




















































 Медицина
МедицинаПохожие презентации:

")






")
")
Первичные иммунодефициты
1. Первичные иммунодефициты
Ещенко Д.О. 405 пед2. Определение понятия
Это врожденные, генетически обусловленныеиммунодефициты.
Проявления
Недостаточность
гуморального
иммунитета
Недостаточность
клеточного
иммунитета
Дефект системы
комплемента
Несостоятельность
клеток
фагоцитирующей
системы
3.
Первичные иммунодефицитыпроявляются в виде различных
синдромов
Синдром дефицита
антител , Тлимфоцитов
Синдромы с
дефицитом
компонентов
комплемента
Дефектов фагоцитов
Дефектов молекул
адгезии и др.
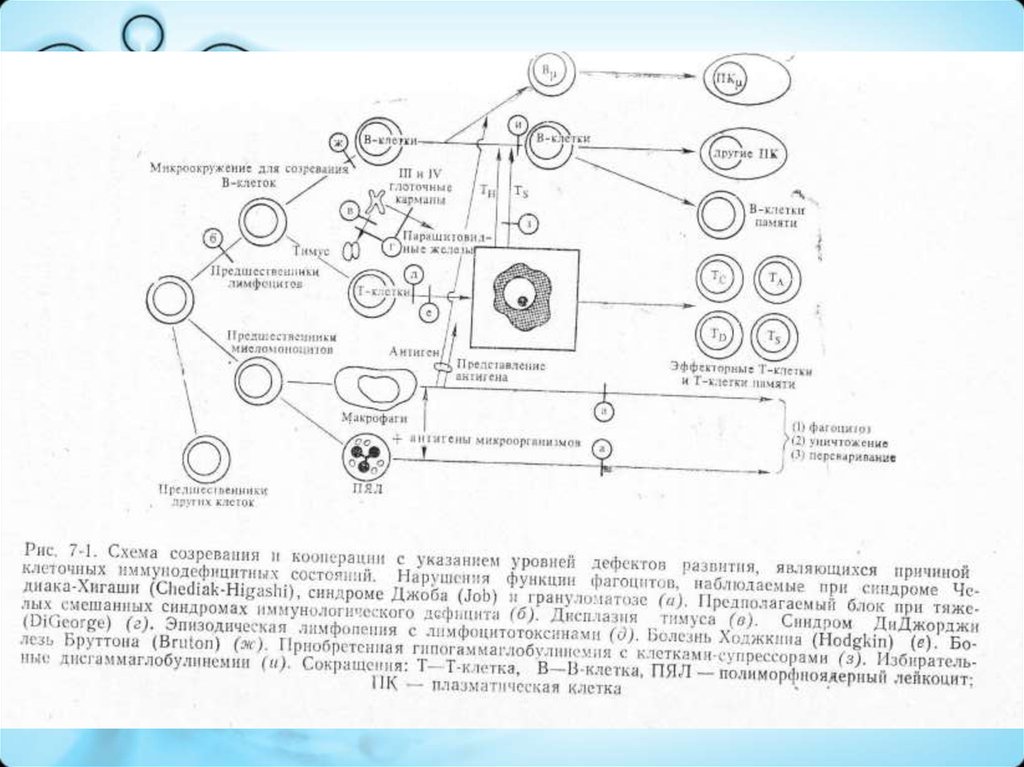
4. Классификация ПИДС
Дефицит гуморального иммунитета:
– Сцепленная с Х-хромосомой агаммаглобулинемия (синдром Брутона)
– Общий вариабельный иммунодефицит
– Транзиторная гипогаммагобулинемия у детей (медленный
иммунологический старт)
– Селективный дефецит иммуноглобулинов (гипо IgA)
Дефицит клеточного звена иммунитета:
– Синдром Ди Джоржди (гипо/аплазия тимуса)
– Хронический слизисто-кожный кандидоз
Комбинированные Т- и В-ИД:
– Тяжелый комбинированный ИД
• Х-сцепленный
• Аутосомно-рецессивный
– Атаксия-телеангиэктазия (синдром Луи-Бар)
– ИД с тромбоцитопенией и экземой ( синдром Вискотта-Олдрича)
Дефицит системы фагоцитов:
– Синдром Чедиака-Хигаси
– Синдром гипериммуноглобулинемии Е (синдром Джоба)
Дефицит системы комплемента:
– Врожденный ангионевротический отек
5.
6.
7. Дефицит В-клеточного иммунитета
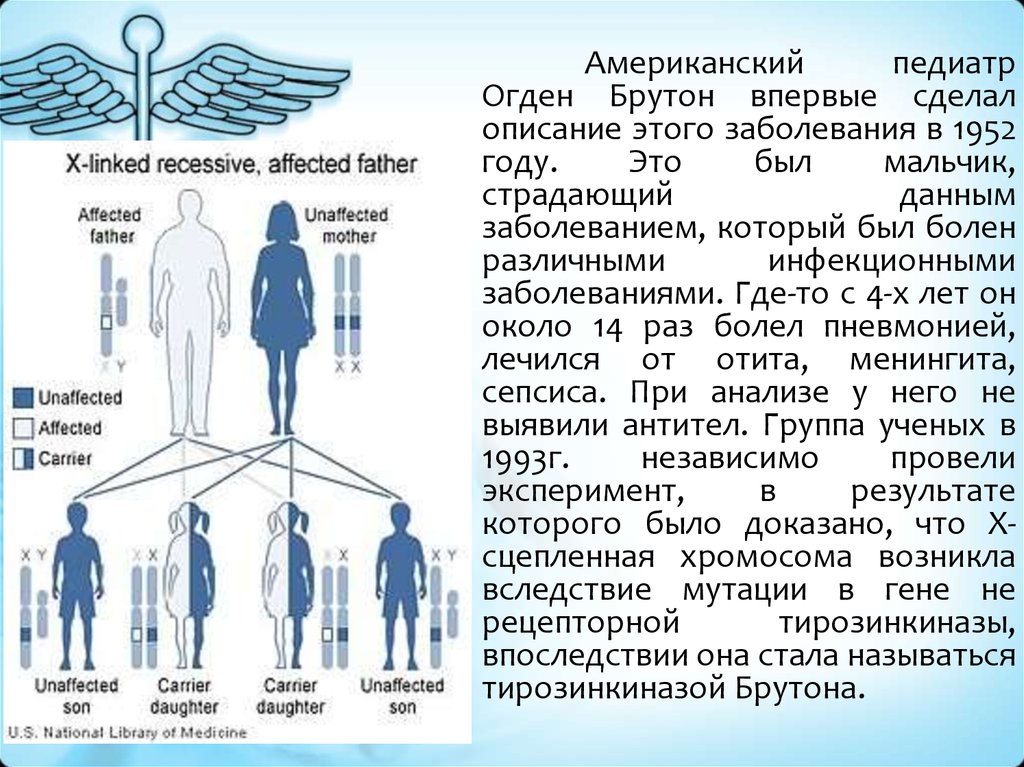
8. Болезнь Брутона
- болезнь, обусловленная дефектом генахромосомы X, кодирующего синтез Bклеточной специфической тирозинкиназы
Брутона (Btk)
9.
Американскийпедиатр
Огден Брутон впервые сделал
описание этого заболевания в 1952
году.
Это
был
мальчик,
страдающий
данным
заболеванием, который был болен
различными
инфекционными
заболеваниями. Где-то с 4-х лет он
около 14 раз болел пневмонией,
лечился от отита, менингита,
сепсиса. При анализе у него не
выявили антител. Группа ученых в
1993г.
независимо
провели
эксперимент,
в
результате
которого было доказано, что Хсцепленная хромосома возникла
вследствие мутации в гене не
рецепторной
тирозинкиназы,
впоследствии она стала называться
тирозинкиназой Брутона.
10. Клиника
• До 9-месячного возраста младенцы защищеиммуноглобулинами матери и не болеют.
• Болеют только мальчики с тяжелыми
пиогенными инфекциями, вызванные
гноеродной флорой.
• После 9-12 месяцев жизни развиваются
менингоэнцефалит, септический артрит,
инфекции дыхательных путей, отит, диарея.
• Уменьшаются лимфоузлы и миндалины.
11. Лабораторная диагностика
Двукратное снижение концентрации вкрови всех типов иммуноглобулинов (IgG —
менее 2 г/л, на 1 году — менее 1 г/л, IgA и IgM у
детей школьного возраста менее 0,2 г/л), низкое
количество В-лимфоцитов(< 5/1000 лимфоцитов).
12. Лечение
Внутривенные иммуноглобулины (ВИГ)вводят в суточной дозе 400 мг/кг в/в капельно по 1
мл/кг/час недоношенным и 4-5 мл/кг/час
доношенным детям.
Недоношенным детям с массой <1500 г и IgG <3 г/л
ВИГ вводят для профилактики инфекций.
ВИГ вводят до достижения концентрации IgG в
крови не ниже 4-6 г/л.
Ежедневно (при гнойно-воспалительных
заболеваниях) или через день 3-5 инъекций до 1-2,5
г/кг, затем – 1 раз в 7 дней или1 раз в месяц.
13. Селективный дефицит IgA
— одна из обычных форм иммунодефицита,которая проявляется недостаточностью IgA в
секретах.
- некоторых пациентов присутствуют мутации
гена TACI (трансмембранный активатор и
партнёр кальциевого модулятора и лиганда
циклофилина). Селективный дефицит IgA также
обычно связан с определенными HLAгаплотипами; часто присутствуют редкие
аллели или делеции генов главного комплекса
гистосовместимости (МНС) региона класса III
14. Клиника
• Рецидивирующие респираторные ижелудочно-кишечные инфекции,
атопические, аутоиммунные и
онкологические заболевания.
• В некоторых случаях возможно
отсутствие клинических проявлений.
15. Лабораторные исследования
• Снижение урованя IgA < 0,7 мг/л принормальных показателях IgG и IgM у
пациентов старше 4 лет в отсутствие
других причин гипогаммаглобулинемии.
• Анафилактические реакции на трасфузию
• Наличие в семейном анамнезе
вариабельного неклассифицируемого
иммунодефицита
16. Лечение
• Агрессивная антимикробная терапия ипрофилактика
• Агрессивная терапия атопических
заболеваний
• Эффект от заместительной терапии в/в
иммуноглобулином наблюдается редко.
• Исключение контактов с продуктами
крови, содержащими IgA
17. Транзиторная гипогаммаглобулинемия у детей
- Исчезновение IgG, переданного плодучерез плаценту матери, вскоре после
рождения, обычно с 3 до 6 мес
- Характер наследования неизвестен
18. Клиника
• Рецидивирующие бактериальныесинопульмонарные инфекции
• Рецидивирующие болезни ЖКТ
• Кандидоз
• Частые ОРВИ в раннем детском возрасте
19. Лабораторные исследования
• Уровень иммуноглобулинов ниженормальных значений при сохранении
продукции специфических АТ и клеточного
иммунного ответа.
20. Лечение:
• Превентивная антибиотикотерапия• Иногда – заместительная терапия гаммаглобулином
21. Общий вариабельный ИД
- Один из наиболее распространенных ПИД,проявляющийся снижением уровня одних
иммуноглобулинов при нормальном содержании
других иммуноглобулинов (дисгаммаглобулинемия)
22. Клиника
• Развиваются рецидивирующиеинфекции респираторного и
желудочно-кишечного трактов у
мальчиков и девочек старше 2 лет.
• Характерна высокая частота
развития лимфоретикулярных и
желудочно-кишечных злокачественных
опухолей
• У 1/3 больных выявляется
спленомегалия или диффузная
лимфаденопатия.
• Большая предрасположенность к
аутоиммунным заболеваниям (20%
аутоиммунная
тромбоцитопеническая пурпура и
аутоиммунная гемолитическая
анемия)
23. Лабораторная диагностика
• Гипогаммаглобулинемия.• ↓IgG, а также в большинстве случаев IgA и
IgM ниже нормы при исключении других
причин агаммаглобулинемии.
• Нарушение продукции специфических АТ.
Отсутствие изогемагглютинатов или
слабый ответ на вакцинацию.
• Нарушение Т-клеточного ответа.
• Содержание В-лимфоцитов в
периферической крови может быть в
норме или снижено.
24. Лечение
• Заместительная терапия гаммаглобулином и антимикробнаяпрофилактика
25. Дефицит Т-клеточного звена иммунитета
• Составляет 5—10% общего количествапервичных иммунодефицитов.
• Селективные дефекты клеточного звена
иммунитета часто свзязаны с
рецидивирующими инфекциями,
вызывающими микроорганизмами
26. Синдром Ди Джорджи
- В основе заболевания лежитгипоплазия или аплазия тимуса и
паращитовидных желез
- Является результатом делеции
локуса 22q11 в 22-й хромосоме,
мутации генов локуса 10р13 в 10-й
хромосоме и мутаций 10р13 в 10-й
хромосоме и мутаций
неизвестных генов
- Поражаются одинаково
мальчики и девочки
27. Клиника
• У младенцев наблюдается низкоерасположение ушных раковин, срединная
расщелина лица, слабое недоразвитие нижней
челюсти, гипертелоризм, короткий губной
желобок, задержка развития и врожденные
заболевания сердца
• У детей имеет место гипо- или аплазия тимуса
и паращитовидных желез, ведущие к Тклеточной недостаточности и
гипопаратиреозу.
• Рецидивирующие инфекции начинаются сразу
после рождения, но степень иммунодефицита
значительно варьирует, функция Тлимфоцитов может спонтанно улучшиться.
28.
• Неизмененная вилочковая железа уноворожденных в рентгеновском
изображении на прямых рентгенограммах.
Контуры железы показаны пунктиром
29. Диагностика
• снижение содержания в периферической кровиТ-клеток (CD3+,CD4+,CD8+)
• Изменения в гуморальном звене иммунитета
не всегда развиваются.
• Чаще определяется нормальное или
повышенное содержание В-лимфоцитов в
крови. Концентрация иммуноглобулинов в
сыворотке в пределах нормы.
• Это свидетельствует в пользу того, что
такие больные могут быть иммунизированы
против ряда бактериальных и вирусных
инфекций (дифтерии, столбняка,
полиомиелита, кори, краснухи и др.).
30. Лечение
• При полной аплазии тимуса трансплантацияжелезы.
• При гипоплазии железы – назначают препараты
тимуса.
• Лечение пороков сердца ведется по
стандартам, принятым в кардиологии, а
недостаточности паращитовидных желез – по
эндокринологическим стандартам.
31. Хронический кожно-слизистый кандидоз
• Являются персистирующими илирецидивирующими инфекциями,
обусловленными врожденными дефектами Тклеток
• Наследование аутосомно-доминантное или
рецессивное
• Кандидозы рецидивируют или персистируют,
обычно начинаясь в младенческом возрасте, а
иногда в раннем детстве.
• У некоторых пациентов также наблюдается
дефицит гуморального иммунитета,
характеризующийся аномальным ответом
антител на полисахаридные антигены,
несмотря на нормальный уровень Ig.
32. Клиника
• Рецессивная форма:– Аутоиммунные проявления
– Эндокринные проявления (гипотиреоз,
недостаточность коры надпочечников,
гипогонадизм, диабет)
– Мелкоочаговая алопеция
– Пернициозная анемия и гепатит
• Общая картина:
– Молочница, инфекции кожи, ЖКТ, утолщение
ногтей, отеки и покраснения околоногтевой
ткани.
– Повреждения кожи покрываются коркой,
нагнаиваются, гиперкератоз.
33. Диагностика
• нормальное содержании в периферическойкрови Т-лимфоцитов (CD3+), нормальная
бласттрансформирующей способность на
ФГА и пролиферативной активности в СКЛ.
• РБТЛ на Candida- антиген и кожная реакция
ГЗТ на этот антиген отрицательная.
• Гуморальный иммунный ответ у таких
больных не нарушен.
34. Лечение
• Назначается противомикозная терапия.• Препараты, стимулирующие активность
клеточного иммунитета.
35. Комбинированные иммунодефициты
• Общим для комбинированныхиммунодефицитов является раннее их
проявление, инфицирование множеством
микроорганизмов, выраженная клиническая
симптоматика, тяжелое течение
инфекционного процесса.
• Дети с ПКИД страдают от тяжело
протекающих инфекций легких, кожи,
слизистой рта и горла, часто наблюдается
кандидоз глотки, пищевода, у больных
развивается хронический понос, сепсис.
36.
• Часто ПКИД сочетается с аномалиямискелета; для них характерна
лимфоцитопения, снижение
количества Т-лимфоцитов и их
функциональной активности,
гипоплазия тимуса. Для некоторых
форм характерна
гипогаммаглобулинемия.
37. Для ТКИД характерно:
• лимфоцитопения• гипоплазия тимуса
• снижение количественного содержания Т- и Влимфоцитов в крови и снижение их
функциональной активности.
• Содержание гранулоцитов и эритроцитов в
периферической крови находится в пределах
нормы.
• отсутствуют кожные реакции замедленного
типа и подавлена продукция антител на
специфические антигены.
• содержание IgА, М, G в крови снижено.
38. Лечение
• Единственным эффективным способомлечения таких болезней является
трансплантация гемопоэтической ткани
(костного мозга, клеток эмбриональной
печени, стволовых клеток периферической
крови) и комплекса – тимус – грудина.
39. Синдром Луи-Барр
• Заболевание связано сдефектностью киназ,
участвующих в регуляции
клеточного цикла.
Заболевание наследуется
аутосомно-рецессивно.
Дефект локализован в
хромосоме 11q22, который
приводит к развитию
врожденной
нейроэтодермальной
дисплазии.
• У пациентов отмечается
недостаток IgA и IgE и
прогрессирующие
нарушения Т-лимфоцитов.
40. Клиника
• Проявляется в возрасте от 5 мес до 3 лет• Признаки мозжечковой атаксии, интенционный
тремор, качание туловища и головы.
• Дизартрия: скандированная речь
• Мышечная гипотония, снижение/исчезновение
сухожильных рефлексов
• Косоглазие
• Телеангиоэктазии начинают появляться с3 до 6
лет на конъюнктиве глазного яблока в виде
паучков, затем в области лица и шеи, локтевых и
коленных сгибов, на поверхности ладоней и стоп
• Может наблюдаться гипетрихоз, ранняя седина
волос, кожные элементы, напоминающие акне или
проявления псориаза
• Хронические заболевания ЛОР-органов
• Злокачественные опухолевые процессы: лейкемия
и лимфома
• Имеют место эндокринные аномалии, такие как
дисгенезия гонад, тестикулярная атрофия,
сахарный диабет.
41. Диагностика
• уменьшение содержания Т-лимфоцитов,сниженный уровень IgА, IgЕ, иногда IgG2,
сниженный ответ в РБТ лимфоцитов на
митогены (ФГА) и бактериальные
антигены.
• Повышение ɑ-фетопротеина
• Анализ крови: снижение уровня лейкоцитов
42. Лечение
• Своевременная антибиотикотерапия при развитииинфекционных заболеваний.
• Ограждение ребенка от контактов с людьми,
болеющими инфекционными заболеваниями (так как
дети с синдромом Луи-Бар очень чувствительны к
инфекционным заболеваниям).
• Увеличение иммунитета:
– пересадка тимуса в раннем возрасте;
– курсы лечения внутримышечным введением
препаратов, содержащих компоненты тимуса
(стимулируют иммунитет);
– введение человеческого иммуноглобулина (содержит
смесь антител).
• Общеукрепляющая терапия: полноценное питание
(употребление мясных продуктов, свежих фруктов и
овощей, прием витаминов), умеренные физические
нагрузки, препараты, улучшающие мозговой кровоток.
43. Синдром Вискотта-Олдрича
• комбинированная недостаточность В- и Тлимфоцитов, которая характеризуетсярецидивирующими инфекциями, экземой и
тромбоцитопенией.
• Является сцепленным с Х-хромосомой
наследственным заболеванием. Причиной
развития синдрома Вискотта – Олдрича
являются мутации гена, который
кодирует цитоплазматический протеин,
необходимый для нормального обмена
сигналами между Т- и В-лимфоцитами.
44. Клиника
В связи с нарушением
функционирования Т- и Влимфоцитов у пациентов
развиваются инфекции, вызванные
гноеродными бактериями и
оппортунистическими
организмами, особенно вирусами
и Pneumocystis jirovecii. Частыми
являются инфекции ветряной оспой
и вирусом герпеса.
• Первыми проявлениями могут быть
геморрагии (обычно кровавая
диарея), затем – рецидивирующие
респираторные инфекции, экзема,
тромбоцитопения.
Злокачественные новообразования,
лимфомы, вызванные вирусом
Эпштейна–Барр, и острая
лимфобластическая лейкемия
развиваются у 10% пациентов
старше 10 лет.
45. Диагностика
• Снижение количества и функции Т-клеток,повышении уровня IgE и IgA, низких уровнях
IgM, и низких или нормальных уровнях IgG,
снижении цитотоксичности
естественных клеток-киллеров и
нарушении хемотаксиса нейтрофилов.
Могут наблюдаться частичные дефекты
антител к полисахаридным антигенам
(например, к антигенам групп крови А и В)
• Тромбоцитопения
46. Лечение
• Лечение заключается в пересадке костного мозга отHLA-идентичного донора.
• При значительном снижении гемоглобина проводят
заместительную терапию эритроцитарной массой. У
больных с частыми кровотечениями целесообразно
удаление селезенки.
• При присоединении бактериальной инфекции назначают
антибактериальную терапию: цефалоспорины
(цефтриаксон, цефатоксим, цефтазидим),
сульфаниламиды (бактрим, септрим), антибиотики
пенициллинового ряда (амоксициллин, амоксиклав),
аминогликозиды (гентамицин, амикацин) и т.д. как для
общего, так и для местного применения в виде мазей и
кремов.
• Для подавления роста грибковой флоры применяют
противогрибковые средства (флуконазол,
интраконазол).
47. Дефицит системы фагоцитов Синдром Чедиака-Хигаси
• редкое аутосомно-рецессивное заболевание,характеризующимся нарушением лизиса
фагоцитированных бактерий, в результате чего
развиваются рецидивирующие бактериальные
респираторные и прочие инфекции, также
отмечается альбинизм
48.
• Он наследуется по аутосомнорецессивному типу. Синдром вызванмутацией гена LYST (регулятор миграции
лизосом; CHS1).
• В нейтрофилах и других клетках
(меланоцитах, шванновских клетках)
формируются огромные лизосомальные
гранулы.
• Аномальные лизосомальные гранулы не
могут слиться с фагосомами, поэтому
поглощенные бактерии не лизируются.
49. Клиника
• Клинические проявления включают альбинизмглаз и кожи, восприимчивость к
респираторным и другим инфекциям.
• У 80% пациентов отмечается острое развитие
заболевания с повышением температуры,
желтухой, гепатоспленомегалией,
лимфаденопатией, панцитопенией,
геморрагическим диатезом и
неврологическими изменениями.
• Острое развитие заболевания при синдроме
Чедиака – Хигаси обычно фатально в течение
30 мес.
50. Диагностика
• Обычно наблюдается нейтропения, снижениеестественной цитотоксичности клетоккиллеров, и гипергаммаглобулинемия.
• Исследуются мазки периферической крови на
наличие гигантских гранул в нейтрофилах и
других клетках; мазки костного мозга
исследуются на наличие гигантских включений
в клетках-предшественниках лейкоцитов.
• Диагноз может быть подтвержден
генетическим исследованием мутаций LYST.
Поскольку это нарушение развивается крайне
редко, нет необходимости осуществлять
скрининг родственников, если клиническое
подозрение не является высоким.
51. Лечение
• Поддерживающая терапия сиспользованием антибиотиков, гаммаинтерферона, в некоторых случаях
кортикостероидов
• Пересадка костного мозга
52. Дефекты комплемента
53. Лечение и диагностика
• Общими принципами лечения этихзаболеваний является введение
«недостающих» ферментов,
антибиотикотерапия, симптоматическое
лечение.
• В анализах: лимфопении, дефицит
определенного комплемента
54. Интернет-источники
• http://immuninfo.ru/immunologiya/pervichnye-immunodeficity/deficit-sistemykomplementa/
• http://interferon.su/php/content.php?id=570&
pr=print
• http://www.msdmanuals.com/ru/профессион
альный/иммунология-аллергическиезаболевания/иммунодефицитныесостояния
55. Литература
• Кондратенко И.В., Медицинская Иммунология,2005 Т.7, №5-6, стр 467-476, СпбРОРААКИ
• Аллергология и иммунология: национальное
руководство / под ред. Хаитова Р.М., Ильиной
Н. И. – М.: ГЭОТАР-Медиа 2009, стр 311-381
• ФЕДЕРАЛЬНЫЕ КЛИНИЧЕСКИЕ
РЕКОМЕНДАЦИИ ПО ДИАГНОСТИКЕ И
ЛЕЧЕНИЮ БОЛЬНЫХ ПЕРВИЧНЫМИ
ИММУНОДЕФИЦИТАМИ C НАРУШЕНИЕМ
ГУМОРАЛЬНОГО ЗВЕНА (РААКИ, Москва 2014)