



")







")
")

")

")
")
.")





")

")
")
")




")
")
 (Гусева М. Н.,Семенов А. В.,2014)")
 (Гусева М. Н.,Семенов А. В.,2014)")
















 иммунодефициты (ВИД)»")
 Медицина
МедицинаПохожие презентации:




")





Клиническая иммунология
1. Государственное бюджетное образовательное учреждение высшего профессионального образования «ПЕРВЫЙ САНКТ-ПЕТЕРБУРГСКИЙ
ГОСУДАРСТВЕННЫЙМЕДИЦИНСКИЙ
УНИВЕРСИТЕТ ИМЕНИ АКАДЕМИКА И.П. ПАВЛОВА»
МИНИСТЕРСТВА ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ
ФЕДЕРАЦИИ
Кафедра
иммунологии
2. «Характеристика некоторых форм первичных иммунодефицитов».
Практическое занятие № 3«Клиническая иммунология»
3. Контроль исходного уровня знаний: фронтальный опрос
Дайте определение понятию первичная иммунологическаянедостаточность.
2. Назовите основные клетки врожденного иммунитета и их основные
функции.
3. Назовите основные рецепторы врожденного иммунитета.
4. Назовите основные гуморальные факторы врожденного иммунитета.
5. Какие процессы происходят в тимусе?
6. Какие процессы происходят в костном мозге?
7. Какие классы иммуноглобулинов вам известны?
8. Какие цитокины вам известны?
9. Что такое сигнал-проводящие пути?
10. Объясните, почему, ПИД более часто проявляются у мальчиков, если
генетические дефекты сцеплены с Х-хромосомой?
1.
4. Характеристика некоторых форм ПИД
1.2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
Тяжелый комбинированный иммунодефицит (ТКИД).
Болезнь Брутона.
Общая вариабельная иммунная недостаточность (ОВИН).
Селективный иммунодефицит IgA.
Транзиторная агаммаглобулинемия.
Синдром Луи-Барр.
Синдром Ди-Джорджи.
Синдром Вискотт-Олдриджа
Гипер-IgE-синдром.
Хронический кожно-слизистый кандидоз.
Врожденные дефекты фагоцитоза.
Хроническая гранулематозная болезнь.
Дефекты рецепторов и сигнальных компонентов врожденного иммунитета.
Аутовоспалительные синдромы.
Дефициты системы комплемента.
Особенности проявления симптомов ПИД.
5. Тяжелый комбинированный иммунодефицит (ТКИД)
Возраст: развивается у детей младше 2-х лет.Генетичекий дефект: выявлено 93 патогенных точечных мутаций, из них
60% картированы в Xq13. Мутации генов, кодирующих общую часть
рецепторов ИЛ-2,4,7,9,15,21.
Наследование: аутосомно-рецессивное. Заболевают и мальчики и
девочки (мальчики-до 75%).
Клиника: тяжелые потенциально смертельные инфекции с первых недель
жизни.
Характерна триада: пневмония, диарея, распространенный микоз
слизистых оболочек.
Патогены: низковирулентная флора.
Прогноз: дети с ТКИД умирают в течение первых 2-х лет жизни, если не
попадают в условия строго стерильного режима ( в котором проводится
адекватная терапия) и не подвергаются трансплантации гемопоэтических
стволовых клеток или терапии, замещающей дефектный ген или его
продукт.
6.
ТКИН,Х-сцепленный
Мутация р.Cys182Tyr
в экзоне 4 гена IL2RG.
7.
КЛАССИФИКАЦИЯ СИНДРОМОВ ТКИНT-B-NK-
T-B-NK+
ADA-SCID
Reticular
dysgenesis
T-B+NK-
T-B+NK+
T+B+
Rag1/Rag2 X-SCID
IL-7Rα
дефицит
ZAP70
дефицит
Omenn’s
syndrome
JAK3дефицит
CD3γ/ε
дефицит
MHC II
дефицит
Navajo
SCID
PNP
дефицит
IL-2
дефицит
8.
ИСТОРИЯ “BUBBLE-BOY”9.
ТЯЖЕЛЫЕ КОМБИНИРОВАННЫЕИММУНОДЕФИЦИТЫ
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ РАЗВИВАЮТСЯ
С ПЕРВЫХ НЕДЕЛЬ ЖИЗНИ:
ПЕРСИСИТИРУЮЩАЯ ДИАРЕЯ
ИНФЕКЦИИ РЕСПИРАТОРНОГО ТРАКТА
ОППОРТУНИСТИЧЕСКИЕ ИНФЕКЦИИ
ГРИБКОВЫЕ ИНФЕКЦИИ СЛИЗИТЫХ И КОЖИ
ЛОКАЛЬНАЯ И ГЕНЕРАЛИЗОВАННАЯ
БЦЖ ИНФЕКЦИЯ
ОТСТАВАНИЕ В РАЗВИТИИ
10. ТЯЖЕЛЫЕ КОМБИНИРОВАННЫЕ ИММУНОДЕФИЦИТЫ Терапия
Единственным методом терапии ТКИН является трансплантация костногомозга.
При наличии HLA-идентичного родственного донора – трансплантация
костного мозга, профилактика реакции трансплантат против хозяина ( РТПХ).
Перед трансплантацией костного мозга проводится заместительная терапия
(внутривенное введение иммуноглобулинов), атибиотикотерапия,
противогрибковая терапия, противотуберкулезная терапия(в случае
вакцинации БЦЖ, противовирусная терапия.
11. Болезнь Брутона: описана в 1952 году
Возраст: проявления у детей с первого полугодия жизни (когдазаканчивается действие материнских иммуноглобулинов).
Генетичекий дефект: Х –сцепленная форма вызвана мутацией в гене,
кодирующем специфическую для В-лимфоцитов протеинкиназу
(названную в честь Брутона Btk –брутоновская тирозинкиназа).
Результат генетического дефекта: задержка созревания В лимфоцитов на
уровне пре-В лимфоцитов. Выраженность ПИД может варьировать от
полного отсутствия иммуноглобулинов в крови до сниженной их
концентрации. Распространенность 1:50 000 -1 000 000.
Наследование: Х-сцепленный вариант встречается в 85% случаев.
Заболевают мальчики.
Клиника: тяжелые бактериальные инфекции с первого полугодия.
Характерны: повторные инфекционные заболевания бронхолегочной
системы, желудочно-кишечного тракта.
Патогены:Streptococcus, Staphylococcus, Haemophilus.
Прогноз: при своевременной диагностике и назначении пожизненной
заместительной терапии иммуноглобулинами- относительно
благоприятный.
12. Иммунодефициты, связанные с преимущественным нарушением синтеза иммуноглобулинов. Агаммаглобулинемия.
13. АГАММАГЛОБУЛИНЕМИЯ С ДЕФИЦИТОМ В-КЛЕТОК клинические проявления
ИНФЕКЦИОННЫЕ
Рецидивирующие бактериальные инфекции: ЛОР-органов, легких, ЖКТ,
кожи, минфекции – ЭНТЕРОВИРУС (менингоэнцефалит,
энцефаломиелит)
Менингиты, артриты, остеомиелиты
Вирусные
НЕИНФЕКЦИОННЫЕ
Гипоплазия лимфоузлов и миндалин (не у всех)
Аутоиммунные проявления: склеродема, ревматоидный артрит,
синдромы, клинически сходные с дерматомиозитом и склередемой,
поражения ЖКТ
14. Клинические примеры: агаммаглобулинемия с дефицитом В-клеток (Гусева М. Н.,Семенов А. В.,2014)
Клинические примеры: агаммаглобулинемия с дефицитом Вклеток (Гусева М. Н.,Семенов А. В.,2014)• Пациенты: однояйцевые сибсы Ш.А. и Ш.В., 2008 г.р.
• Предварительный диагноз установлен 07.2013 г.
• Клиническая картина:
Ш.В.
Ш.А.
рецидивирующие отиты
рецидивирующие отиты
рецидивирующие бронхиты
реактивный артрит
синовиит коленного сустава
коксартрит
остеомиелит (3-х кратно прооперирован в 2012 г.)
пневмония (2 раза за 2013 г.)
Данные лабораторного обследования:
Ш.В.
Ш.А.
IgA, г/л (N - 0,70-4,00)
0,01
0,01
IgM, г/л (N - 0,20-2,00)
0,02
0,03
IgG, г/л (N - 7,00-16,00)
<0,004
<0,004
15. Данные лабораторного обследования (Гусева М. Н.,Семенов А. В.,2014)
Ш.В., 2008 г.р.Ш.А., 2008 г.р.
16.
ФБУН "Санкт-Петербургский НИИ эпидемиологии и микробиологии имени Пастера" Роспотребнадзора197101, Санкт-Петербург, ул. Мира, 14 Тел: (812) 232-31-55.
Лицензия на медицинскую деятельность № ФС-78-01-002201 от 14.07.2011 г
ФБУН "Санкт-Петербургский НИИ эпидемиологии и микробиологии имени Пастера" Роспотребнадзора
197101, Санкт-Петербург, ул. Мира, 14 Тел: (812) 232-31-55.
Лицензия на медицинскую деятельность № ФС-78-01-002201 от 14.07.2011 г
ЦЕНТРАЛЬНАЯ КЛИНИКО-ДИАГНОСТИЧЕСКАЯ ЛАБОРАТОРИЯ
ЦЕНТРАЛЬНАЯ КЛИНИКО-ДИАГНОСТИЧЕСКАЯ ЛАБОРАТОРИЯ
Ф.И.О.:______ Ш.В. ________________ Пол: муж Возраст__4 года 9 мес.___
Ф.И.О.:____Ш.А._______ Пол: муж Возраст___4 года 9 мес._________
Ф.И.О. направляющего врача: __________Гусева М.Н.__________ Дата «__09__»___июля______2013 г
Ф.И.О. направляющего врача: ___________Гусева М.Н.______________ Дата «_09_»___июля_______2013г
1.
1.
Фенотипирование лимфоцитов (основных и малых субпопуляций) на проточном
цитофлюориметре FACS Canto II
Определение лимфоцитов, основных и малых
субпопуляций
Относительное (%)
абсолютное (x109)/л
Границы нормы
Границы нормы
2,9-5,1
1,8-3,0
0,9-2,091
43,0
23,0-40,0
2,021
0,667-2,040
0,1
0,1-1,5
0,005
0,003-0,077
1,1
11,9
1,0-1,6
8,0-15,0
0,559
0,232-0,765
10,1
7,8-17,0*
0,475
0,120-0,347
1,8
0,2-1,0*
0,085
0,003-0,022
0,007-0,165
Т-цитотоксические (CD45+ CD3+CD8+)
Дубль-позитивные Т-клетки (CD45+
CD3+CD4+CD8+)
Cоотношение CD4/CD8
NK-клетки (CD45+ CD3-CD16+ CD56+)
NK-клетки цитолитические (CD45+ CD3CD16+CD56dim)
NK-клетки цитокин-продуцирующие (CD45+
CD3-CD16-(or lоw)CD56bright)
ТNK-клетки (CD45+ CD3+CD16- CD 56+)
3,5
0,1-8,0
0,165
0,007-0,165
0,348-1,428
0,174-0,612
0,050-0,450
В-лимфоциты (CD45+ CD19+)
B1-клетки (CD45+ CD19+CD5+CD27-)
B2-клетки - (CD45+ CD19+CD5-CD27-)
0,0
0,0
0,0
12,0-28,0
6,0-12,0
2,0-9,0
0,000
0,000
0,000
0,348-1,428
0,174-0,612
0,050-0,450
0,000
0,007-0,070
0,0
2,0-5,0
0,000
0,007-0,070
0,256
0,068-0,702
B-клетки памяти (CD45+ CD19+CD5-CD27+)
T-хелперы активированные/памяти (CD45+
CD4+CD45RO+CD45RА+)
T-хелперы наивные (CD45+ CD4+CD45ROCD45RA+)
αβT-клетки (CD45+ CD3+ αβTcR+)
γδT-клетки (CD45+ CD3+ γδTcR+)
Регуляторные T-клетки (CD45+
CD4+CD25+CD127-)
CD25 позитивные лимфоциты (Рецептор ИЛ2) (CD45+ CD25+)
HLA-DR позитивные лимфоциты (CD45+
HLA-DR+)
2,9-5,1
1,8-3,0
0,9-2,091
Т-цитотоксические (CD45+ CD3+CD8+)
Дубль-позитивные Т-клетки (CD45+
CD3+CD4+CD8+)
Cоотношение CD4/CD8
NK-клетки (CD45+ CD3-CD16+ CD56+)
NK-клетки цитолитические (CD45+ CD3CD16+CD56dim)
NK-клетки цитокин-продуцирующие (CD45+
CD3-CD16-(or lоw)CD56bright)
ТNK-клетки (CD45+ CD3+CD16- CD 56+)
43,8
23,0-40,0
1,923
0,667-2,040
0,1
0,1-1,5
0,004
0,003-0,077
1,1
12,9
1,0-1,6
8,0-15,0
0,566
0,232-0,765
11,0
7,8-17,0*
0,483
0,120-0,347
1,9
0,2-1,0*
0,083
0,003-0,022
2,5
0,1-8,0
0,110
В-лимфоциты (CD45+ CD19+)
B1-клетки (CD45+ CD19+CD5+CD27-)
B2-клетки - (CD45+ CD19+CD5-CD27-)
0,0
0,0
0,0
12,0-28,0
6,0-12,0
2,0-9,0
0,000
0,000
0,000
B-клетки памяти (CD45+ CD19+CD5-CD27+)
T-хелперы активированные/памяти (CD45+
CD4+CD45RO+CD45RА+)
T-хелперы наивные (CD45+ CD4+CD45ROCD45RA+)
αβT-клетки (CD45+ CD3+ αβTcR+)
γδT-клетки (CD45+ CD3+ γδTcR+)
Регуляторные T-клетки (CD45+
CD4+CD25+CD127-)
CD25 позитивные лимфоциты (Рецептор ИЛ2) (CD45+ CD25+)
HLA-DR позитивные лимфоциты (CD45+
HLA-DR+)
0,0
2,0-5,0
5,8
5,0-25,0*
45,5
20,0-40,0*
1,786
0,272-1,123
87,1
2,7
60,0-80,0
2,0-7,0
3,823
0,106
0,925-1,965
0,020-0,115
3,6
0,3-10,0
0,417
0,004-1,100
2,8
1,5-6,0
0,123
0,043-0,306
1,8
12,0-30,0
0,079
0,348-1,530
2. Определение сывороточных иммуноглобулинов методом иммунотурбидиметрии на
анализаторе «Daytona»
0,023
0,183
0,162
Границы нормы, г/л
0,52 – 2,20
0,4 – 1,8
6,4 – 14,2
0,0 МЕ/мл (Ниже порога
0 - 50 МЕ/мл
Определение подклассов иммуноглобулинов G методом ИФА
Подклассы иммуноглобулинов G
Иммуноглобулины подкласса G1 (IgG1)
Границы нормы
4,700
4,207
2,157
4,390
3,929
2,046
определения метода)
абсолютное (x109)/л
Границы нормы
38,0-53,0
62,0-69,0
33,0-41,0
38,0-53,0
62,0-69,0
33,0-41,0
Показатели, г/л
Относительное (%)
40,0
89,5
45,9
37,9
89,5
46,6
Классы иммуноглобулинов
Определение лимфоцитов, основных и малых
субпопуляций
Лимфоциты
Т-лимфоциты (CD45+ CD3+)
Т-хелперы (CD45+ CD3+CD4+)
Лимфоциты
Т-лимфоциты (CD45+ CD3+)
Т-хелперы (CD45+ CD3+CD4+)
Иммуноглобулины класса А (Ig A)
Иммуноглобулины класса М (Ig M)
Иммуноглобулины класса G (Ig G)
Иммуноглобулины класса Е (Ig E)
Фенотипирование лимфоцитов (основных и малых субпопуляций) на проточном
цитофлюориметре FACS Canto II
7,1
5,0-25,0*
0,334
0,068-0,702
43,2
20,0-40,0*
1,819
0,272-1,123
84,7
5,4
60,0-80,0
2,0-7,0
3,979
0,227
0,925-1,965
0,020-0,115
2,9
0,3-10,0
0,341
0,004-1,100
2,3
1,5-6,0
0,108
0,043-0,306
3,2
12,0-30,0
0,150
0,348-1,530
2. Определение сывороточных иммуноглобулинов методом иммунотурбидиметрии на
анализаторе «Daytona»
Классы иммуноглобулинов
Иммуноглобулины класса А (Ig A)
Иммуноглобулины класса М (Ig M)
Иммуноглобулины класса G (Ig G)
Иммуноглобулины класса Е (Ig E)
Показатели, г/л
0,037
0,205
0,224
Границы нормы, г/л
0,52 – 2,20
0,4 – 1,8
6,4 – 14,2
0,0 МЕ/мл (Ниже порога
0 - 50 МЕ/мл
определения метода)
Определение подклассов иммуноглобулинов G методом ИФА
Показатели, г/л
Границы нормы, г/л
0,04
2,3 - 6,4
Показатели, г/л
Границы нормы, г/л
Иммуноглобулины подкласса G1 (IgG1)
Подклассы иммуноглобулинов G
0,16
2,3 - 6,4
Иммуноглобулины подкласса G2 (IgG2)
0,013
0,7 – 4,5
Иммуноглобулины подкласса G2 (IgG2)
0,03
0,7 – 4,5
Иммуноглобулины подкласса G3 (IgG3)
0,014
0,1 - 1,1
Иммуноглобулины подкласса G3 (IgG3)
0,02
0,1 - 1,1
Иммуноглобулины подкласса G4 (IgG4)
0,002
0,1 - 0,8
Иммуноглобулины подкласса G4 (IgG4)
0,006
0,1 - 0,8
Определение компонентов системы комплемента методом иммунотурбидиметрии на анализаторе
«Daytona»
Компоненты системы комплемента
Показатели, г/л
Границы нормы, г/л
Компонент С3 комплемента (С3)
Компонент С4 комплемента (С4)
1,14
0,28
0,75 – 1,35
0,09 – 0,36
Определение компонентов системы комплемента методом иммунотурбидиметрии на анализаторе «Daytona»
Компоненты системы комплемента
Компонент С3 комплемента (С3)
Компонент С4 комплемента (С4)
Показатели, г/л
Границы нормы, г/л
1,19
0,32
0,75 – 1,35
0,09 – 0,36
17. Поствакцинальные антитела (Гусева М. Н.,Семенов А. В.,2014)
Ш.В.Ш.А.
Противодифтерийные
антитоксические АТ
0,003 МЕ/мл
(восприимчив к
дифтерии)
0,002 МЕ/мл
(восприимчив к
дифтерии)
Противостолбнячные
антитоксические АТ
1:10
1:10
18. Генетический анализ
• Проведен генетический анализ образца крови методами ПЦР иSSCP с последующим секвенированием фрагментов (19
экзонов).
• Выявлена точечная мутация в 17 экзоне в положении 1877 C>T.
• Данная мутация представляет собой миссенс-мутацию с
заменой кодона CGG->TGG (Arg->Trp).
19. Схема введения в/в иммуноглобулинов (октагам, 5%)
Концентрация IgG, г/лНасыщение – 4 недели,
Далее –
поддерживающая доза
(1 раз в 3-4 недели).
На фоне терапии:
Недели наблюдения
Купирование клинической
симптоматики артрита ,
Отсутствие пневмоний.
Нормальное развитие.
Посещение
общеобразовательной
школы.
20. Общая вариабельная иммунная недостаточность (ОВИН)
Возраст: начало болезни в возрасте старше 2-х лет. Плохой ответ навакцинацию.
Генетичекий дефект: мутации в генах 6 хромосомы, кодирующих белки,
вовлеченные в В-лимфопоэз и гомеостаз В-клеток (включая CD 19 и
др.). Результат генетического дефекта: снижение в сыворотке крови
уровней иммуноглобулинов как минимум 2 изотипов (IgA,IgG и/или
IgM) в 2 раза и более по сравнению с возрастными нормами.
Распространенность 1:7 000-200 000.
Клиника: рецидивирующие инфекции респираторного и
пищеварительного трактов, вызванные бактериями, вирусами, грибами и
паразитами.
Характерно: повышение вероятности развития аутоиммунных и
онкологических заболеваний.
Прогноз: при своевременной диагностике и назначении пожизненной
заместительной терапии иммуноглобулинами- относительно
благоприятный.
21. ОВИН: мутации в генах , кодирующих белки, вовлеченные в В-лимфопоэз и гомеостаз В-клеток (включая CD 19 и др.).
22. Селективный иммунодефицит IgA
Возраст: встречается у пациентов старше 4 лет.Генетичекий дефект: результат генетического дефекта: снижение
концентрации IgA менее 0.07 г/л при нормальных уровнях IgM и IgG в
сыворотке крови и при допустимом поствакцинальном синтезе IgG.
Распространенность: 1:300-700.
Клиника: бактериальные инфекции.
Характерны: предрасположенность к инфекционным заболеваниям
бронхолегочной системы, желудочно-кишечного тракта.
Патогены:Streptococcus, Staphylococcus и др.
Прогноз: при своевременной диагностике и назначении адекватной
благоприятный.
23. Гипер-IgE-синдром
Характеристики: в крови существенно повышены уровнииммуноглобулина класса Е.
Возраст: врожденные аномалии строения лицевого скелета.
Генетичекий дефект: предполагают роль гомозиготных делеций в гене
TYK2, кодирующем тирозинкиназу человека.
Результат генного дефекта: дефект сигнального пути цитокинов,
включая интерфероны 1 типа. ИЛ-6,10,12,23. нарушение ответа Th1
лимфоцитов. Аутосомно-рецессивная форма наследования.
Описаны также мутации в гене, кодирующем STAT 3 (аутосомнодоминантное наследование). Результат –полиорганная патология.
Характерны: повторные абсцессы кожи и подкожной клетчатки,
пневмонии, атопические дерматиты.
Лечение : заместительная терапия иммуноглобулинами; гормонами
тимуса. Лечение антибиотиками, противогрибковыми препаратами.
24. Гипер-IgE-синдром
25. Транзиторная агаммаглобулинемия
Возраст: диагностируется у пациентов в возрасте от 6 месяцев до 2-3 лет.Генетичекий дефект: результат генетического дефекта: снижение
концентрации IgG не менее, в 2 раза по сравнению с возрастной нормой
при нормальных или сниженных показателях IgА и IgМ в сыворотке
крови.
Клиника: бактериальные инфекции.
Характерны: предрасположенность к инфекционным заболеваниям
ЛОР-органов, бронхолегочной системы, желудочно-кишечного тракта.
Патогены:Streptococcus, Staphylococcus и др.
Прогноз: при своевременной диагностике и назначении адекватной
терапии - благоприятный.
Купирование транзиторной гипогаммаглобулинемии у детей раннего
возраста происходит в период между 18 – и 36 –м месяцем жизни (т.е. к
3-м годам).В большинстве случаев прогноз-благоприятный.
Дифференциальная диагностика: с патологической
гипогаммаглобулинемией.
26. Синдром Ди-Джорджи
Возраст: с рождения - гипоплазия или аплазия тимуса, множественныедефекты артериального ствола, гипопаратиреоз; с 1,5 лет –появляется
шаткая походка (атаксия), к 6 годам развивается расширение капилляров
(телеангиоэктазия).
Генетичекий дефект: гомозиготная делеция хромосомы 22q 11.2 ,
выявляемая у 90 % больных с синдромом Джорджи, ломкость и разрывы
в 7 и 14 хромосомах.
Результат генетического дефекта: стволовые клетки не
дифференцируются в Т-лимфоциты.
Т-иммунодефицит: число и активность Т –лимфоцитов резко снижены.
Способность к синтезу антител сохранена, но на более низком уровне. У
70% больных –недостаток иммуноглобулинов класса А.
Наследование: аутосомно-доминантное.
Клиника: тяжелые рецидивирующие инфекции.
Характерны: предрасположенность к инфекционным ЛОР –
заболеваниям и заболеваниям бронхолегочной системы.
27.
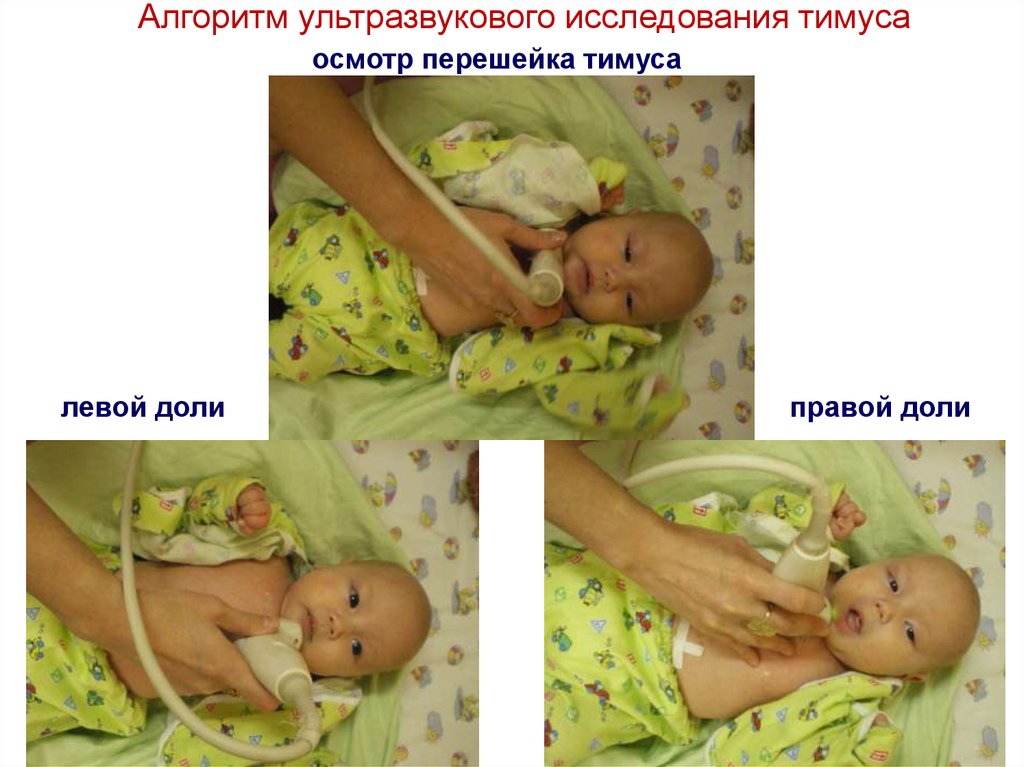
Алгоритм ультразвукового исследования тимусаосмотр перешейка тимуса
левой доли
правой доли
28. Синдром Ди-Джорджи - нарушение развития тимуса в эмбриональном периоде (гипоплазия или аплазия тимуса)
29. Синдром Ди-Джорджи
30. Атаксия –телеангиоэктазия (синдром Луи-Барр)
Характеристики: прогрессирующая мозжечковая атаксия, сочетающаясяс задержкой роста и отсутствием иммуноглобулинов классов А и М.
Возраст: проявляется у пациентов от 2 до 6 лет (появляются
телеангиоэктазии на конъюктиве, открытых участках тела,слизистых
оболочках).
Генетичекий дефект: гена, расположенного на длинном плече 11
хромосомы. Результат генетического дефекта: нарушение синтеза белка
АТМ, ответственного за контроль клеточного роста, распознавание
клеткой поврежденных участков ДНК и репарации поврежденных
участков ДНК или блокирование клеточного цикла в таких клетках.
В крови повышено число циркулирующих Т-лимфоцитов с
преобладанием незрелых Т-лимфоцитов.
Распространенность: 1:500 000 – 1000 000 (с одинаковой частотой у
мальчиков и девочек).
Характерны: предрасположенность к инфекционным заболеваниям
бронхолегочной системы, желудочно-кишечного тракта.
Лечение: заместительная терапия иммуноглобулинами; гормонами
тимуса, лечение антибиотиками, противогрибковыми препаратами.
31. Атаксия –телеангиоэктазия (синдром Луи-Барр)
32. Атаксия –телеангиоэктазия (синдром Луи-Барр)
Атаксическая ПОХОДКА33. Синдром Вискотт-Олдриджа
Характеристики: тромбоцитопения.Возраст: врожденные аномалии тромбоцитарного звена: резко
выраженное изменение морфологии тромбоцитов и существенно
сниженное их число.
Генетический дефект: сцеплен с Х-хромосмой. Страдают мальчики.
Результат генного дефекта: склонность к кровоточивости из-з
сниженного абсолютного числа тромбоцитов, нарушение морфологии Тлимфоцитов, нарушение процессов кооперации Т- и В-лимфоцитов,
сниженные уровни иммуноглобулинов класса М.
Характерны: экзема, гнойные и оппортунистические инфекции.
Возможно развитие злокачественных новообразований.
Лечение : заместительная терапия иммуноглобулинами.
Лечение антибиотиками, противогрибковыми препаратами.
34. Хронический кожно-слизистый кандидоз
Генетичекий дефект: не установлен.Характерны: частые поражения Candida Albicans кожи и слизистых
оболочек.
В 50% случаев обнаруживают сочетание с аутоиммунной
полиэндокринопатией в виде патологии паращитовидных, щитовидной,
половых желез, гипофиза.
Лечение: заместительная терапия иммуноглобулинами; антибиотики,
противогрибковые препараты.
35. Клеточный иммунный ответ
36. Врожденные дефекты фагоцитоза
В настоящее время выделяют более 20 нозологических форм.К ним относятся дефекты числа и функций нейтрофилов, в частности:
• Нарушение хемотаксиса.
• Нарушения адгезии.
• Нарушение эндоцитоза.
• Нарушение киллинга.
• Нарушения деградации фагоцитированных частиц.
• Нарушение секреции цитокинов и др.
Примеры –синдром «ленивых» лейкоцитов (синдром Шварцмана) дефекты генов мембранных молекул адгезии вследствие чего развивается
дефект хемотаксиса нейтрофилов и макрофагов.
37. Хроническая гранулематозная болезнь (ХГБ)
Характеристики: фагоциты не способны генерировать радикалыкислорода, нарушена способность к «кислородному взрыву».
Возраст: проявляется у пациентов практически с рождения (1 месяц).
Генетичекий дефект: генов, кодирующих любой из компонентов
NADFH, состоящей из 4 субъединиц: gp 91 phox; p22 phox,
составляющих цитохром b 558 и двух цитозольных компонгентов - p47
phox и p 67 phox.
Наследование: в 70-85% случаев сцеплено с Х-хромосомой (болеют
мальчики); в остальных случаях –аутосомно-рецессивное.
Характерны: рецидивирующие инфекционные заболевания
бронхолегочной системы, поражающие также кожу, лимфатические
узлы, печень, почки. Характерно возникновение лимфаденита, BCG,
абсцессов с гигантоклеточными гранулемами, гепатоспленомегалия.
Лечение: пожизненная антибактериальная терапия, с включением
иммунотерапии (в ряде случаев- препараты интерферона гамма), терапия
противогрибковыми препаратами.
Генная терапия- введение в стволовые клетки гена gp 91 phox.
38. Хроническая гранулематозная болезнь (ХГБ)
39. Хроническая гранулематозная болезнь (Клинический пример) (Гусева М. Н.,Семенов А. В.,2014)
• Б.М., 2010 г.р. Ребенок от четвертой беременности стоксикозом второй половины (отеки беременных). 1БМальчик 8 лет, здоров. 2Б- мед. аборт, 3Б- мед. аборт.
Роды срочные. По Апгар 8/9 баллов. Закричал сразу.
• Масса тела при рождении 3250 г.
• В 1месяц перенес везикулопустелез.
• В 3,5 месяца - вскрывшийся парапроктит. Внутренний
ректальный свищ. Дисбактериоз. Анемия легкой степени.
• В 4,5 месяца (декабрь 2010 г.)- гнойный лимфаденит
подчелюстной области. Хирургическое лечение.
• В 7 меяцев - правосторонняя пневмония.
40. Хроническая гранулематозная болезнь (Клинический пример) (Гусева М. Н.,Семенов А. В.,2014)
• В 1 год – пневмония.• В 1 год 2 месяца – постпневмонический пневмофиброз S5
справа. Локальные бронхоэктазы.
• В 1 год 7 месяцев – правосторонняя н/долевая (S8-9)
пневмония.
• В 1год 8 месяцев - хронический парапроктит.
• В 1 год месяцев - аденофлегмона подбородочной области.
Аденофлегмона правой подчелюстной области.
• В 2 года 3 месяца – бронхит (при поступлении в стационар
СПбГПМУ)
• В 2 года 4 месяца – положительный квантифероновый
тест QuantiFERON-TB test (QFT)
41. Клинический анализ крови с 23.10.12 по 20.11.12
ДатаEr
1012/л
Hb
Ц.п.
г/л
Tr
L
109/л
109/л
Нейтрофи Лим Мон Эоз. Баз.
лы
ф.
.
%
%
п/я% с/я%
%
%
Сверт.
по
Сухар
еву
СОЭ
мм/ч
23.10.
12
5,05 106
-
533
12,8 -
56
40
4
-
-
-
37
31.10.
12
4,89 96
0,58 563
10,2 -
40
48
12
-
-
-
25
07.11.
12
4,43 93
0,63 370
4,5
4
36
52
7
1
-
3’29’ 5
20.11.
12
4,91 101
0,62 497
9,7
-
21
74
1
4
-
-
11
42. Содержание иммуноглобулинов в сыворотке крови в 2 г. 3 мес.
IgAIgM
IgG
Значение (г/л)
2,3
1,15
8,75
Норма (г/л)
0,34-1,08
0,42-0,80
7,09-10,75
43. Результаты Burst-теста
ФБУН "Санкт-Петербургский НИИ эпидемиологии и микробиологии имени Пастера" Роспотребнадзора197101, Санкт-Петербург, ул. Мира, 14 Тел: (812) 232-31-55.
Лицензия на медицинскую деятельность № ФС-78-01-002201 от 14.07.2011 г
ЦЕНТРАЛЬНАЯ КЛИНИКО-ДИАГНОСТИЧЕСКАЯ ЛАБОРАТОРИЯ
Ф.И.О.:_Б. М._______ __________ Пол: муж Возраст_3г. 2 мес.
Ф.И.О. направляющего врача:__________Гусева М.Н._________________Дата «04»
октября_2013г
FAGOBURST определение количества гранулоцитов с
внутриклеточной продукцией активных форм кислорода на
проточном цитофлюориметре FACS Canto II
Стимуляторы
E. coli
fMLP
PMA
% клеток, продуцирующих
активные формы кислорода
Границы нормы, %
Индекс
стимуляции
9,6
12,3
8,7
97-100
1-10
98-100
6,5
6,8
5,7
44. Дефекты рецепторов и сигнальных компонентов врожденного иммунитета
• Известны ПИД с нарушениями в TLR-сигнальнойсистеме, вызванные дефектами:
• Киназы IRAK 4 (повышается чувствительность к
пневмококковой инфекции).
• Трансмембранного белка эндоплазматического
ретикулума UNC-93B, участвующего в процессе
проведения сигналов с TLR3,7,8,9, вовлеченных в ответ на
вирусную инфекцию(повышается чувствительность к
герпесвирусной и другим типам инфекции).
• Дефицит TLR3 (повышается чувствительность к
герпесвирусной инфекции).
45. Типы иммунного ответа
СвойстваКлеточный тип ответа
Воспалительный
Клеточная
цитотоксичность иммунный ответ
Гуморальный
тип ответа
(гиперчувствитель
ность
замедленного типа
–ГЗТ)
Макрофаги, гипер В-лимфоциты
КлеткиКлон
эффекторы цитотоксическ активирован превращаются в
плазматичес
ные
их CD8+
интерфероном - , кие клетки и в В
лимфоцитов- синтезируемым
клетки памяти
(CTL)
Т хелперами 1
типа
46. Типы иммунного ответа
СвойстваКлеточный тип ответа
Клеточная
цитотоксичность
Локализация
антигена
Гуморальный
тип ответа
Воспалительный
иммунный ответ
(гиперчувствительно
сть замедленного
типа –ГЗТ)
В цитозоле, между В фагоцитарных
органеллами
вакуолях
Вне клетки
АПК
дендритные
клетки
макрофаги
дендритные
клетки
дендритные
клетки
В лимфоциты
Представле
ние АГ
MHC I
MHC II
MHC II
47. Аутовоспалительные синдромы, связанные с дисфункцией врожденного иммунитета за счет изменений в системе NOD-подобных рецепторов
Группа заболеваний, в основе которых лежит генетическидетерминированная активация врожденного иммунитета, и в первую
очередь, NOD - подробных рецепторов (NOD - LR).
Основные клинические проявления:
• Эпизоды гектической лихорадки, сопровождающиеся высыпаниями,
миалгиями, артралгиями.
• При некоторых синдромах возможно тяжелое поражение опорнодвигательного аппарата, центральной нервной системы (ЦНС),
желудочно-кишечного тракта (ЖКТ).
Следствием этих нарушений является повышенный риск амилоидоза.
(Амилоидоз характеризуется нарушением обмена веществ, в результате чего образуется новое для
организма вещество (амилоид), которое откладывается вокруг ретикулярных и коллагеновых волокон и
вызывает нарушение функций внутренних органов).
48. Дефициты системы комплемента
Редкие формы ПИД (в подавляющем большинстве случаев дефектысистемы комплемента являются вторичными).
В клинической практике наблюдаются рецидивирующие пиогенные
инфекции, вплоть до сепсиса
Наследование –по аутосомно-рецессивному типу.
Проявления: нарушение опсонизации, фагоцитоза и разрушения
микроорганизмов.
Наиболее тяжело протекает ПИД с дефектом ингибитора компонента
системы комплемента С1.
Наследование - по аутосомно-доминантному типу.
Развитие при такой форме ПИД ангиогенных отека связано с
накоплением избыточных количеств С3а и С5а компонентов, которые
вызывают повышение проницаемости капилляров.
49. Особенности проявления симптомов ПИД
Характерис
тика
Комбиниро
ванные ПИД
Преимущественно Вдефекты
Дефекты
фагоцитарного звена
Дефекты
комплемента
Начало
заболева
ния
Раннее начало
(1-3 месяц жизни)
5-7 месяц жизни
Раннее начало
(1-3 месяц жизни)
В любом
возрасте
Спектр
патогенов
Бактерии; вирусы; грибы;
паразиты;
оппортунистические
инфекции (пневмоцисты)
Бактерии; вирусы;
паразиты (лямблии)
Бактерии; грибы и
паразиты
Бактерии:нейсе
рии; кишечная
палочка
Патология
органов
Отставание в физическом
развитии; поражение
легких, диарея, кандидоз
кожи и слизистых оболочек
Повторные
инфекционные
заболевания
респираторного и
пищеварительного
трактов,
энтеровирусный
энцефалит
Гнойные
инфекционные
заболевания кожи,
гнойный
лимфаденит,
периодонтит,
язвенный стоматит,
абсцессы,
остеомиелит
Менингит,
повторные
инфекционные
заболевания
респираторного
тракта
50. Сруктура летальности ПИД
ЧИСЛОУМЕРШИХ
Сруктура летальности ПИД
Число пациентов
в регистре
(27 б-х - 15,9%
регистра)
Синдром Чедиака-Хигаши
2
2
ТКИН (тяжелый комбинированный иммунодефицит)
8
7
Аутоиммунный
лимфопролиферативный синдром
3
2
Синдром Ди Джорджи
2
1
Синдром Ниймеген
2
1
ХКСК
3
2
Хроническая гранулемотозная болезнь
6
4
Врожденная нейтропения
2
1
Синдром Вискотта-Олдрича
2
1
Атаксия-телеангиэктазия
6
3
Агаммаглобулинемия Брутона
11
2
ОВИН(общая вариабельная иммунная недостаточность)
12
1
Селективный дефицит IgА
86
1
Форма
ПИД в регистре (170 б-х)
51. Вопросы
1.Каковы основные клинические проявления ПИД? В каком возрасте проявляются симптомы при
комбинированных ПИД и при ПИД с преимущественным поражением В звена, фагоцитарного
звена, системы комплемента?
2. Дайте характеристику селективному дефициту IgA .
3. Дайте характеристику болезни Брутона.
4. Что такое ОВИН?
5. Дайте характеристику ТКИД.
6. Дайте характеристику синдрому Ди Джорджи.
7. Дайте характеристику синдрому Луи-Бар.
8. Что такое транзиторная агаммаглобулинемия?
9. Что такое гипер-IgE –синдром?
10. Приведите примеры поражения органов и систем при комбинированных ПИД и при ПИД с
преимущественным поражением В звена, фагоцитарного звена, системы комплемента.
52. Тестовые вопросы
Изолированное снижение уровня IgA в сыворотке крови определяют при:1. ТКИД.
2. Транзиторной гипогаммаглобулинемии.
3. Общей вариабельной иммунной недостаточности.
4. Селективном дефиците IgA.
5. Атаксии-телеангиоэктазии.
_________________________________________________________________
Генетические дефекты в TLR –сигнальной системе характерны для:
1. ТКИД.
2. Дефектов врожденного иммунитета.
3. Общей вариабельной иммунной недостаточности.
4. Селективном дефиците IgA
5. Атаксии-телеангиоэктазии.
53. Тестовые вопросы
Транзиторная гипогаммаглобулинемия диагностируется в возрасте:1. С 6 месяцев до 3 лет.
2. С рождения до 6 месяцев.
3. С 3 до 5 лет.
4. В подростковом возрасте.
5. У взрослых.
_________________________________________________________________
Врожденное отсутствие тимуса (синдром Ди Джорджи) сопровождается:
1. Отсутствием стволовых клеток.
2. Отсутствием антител в сыворотке крови.
3. Отсутствием зрелых Т-лимфоцитов.
4. Отсутствием В-лимфоцитов.
5. Отсутствием естественных киллеров (NK).
54. Тестовые вопросы
При аутовоспалительных нарушениях имеет место дисфункция врожденного иммунитетаза счет изменения в системе NOD-подобных рецепторов. Клинически следствием это
является:
1. Онкопатология.
2. Повышенная ломкость костей.
3. Хронический пиелонефрит.
4. Аллергия.
5. Развитие амилоидоза.
_________________________________________________________
Задержка созревания В–лимфоцитов на уровне пре-В клеток за счет генетических
нарушений в системе тирозинкиназы имеет место при:
1. Х-сцепленной агаммаглобулинемии (болезнь Брутона).
2. Селективном дефиците IgA.
3. Дефектах врожденного иммунитета.
4. Общей вариабельной иммунной недостаточности.
5. Атаксии-телеангиоэктазии.
55. Тестовые вопросы
С наибольшей частотой в популяции встречается:1. Кожно-слизистый кандидоз
2. Транзиторная гипогаммаглобулинемия.
3. Общая вариабельная иммунная недостаточность
4. Селективный дефицит IgA.
5. Атаксия-телеангиоэктазия.
_________________________________________________________________
Для ПИД с преимущественным поражением В лимфоцитов характерным является:
1. Редкая заболеваемость грибковыми и вирусными инфекциями (за исключением
энтеровирусов).
2. Частая заболеваемость грибковыми и вирусными заболеваниями.
3. Гельминтозы.
4. Онкопатология.
5. Ангиогенные отеки.
56. Тестовые вопросы
К врожденным дефектам фагоцитарного звена относятся:1. Нарушение хемотаксиса.
2. Нарушения адгезии.
3. Нарушение эндоцитоза.
4. Нарушение киллинга.
5. Нарушения деградации фагоцитированных частиц.
_________________________________________________________________
Фагоциты не способны генерировать радикалы кислорода, нарушена способность к
«кислородному взрыву» при:
1. ТКИД.
2. Общей вариабельной иммунной недостаточности.
3. Селективном дефиците IgA
4. Атаксии-телеангиоэктазии.
5. Хронической гранулематозной болезни (ХГБ).
57. Домашнее задание к занятию № 4 «Приобретенные (вторичные) иммунодефициты (ВИД)»
1. Ознакомиться с возможными причинами ВИД.2. Ознакомиться с наиболее общими проявлениями ВИД.
3. Вспомнить материал практического занятия из цикла
«Общая иммунология», посвященного методам
иммунного анализа.
4. Уметь проводить анализ возможного последствия того
или иного дефекта в иммунной защите: при дефекте
какого звена иммунной системы - какие инфекции и
какие клинические проявления ИД наиболее вероятны.