введения правил GMP
15.
Качество должно быть «встроено»?• Первоначально понималось, что качество должно быть
«встроено» в процессе производства (GMP)
• Теперь считается, что встраивать нужно начиная с
дизайна продукта (концепции, проекта, задумки) и в
ходе фармацевтической (и аналитической) разработки
• В дальнейшем качество фиксируется через процедуру
регистрации продуктов
• И поддерживается на всех этапах оборота путем
соблюдения правил GMP, GDP, GPP, GSP и др.
16.
Новые понятия• Целевой профиль качества: желаемые
(фармакокинетические) свойства продукта
(ориентир - фармакопея или потребитель?)
• Качество через дизайн
• Пространство дизайна (PAR & NOR)
• Гибкие регуляторные подходы
• Уточнение понятий: «критические
характеристики и параметры»
16
17. О качестве
• Важно признать, что качество не может бытьдостигнуто путем испытания продуктов, иначе говоря,
качество должно cоздаваться, начиная с дизайна
(проекта, задумки) продукта
• Изменения в прописи и в производственных
процессах в ходе разработки и управления жизненным
циклом должны рассматриваться как возможность
получать дополнительные знания, поддерживающие
установление пространства дизайна.
18.
Гибкие регуляторные подходы• регуляторные решения на основе анализа риска
(рассмотрение заявки и инспектирование);
• усовершенствование производственного процесса в
пределах
одобренного
пространства
дизайна,
описанного в досье, без дальнейшего рассмотрения;
• сокращение заявок на изменение регистрационных
условий;
• проверка качества в реальном времени, ведущая к
сокращению испытаний при выпуске конечного
продукта.
19. Обосновать выбор лекформы, прописи и технолгии
Этот раздел должен содержать данные, подтверждающие, что
выбранная
дозированная форма и предложенная пропись
подходят для предназначенного использования.
Могут прилагаться
таблицы и графики,
облегчающие
рассмотрение заявки.
Должны быть определенны те аспекты лекарственных
субстанций, вспомогательных веществ, упаковочно-укупорочной
системы и производственных процессов, которые являются
критическими для качества продукта.
Критические характеристики прописи и параметры процесса
выявляются через оценку степени влияния их изменений на
качество лекарственного продукта.
20. Кроме того
• Заявительможет проводить исследования по
фармацевтической разработке, ведущие к увеличению
знаний о действии
продукта в более широком
диапазоне свойств материалов, вариантов выбора
процессов и их параметров.
• Включение этой дополнительной информации дает
возможность демонстрировать более высокую степень
понимания свойств материалов, характеристик
производственных процессов и их контроля.
• В этих ситуациях имеется возможность для введения
более гибких регуляторных подходов.
21. Лекарственная субстанция
Физико-химические и биологические свойства лекарственной
субстанции, могущие повлиять на действие продукта и его
технологичность, или
специально
заложенные в проект
(например,
свойства
твердого
тела),
должны
быть
идентифицированы и обсуждены.
Примеры: физико-химические и биологические свойства растворимость, содержание влаги, размер частиц,
свойства
кристаллов, биологическая активность, и способность проникать
сквозь биологические мембраны.
Эти свойства могут быть взаимосвязаны, в связи с чем может
потребоваться их совместное рассмотрение.
22. Вспомогательные вещества
Необходимо оценить совместимость лекарственной субстанции
с вспомогательными веществами в прописи,
Обсуждение функций каждого вспомогательного вещества, их
концентрации, характеристик, могущих влиять на действие
лекарственного
продукта
(например,
стабильность,
биодоступность) или технологичность,
Должна
также
быть
продемонстрирована
способность
вспомогательных веществ (например, антиоксидантов, веществ
увеличивающих всасывание, способствующих распадаемости,
контролирующих
высвобождение)
обеспечить
их
предназначенные функциональные возможности, и действовать в
течение всего предполагаемого срока годности продукта.
23. Лекарственный продукт
Необходимо представить резюме (обзор) с описанием разработки
прописи, включая идентификацию тех характеристик, которые являются
критическими в отношении качества лекарственного продукта, учитывая
(его) предназначенное использование и способ введения.
Акцент на ходе разработки дизайна прописи, от первоначального
проекта до окончательного варианта.
Выбор компонентов лекарственного продукта (например, свойства
лекарственной субстанции, вспомогательных веществ,
упаковочноукупорочной
системы
и
любых
дозирующих
устройств),
производственный процесс,
Обоснования интервалов в закладке вспомогательных веществ, вошедших
в регламент производства (3.2.P.3.2). Это обоснование часто может
базироваться на опыте, полученном в процессе разработки или
производства.
24. Избыток
• Как правило, не рекомендуется использование избыткалекарственной субстанции для компенсации разрушения в процессе
производства или хранения продукта, либо для продления срока
годности.
• Любые избытки в производстве лекарственного продукта,
появляются ли они в готовой дозированной форме или нет, должны
быть обоснованы с точки зрения безопасности и эффективности
продукта.
• Должна быть предоставлена информация о 1) количестве избытка, 2)
причинах использования (например, чтобы компенсировать
ожидаемые и документированные производственные потери), и 3)
обоснование величины избытка.
• Избыток должен быть включен в указанное в описании технологии
количество лекарственной субстанции
25. Производственный процесс
Объяснение выбора производственного процесса и видов
контроля.
Рассмотрение критических характеристик прописи, вместе с
возможными вариантами производственного процесса, чтобы
выбрать производственный процесс и подтвердить пригодность
компонентов.
Пригодность оборудования применительно к запланированным
продуктам.
Исследования по разработке производственного процесса
должны обеспечить основание для усовершенствования процесса,
его валидации, непрерывной верификации процесса (где это
применимо) и любых требований в отношении контроля
процесса.
26. Производственный процесс -2
• В соответствующих случаях такие исследования должны бытьнаправлены на микробиологические, а также на физические и
химические характеристики. Знания, полученные в результате
исследований по разработке производственного процесса, могут
использоваться, при необходимости, для обоснования
спецификации лекарственного продукта
• Программа разработки (или усовершенствования)
производственного процесса должна выделять любые
критические параметры процесса, подлежащие мониторингу или
контролю (например, время окончания грануляции) с тем, чтобы
гарантировать желаемое качество продукта.
27. Упаковочно-укупорочная система
Выбор и основания для выбора
упаковочно-укупорочной
системы закрытия.
Внимание намечаемому использованию лекарственного продукта
и пригодности соответствующей упаковки для хранения и
транспортировки, включая, при необходимости, хранение и
транспортную упаковку для нерасфасованного лекарственного
продукта (балк-продукта).
Выбор материалов для первичной упаковки должен быть
обоснован. Обсуждение должно описать исследования,
выполненные, чтобы продемонстрировать целостность упаковки
(контейнера) и её укупорки. Должно быть рассмотрено возможное
взаимодействие между продуктом и упаковкой или этикеткой.
28. Выбор упаковочных материалов
• Выбор первичных упаковочных материалов долженбыть обоснован, например, выбор материалов,
защищающих от влажности и света, совместимость
упаковки с дозированной формой (включая сорбцию,
извлечение); также необходимо принять во внимание
безопасность материалов упаковки.
• В случаях, когда это имеет значение, необходимо
обосновать выбор вторичной упаковки.
29. Микробиологические свойства
В соответствующих случаях в этом разделе должны быть
рассмотрены микробиологические свойства лекарственного
продукта. Необходимо обосновать, например:
- для нестерильных форм выполнение или невыполнение
испытаний на микробиологическую чистоту
- выбор и эффективность
систем консервантов в продуктах
содержащих антимикробный агент или антибактериальная
эффективность для продуктов, обладающих естественными
антибактериальными свойствами;
• Для стерильных продуктов целостность упаковки, в связи с
предотвращением микробного загрязнения.
30. Совместимость окончательной лекформы
Необходимо обратить внимание на совместимость активного
вещества с растворами для приготовления лекарственной формы
перед применением (например, осаждение, стабильность), чтобы
предоставить соответствующую подтверждающую информацию
для маркировка.
Эта информация должна отображать рекомендованный срок
хранения приготовленной перед использованием лекформы, при
рекомендованной
температуре
хранения
и
возможных
максимальных концентрациях.
Может
возникнуть необходимость рассмотреть варианты с
добавкой (например, «подкалыванием» в инъекционные растворы
большого объема) или разведением перед применением.
31.
Разработка методов контроля• Отправная точка: желаемые
(фармакокинетические) свойства продукта
• Лабораторный и полупроизводственный
масштаб
• Решение вопросов:
(Состав)
Параметры процесса и ВПК
Спецификация готового продукта
Аналитические методы
32.
ФАРМАЦЕВТИЧЕСКАЯРАЗРАБОТКА
Практические аспекты:
опыт производителей
33. Обсуждение руководства
• Практические аспекты• Опыт производителей
34.
Фармацевтическая разработка:три вида подхода
Инновационные препараты
Дженерики с улучшенными свойствами
Дженерики – точные копии
инновационных препаратов
34
35.
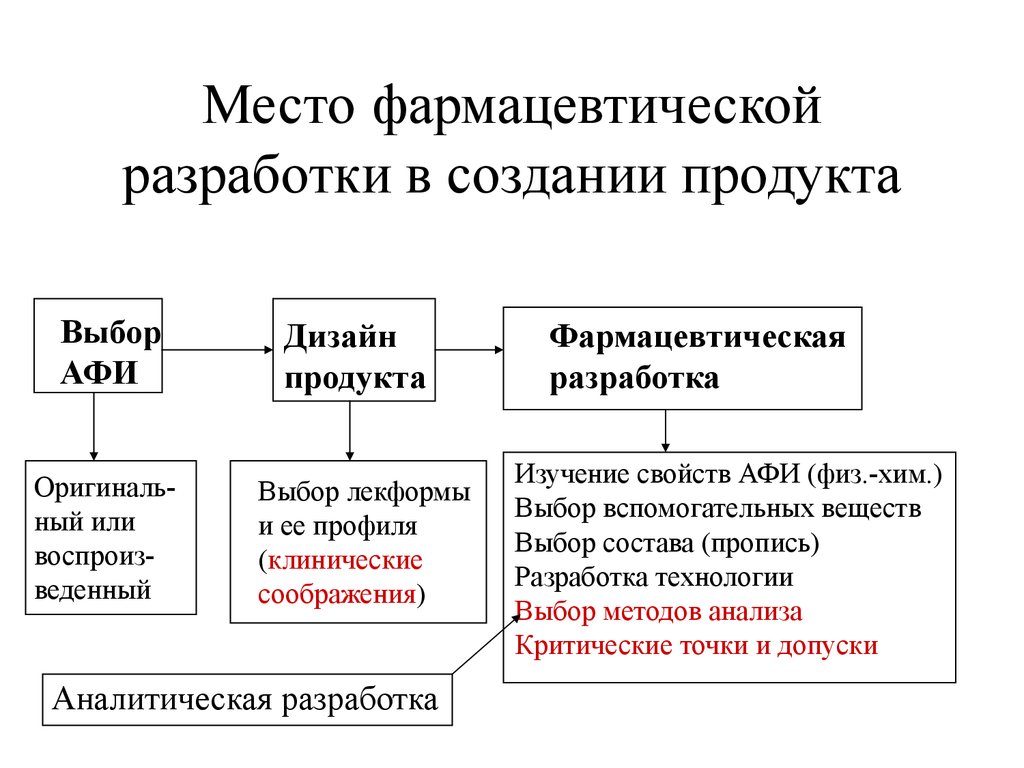
Место фармацевтическойразработки в создании продукта
Выбор
АФИ
Оригинальный или
воспроизведенный
Дизайн
продукта
Выбор лекформы
и ее профиля
(клинические
соображения)
Аналитическая разработка
Фармацевтическая
разработка
Изучение свойств АФИ (физ.-хим.)
Выбор вспомогательных веществ
Выбор состава (пропись)
Разработка технологии
Выбор методов анализа
Критические точки и допуски
36.
Информация, получаемая в ходефармацевтической разработки,
• Условно делится на три части:
Раздел «Характеризация» (изучение
свойств)
Раздел «Оптимизация»
Раздел «Верификация»
37.
Раздел «Характеризация»(изучение свойств)
Предварительное изучение субстанции
(preformulation - физико-химические свойства
АФИ )
Совместимость (compatibility – выбор
вспомогательных веществ)
Пропись (состав)
Процесс (технология и контроль – ВПК)
Аналитические методы
38.
Раздел «Оптимизация»Устойчивость
Критические параметры
Реалистичные допуски
39.
Раздел «Верификация»Валидация (включая квалификацию)
Масштабирование
Перенос технологии (на другую
площадку)
Текущее усовершенствование
технологии (оптимизация)
40.
Варианты оптимизациипрописи и технологии
• Целенаправленные исследования по отработке
технологии, включая уточнение прописи
(ограниченное время, до валидации)
• Непрерывное совершенствование на основе
накопляемого опыта, в т.ч. за счет выявления и
устранения причин инцидентов (после
валидации, постоянно, через систему
управления изменениями)
41. Валидация и фармацевтическая разработка
Фармацевтическая разработкапозволяет получить обоснованные
исходные данные для валидации
технологических процессов и
аналитических методик
42.
Пример изучения свойств активнойсубстанции Х (preformulation)
• Форма и размер кристаллов: кубическая; D10 2,6 μm; D50 -15 μm; D90 - 48 μm
• Удельная поверхность – 1,5 м2/г
• Сыпучесть: слабая
• Прессуемость: плохая
• Угол контакта: 77º
• Скорость растворения: 1,5 μг/см2/мин.
• Полиморфизм: нет
• Термическая взрывоопасность: нет
43.
Изучение свойств активнойсубстанции (раствор)
• Профиль растворимости: макс. 10 μг/мл при рН
1-4 (добавление буферного р-ра)
• Зависимость стабильности от рН: деградация
выше рН 5,5
• Влияние растворителей: добавка ПЭГ 400
(20/80) при рН 4 – 150 μг/мл
• Стерилизация: термическая стерилизация
невозможна – только γ-стерилизация
44.
От свойств АФИ к выборувспомогательных веществ
• Для твердых пероральных форм: если
растворимость АФИ менее 1% требуются комплексообразующие или
поверхностно активные вещества
(циклодекстрины)
• При недостаточной стабильности –
использование антиоксидантов
44
45.
Вспомогательные веществадобавляются в целях:
• Увеличения стабильности лекформы
(антиоксиданты)
• Оптимизации биофармацевтических
свойств (дезинтегранты, оболочка)
• Улучшения технологичности
(наполнители, скользящие в-ва)
• Улучшение принятия пациентами
(вкусовые добавки, красители)
45
46.
Стресс-испытания субстанций ипрописи: примеры
• Температура: в открытом виде увлажненный
материал – 4 недели при 80°C, анализ
еженедельно
• Влажность: в открытом виде 4 недели при 40°C
/100% о.в., анализ через 2 недели
• Окисление: 24 часа, через насыщенный водный
раствор (суспензию) материала пропускается
кислород при перемешивании, анализ каждые 8
часов.
46
47.
Стресс-испытания субстанций ипрописи: примеры
• Температура: в открытом виде увлажненный
материал – 4 недели при 80°C, анализ
еженедельно
• Влажность: в открытом виде 4 недели при 40°C
/100% о.в., анализ через 2 недели
• Окисление: 24 часа, через насыщенный водный
раствор (суспензию) материала пропускается
кислород при перемешивании, анализ каждые 8
часов.
47
48.
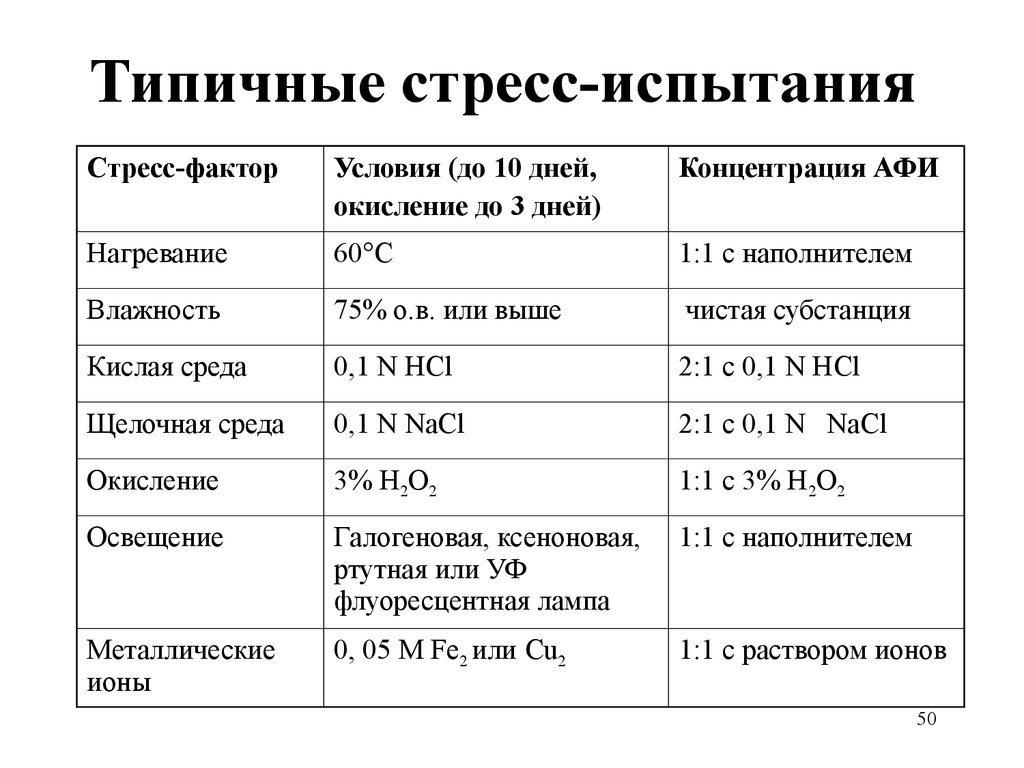
Типичные стресс-испытанияСтресс-фактор
Условия (до 10 дней,
окисление до 3 дней)
Концентрация АФИ
Нагревание
60°С
1:1 с наполнителем
Влажность
75% о.в. или выше
чистая субстанция
Кислая среда
0,1 N HCl
2:1 с 0,1 N HCl
Щелочная среда
0,1 N NaCl
2:1 с 0,1 N NaCl
Окисление
3% Н2О2
1:1 с 3% Н2О2
Освещение
Галогеновая, ксеноновая,
ртутная или УФ
флуоресцентная лампа
1:1 с наполнителем
Металлические
ионы
0, 05 М Fe2 или Cu2
1:1 с раствором ионов
48
49.
Типичные стресс-испытанияСтресс-фактор
Условия (до 10 дней,
окисление до 3 дней)
Концентрация АФИ
Нагревание
60°С
1:1 с наполнителем
Влажность
75% о.в. или выше
чистая субстанция
Кислая среда
0,1 N HCl
2:1 с 0,1 N HCl
Щелочная среда
0,1 N NaCl
2:1 с 0,1 N NaCl
Окисление
3% Н2О2
1:1 с 3% Н2О2
Освещение
Галогеновая, ксеноновая,
ртутная или УФ
флуоресцентная лампа
1:1 с наполнителем
Металлические
ионы
0, 05 М Fe2 или Cu2
1:1 с раствором ионов
49
50.
Типичные стресс-испытанияСтресс-фактор
Условия (до 10 дней,
окисление до 3 дней)
Концентрация АФИ
Нагревание
60°С
1:1 с наполнителем
Влажность
75% о.в. или выше
чистая субстанция
Кислая среда
0,1 N HCl
2:1 с 0,1 N HCl
Щелочная среда
0,1 N NaCl
2:1 с 0,1 N NaCl
Окисление
3% Н2О2
1:1 с 3% Н2О2
Освещение
Галогеновая, ксеноновая,
ртутная или УФ
флуоресцентная лампа
1:1 с наполнителем
Металлические
ионы
0, 05 М Fe2 или Cu2
1:1 с раствором ионов
50
51.
Типичные стресс-испытанияСтресс-фактор
Условия (до 10 дней,
окисление до 3 дней)
Концентрация АФИ
Нагревание
60°С
1:1 с наполнителем
Влажность
75% о.в. или выше
чистая субстанция
Кислая среда
0,1 N HCl
2:1 с 0,1 N HCl
Щелочная среда
0,1 N NaCl
2:1 с 0,1 N NaCl
Окисление
3% Н2О2
1:1 с 3% Н2О2
Освещение
Галогеновая, ксеноновая,
ртутная или УФ
флуоресцентная лампа
1:1 с наполнителем
Металлические
ионы
0, 05 М Fe2 или Cu2
1:1 с раствором ионов
51
52.
Лабораторный уровень• Предварительные исследования
(preformulation)
• Масштаб – 1х
• Изучение физико-химических характеристик
активной субстанции и вспомогательных
веществ (раздельно)
• Выбор и отработка аналитических методик
• Определение критических характеристик
• Выбор стратегии контроля
52
53.
Крупно-лабораторный уровеньВыбор прописи (formulation)
Масштаб – 10х
Совместимость (изучение смесей)
Стресс-испытания выбранной прописи
Стабильность – скрининг (открытое состояние,
4 недели при 40°С/75% о.в.)
• Выбор и оптимизация процесса (технологии)
• Наработка серий для испытания стабильности
53
54.
Уровень опытнонаработочного цеха• Масштаб – от 100х до 1/10 серийного производства; не
меньше 100 тыс. таблеток (капсул)
• Оборудование – того же типа, что в серийном
производстве
• Отработка технологии (оптимизация)
• Наработка «биосерий» (для клиники, испытания
биоэквивалентности)
• Наработка серий для испытания стабильности (до 12
мес. при 25ºC/60% о.в. и до 6-9 мес. при 30ºC/60% о.в.
по 2-3 сериям)
• Предварительная валидация процесса (десятки серий)
54
55.
Серийное производство• Первые три (две) серии – валидационные
(подтверждающие – conformance batches)
• Отчет о валидации, утверждение отчета
• Утверждение/отклонение прописи и технологии
• Для регистрационного досье (если не сделано ранее):
результаты анализа серий по предлагаемой
спецификации (5 серий или меньше) с указанием
объема, даты и цели производства
• Оптимизация процесса (постоянно)
55
56.
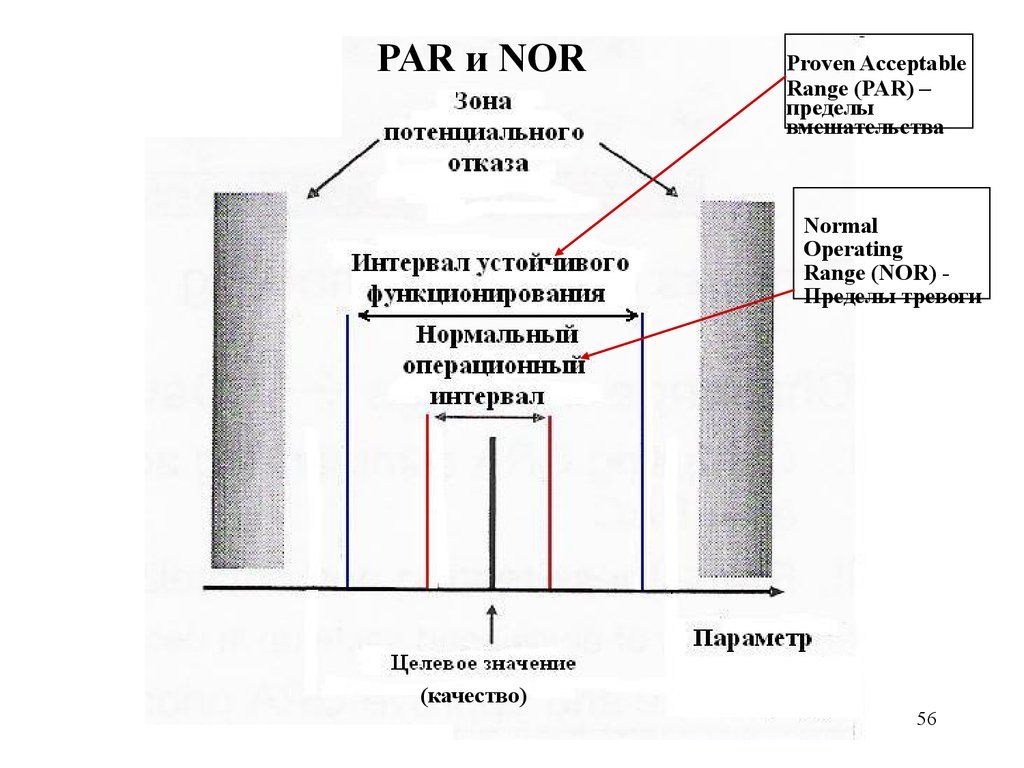
PAR и NORProven Acceptable
Range (PAR) –
пределы
вмешательства
Normal
Operating
Range (NOR) Пределы тревоги
(качество)
56