







































 Медицина
Медицина Биология
БиологияПохожие презентации:






 изменчивость")



Изменчивость, ее формы и проявления. Наследственные болезни
1.
Крымский федеральный университетимени В.И. Вернадского
Медицинская академия имени С.И. Георгиевского
Кафедра биологии медицинской
Изменчивость,
ее формы и проявления.
Наследственные болезни
Лекция №5 для студентов
Лечебного дела
Агеева Елизавета Сергеевна
доктор медицинских наук,
доцент по кафедре патофизиологии,
Заведующий кафедрой биологии медицинской
Стоматологического факультета
2.
Изменчивость – фундаментальное свойство живыхорганизмов приобретать новые признаки и
свойства в процессе онтогенеза.
Выделяют модификационную, комбинативную и
мутационную изменчивость
3.
Фенотипическая ненаследственнаяПризнаки для которых характерен этот вид
изменчивости: рост, масса , окраска
Механизм: условия среды воздействуют на реакции и
приводят к их изменению
Факторы:
1. определяются влиянием среды. Имеют
направленный характер
2. изменения не наследуются потомками, связаны
только с фенотипом
3. изменения появляются массово
4. изменения носят адаптивный характер
(приспособительный)
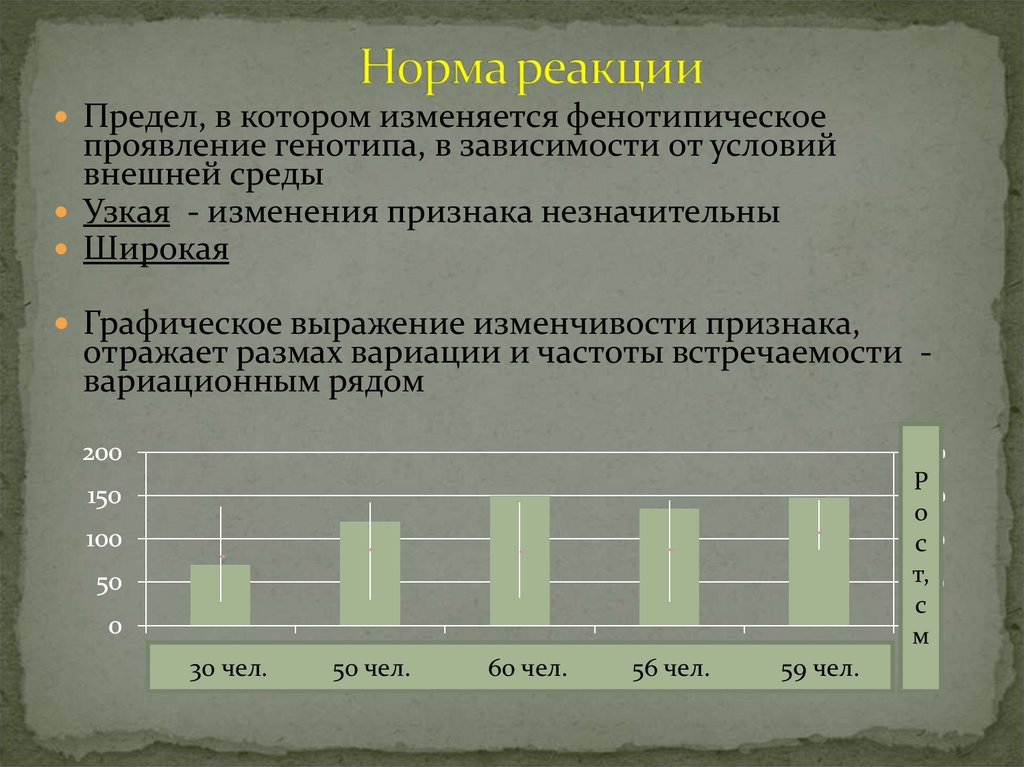
4.
Предел, в котором изменяется фенотипическоепроявление генотипа, в зависимости от условий
внешней среды
Узкая - изменения признака незначительны
Широкая
Графическое выражение изменчивости признака,
отражает размах вариации и частоты встречаемости вариационным рядом
Р
о
с
т,
с
м
30 чел.
50 чел.
60 чел.
56 чел.
59 чел.
5.
Генотипическая наследственнаяПолучение новых сочетаний генов в генотипе
Механизм:
1. независимое расхождение хромосом в анафазе
мейоза-1, хроматид в анафазе мейоза-2
2. перекомбинация генов при кроссинговере
3. случайное сочетание гамет при оплодотворении
6.
Генотипическая наследственнаяМутации генов в генотипе
Свойства мутаций:
1. возникают внезапно
2. наследуются
3. ненаправлены
4. могут возникать повторно
NB! В ходе репликации и рекомбинации постоянно возникают
различные нарушения в структуре ДНК и хромосом, которые
распознаются и исправляются системами репарации.
Нарушения во время реализации этого процесса может
приводить к мутациям.
7.
Наследственная патология связана с мутациями(т.е. с изменением генетического материала).
-6
Средняя частота мутаций 10 на 1 гамету в
поколение.
Причины мутаций - мутагены.
8.
физические - ионизирующее излучение,температурный фактор,
химические – АФК, ароматические
углеводороды, цитостатики, органические
растворители, пестициды, препараты ртути,
биологические – вирусы кори, краснухи,
гриппа, антигены некоторых
микроорганизмов
9.
Если по происхождению мутагеныотносятся к факторам окружающей
среды, то они называются
экзогенные.
Если они образуются во время
жизнедеятельности организма, то эндогенные.
Мутагенез - это, соответственно,
процесс.
10.
в соматических клеткахзлокачественные опухоли
врожденные пороки развития
в половых (генеративные)
наследственные болезни
болезни с наследственной
предрасположенностью ( МФЗ)
врожденные пороки развития (мутации во
время органогенеза)
11.
Спонтанные (самопроизвольные)Под действием естественных мутагенных
факторов внешней среды без вмешательства
человека
Индуцированные
Появляются в результате направленного
воздействия определенных мутагенных
факторов
12.
ЛетальныеВызывают гибель организма
Сублетальные
Снижают жизнедеятельность
Нейтральные
Не влияют жизнедеятельность
13.
ДоминантныеСубдоминантные (проявляющиеся частично)
Рецессивные
14.
Генные – изменение молекулярной структурыгена (последовательности нуклеотидов) в
пределах одного гена
Хромосомные – изменение структуры хромосом
Геномные – изменение количества хромосом
15.
ГеныСтруктурные
Регуляторные
в кодирующей части –может качественно
измениться синтез белка
в регуляторной части – например, в промоторе –
количественно измениться
в интронах – ничего не будет – нейтральная
(сайлент) мутация
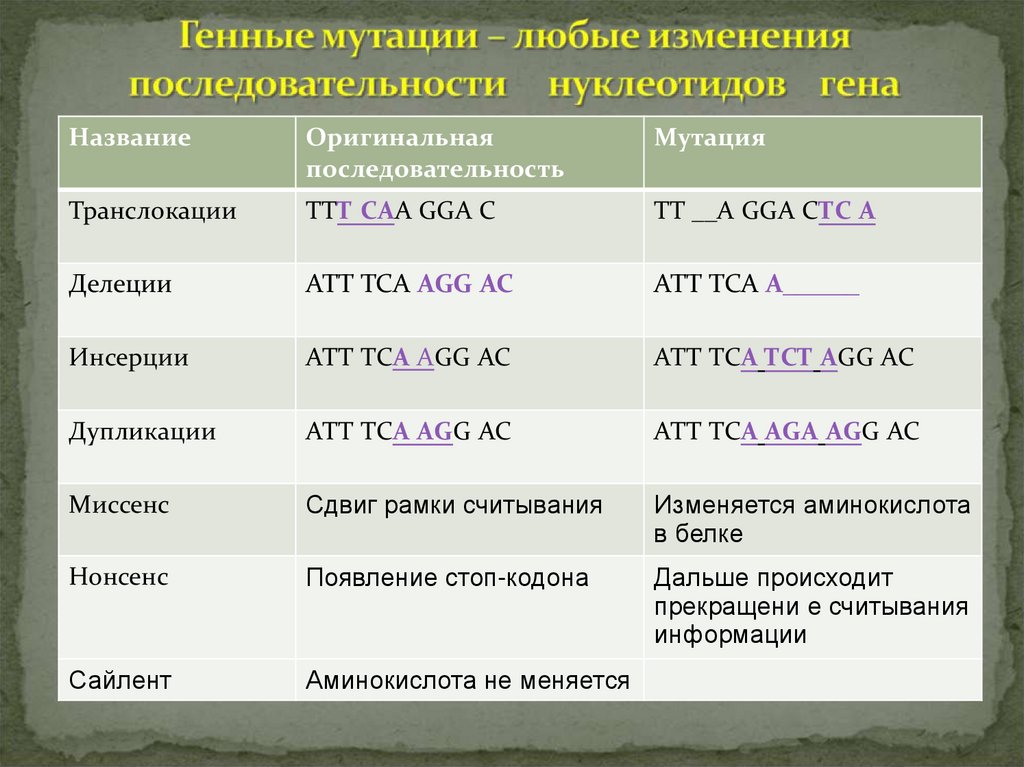
16.
НазваниеОригинальная
последовательность
Мутация
Транслокации
ТТТ САА GGA C
ТТ __А GGA CТС А
Делеции
АТТ ТСА АGG AC
АТТ ТСА А______
Инсерции
АТТ ТСА АGG AC
АТТ ТСА ТСТ АGG AC
Дупликации
АТТ ТСА АGG AC
АТТ ТСА АGА АGG AC
Миссенс
Сдвиг рамки считывания
Изменяется аминокислота
в белке
Нонсенс
Появление стоп-кодона
Дальше происходит
прекращени е считывания
информации
Сайлент
Аминокислота не меняется
17.
Заболевания связанные с генными мутациями могутбыть – моногенными (1 ген – 1 заболевание)
Пример – фенилкетонурия –
аутосомно-рецессивное заболевание,
обусловленное нарушением обмена
незаменимой аминокислоты
фенилаланина, поступающей в
организм человека с белковой пищей
ФКУ 1 типа – дефицит фенилаланин-4гидроксилазы
ФКУ 2 и 3 типа – дефект птеринового
кофактора (1-3%)
https://zen.yandex.ru/media/kak_to_tak/fenilketonuriia-chtohttps://zen.yandex.ru/media/kak_to_tak/fenilketonuriia-chto-eto-za-bolezneto-za-bolezn-prichiny-simptomy-lechenieprichiny-simptomy-lechenie-5cee2cd0c57ced00ae10b11f
5cee2cd0c57ced00ae10b11f
18.
Эпидемиология фенилкетонурияПричина – рецессивная мутация
Частота ФКУ составляет 1 на 10 000
новорожденных
(1 : 4370 в Турции, 1 : 80500 в
Японии, 1 : 12280 Италия)
(1 : 4735 Курская область, 1 :
18000 Республика Тыва, 1 : 7600
Санкт-Петербург, 1 : 6772 Москва)
Частота носителей гена
(гетерозиготы) 1 на 50
https://zen.yandex.ru/media/kak_to_tak/f
enilketonuriia-chto-eto-za-boleznprichiny-simptomy-lechenie5cee2cd0c57ced00ae10b11f
19.
Фенилкетонурия1 тип - дефицит фермента
фенилаланингидроксилазы ведет к
накоплению фенилаланина и продуктов его
распада в биологических жидкостях – 12q22q24.1
2 тип – дефицит цитозольной
дигидроптеринредуктазы, который приводит к
блоку на пути превращения фенилаланина в
тирозин, а также предшественников
образованиякатехоламинов и серотонина –
4р15.3
3 тип – недостаточность цитозольной 6пирувоилтетрагидроптеринсинтетазы в печени
и эритроцитах, участвующей в процессе
синтеза тетрогидробиоптерина из
дигидронеоптерина трифосфата – q22.3-23.3
20.
ЗаболеваниеНа первом году жизни (2-6 месяцев) –
вялость, отсутствие интереса к
окружающему, иногда повышенная
раздражительность, беспокойство,
срыгивание, гипотония, судороги,
явления дерматита
Задержка мышечного и психического
развития
Микроцефалия и гидроцефалия
Гипопигментация кожи
Эпилептические приступы
Течение прогрессирующее,
приводящее к смерти на 2-3 году
жизни
21.
Серповидноклеточная анемияМутация в гене β-субъединицы
гемоглобина
Дефект гена HBB - 11p15.5.
Дефектный гемоглобин HbS
образуется в результате замены
валина на глутаминовую кислоту
Отдавая тканям кислород гемоглобин полимеризуется с
образованием волокон, которые деформируют эритроциты с
формированием длинных цепей, эритроциты становятся
серповидными
Это вызывает увеличение вязкости крови, стаз; создается
механическая преграда в мелких артериолах и капиллярах, что
приводит к тканевой ишемии (с чем связаны болевые кризы),
образуются тромбы, возникает анемия.
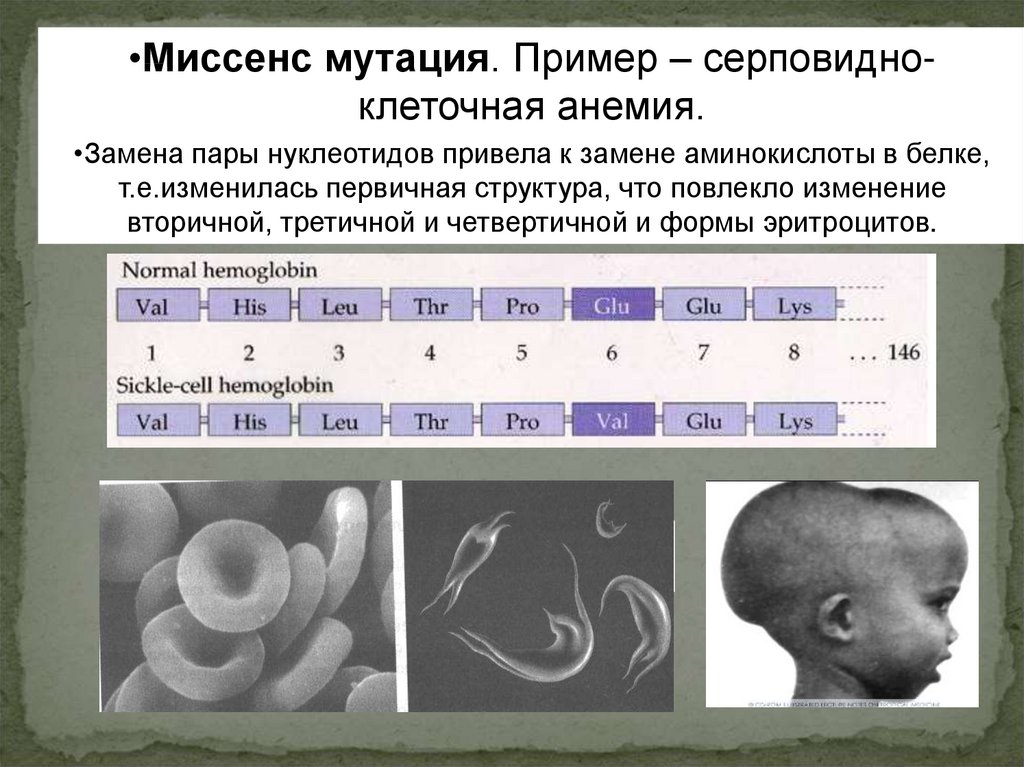
22.
•Миссенс мутация. Пример – серповидноклеточная анемия.•Замена пары нуклеотидов привела к замене аминокислоты в белке,
т.е.изменилась первичная структура, что повлекло изменение
вторичной, третичной и четвертичной и формы эритроцитов.
ЦТТ в ДНК
ГАА в РНК
ЦАТ в ДНК
ГУА в РНК
23.
24.
Нонсенс мутация может возникнуть как в
результате замены нуклеотида, так и при
сдвиге рамки считывания. Пример: группа
крови О.
У людей с данной группой крови в гене
произошло выпадение (делеция) одного
нуклеотида – в результате возник стоп-кодон.
Синтезируется короткий и неактивный белокфермент.
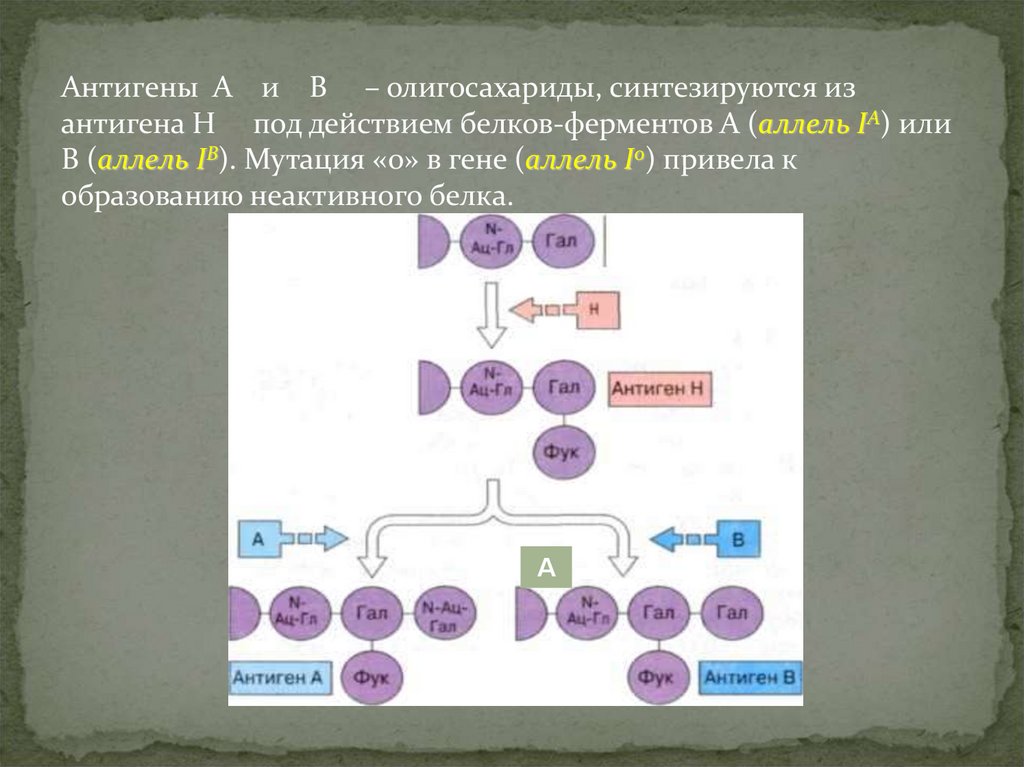
25.
Антигены А и В – олигосахариды, синтезируются изантигена Н под действием белков-ферментов А (аллель IA) или
В (аллель IВ). Мутация «0» в гене (аллель I0) привела к
образованию неактивного белка.
А
26.
Прогерия – генетическое заболевание,характеризующееся быстрым постарением.
Быстрое старение молодого организма
Описана независимо двумя врачами
Дж. Хатчинсоном (1886 г.) и Х.
Гилфордом (1904 г.).
В их честь это заболевание называется
синдромом Хатчинсона-Гилфорда.
За 125 лет в медицинской практике
было описано более ста случаев
заболевания детской прогерией
Взрослая форма (синдром Вернера)
Слева направо: 15, 12 и 26 лет
27.
Детская формамутация в гене LMNA, возникшей
ещё на стадии внутриутробного
развития.
Ген LMNA кодирует белок ламин А,
который играет важную роль в
формировании структуры ядра
клеток. При прогерии белок не
выполняет свою функцию, что
приводит в конечном счёте к
преждевременной гибели клеток.
28.
При взрослой форме заболевания (синдромВернера) мутация не возникает впервые, а
наследуется. Это аутосомно-рецессивное
заболевание, то есть ребёнок должен унаследовать
по одной копии дефектного гена RECQL2 от
каждого из родителей. Эта форма встречается
чаще, чем
https://www.wonderzine.com/wonderzine/health/wel
lness/242771-progeriaдетская
29.
30.
• Соматические мутации в специфических генах,участвующих в контроле клеточного деления,
дифференцировки, пролиферации, являются
причиной развития многих онкологических
заболеваний.
• Эти гены делятся на два класса: доминантные и
рецессивные онкогены.
• Таким образом, рак – это болезнь генов,
обусловленная мутациями, чаще всего
возникающими de novo в тех соматических клетках
и тканях.
31.
Основная сложность состоит в понимании генетических основмультифакториальных заболеваний, к которым относится
большинство распространенных болезней человека, в том
числе артериальная гипертония, коронарный атеросклероз,
сердечная недостаточность, сахарный диабет и т.д.
Моногенные болезни
Мутации
дефект (мутация) одного гена, в силу
особенностей его участия в
метаболических процессах
организма, вызывает заболевание
Мультфакториальные болезни
Полиморфизмы
Распространенные
генетические варианты
(частота >1%)
•Участвует множество генов
•Факторы внешней среды
•Межгенные взаимодействия
32.
Нарушение структуры хромосомымежхромосомные, изохромосомные
33.
Большинство мутаций летальны.Патологический кариотип характерен для всех
клеток.
Если мутации во время органогенеза- мозаицизм
34.
Механизмы возникновенияхромосомного мозаицизма
КОРРЕКЦИЯ ТРИСОМИИ
(анеуплоидная зигота)
МИТОТИЧЕСКИЕ ОШИБКИ
(эуплоидная зигота)
47 47
46 46
47 47
46 46
47 4747
47 47
47
47
46 48
47
47
47
Нерасхождение
47 4747
47 47
47
47
46 47
47
47
47
Анафазное
отставание
46 4646
46 46
46
46
45 47
46
46
46
Нерасхождение
46 4646
46 46
46
46
46 45
46
46
46
Анафазное
отставание
35.
Аутосомные – в аутосомах (неполовыххромосомах)
Сцепленные с Х-хромосомой
Сцепленные с Y-хромосомой
36.
Моносомии, ХОПолисомии , ХХХ, синдром Дауна
Анеуплоидии,
Полиплоидии (1п, 3п, 4п).
Геномные – изменение числа хромосом
. Полиплоидии (изменение числа
хромосом в кариотипе кратное
гаплоидному набору(1п, 3п, 4п)) и
гетероплоиди
37.
Генотип 47, ХХ / ХУ, 21+38.
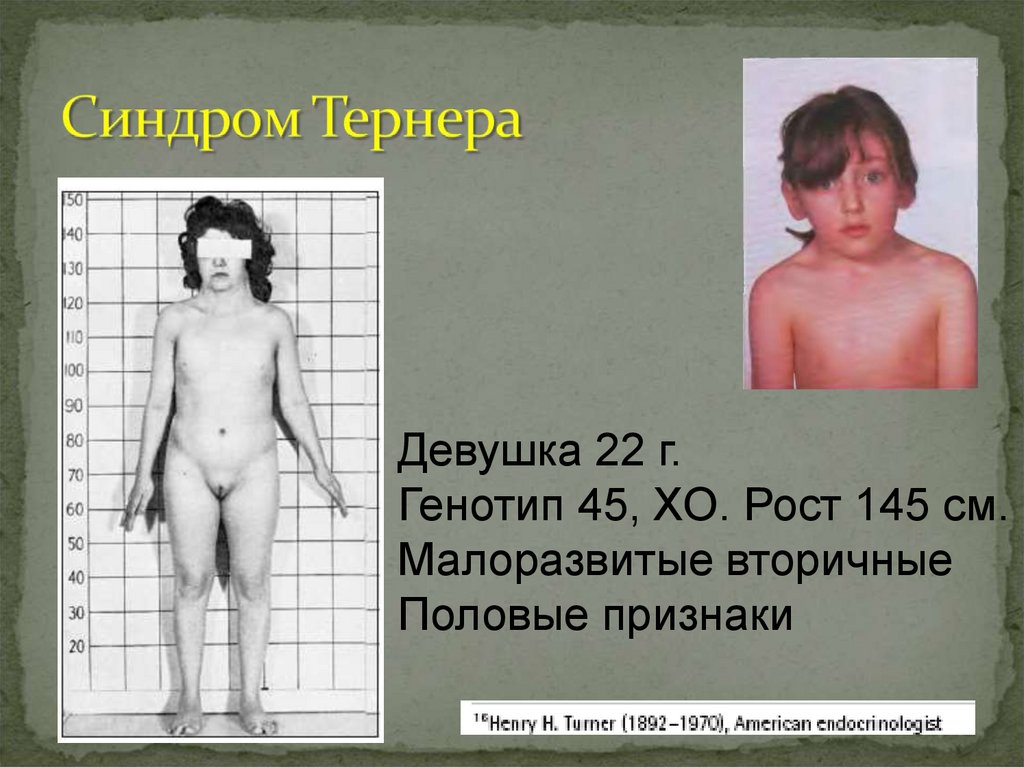
Девушка 22 г.Генотип 45, ХО. Рост 145 см.
Малоразвитые вторичные
Половые признаки
39.
Синдром Кляйнефелтера (ХХУ, ХХХУ, ХХХХУ) 2-2,5случая на 1000.
Трисомия Х (ХХХ) 1:1000.
Синдром Шерешевского- Тернера (ХО) 1:3000.
40.
41.
ЯдерныеЦитоплазматические (немногочисленные, но
тяжелые митохондриальные болезни)
Митохондрии имеют
свою кольцевую ДНК
42.
Происхождение и дифференцировка эмбриональныхи внезародышевых структур в раннем эмбриогенезе человека
Инвазивный трофобласт
Зигота
Якорный трофобласт
Синцитиотрофобласт
Морула
Трофобласт
Цитотрофобласт
Плацента
Хорион
Цитотрофобласт
Экстраэмбриональная
мезодерма
Эмбриональная
эктодерма
Эмбриональная
эктодерма
Эмбриональная
мезодерма
Эпибласт
Бластоциста
Эмбриональная
эндодерма
Эмбриональная
эндодерма
Эмбриобласт
Экстраэмбриональная
эктодерма
Гипобласт
Экстраэмбриональная
эндодерма
Эктодерма
амниона
Эндодерма
желточного мешка
Амнион
Желточный
мешок
43.
ГАМЕТОПАТИИФАКТОР
Заболевание или
ДЕЙСТВУЮЩИЙ НА патологическое
ПОЛОВЫЕ КЛЕТКИ
состояние
Бластопатии
1-15
после Сросшие
оплодотворения
Циклопия
Эмбриопатии
С 16 дней до 8 недель Талитомидные,
берем
диабетические,
алкогольные,
медикаментозные,
вирус краснухи.
Фетопатии
От 9 недель берем до Крипторхизм, открытый
рождения
боталлов
проток
гипоплазия органа
близнецы
44.
Формы наследственной патологииЧастота на 1000
1. МОНОГЕННЫЕ
4.5 – 15.0
Аутосомно-доминантные
2.0 – 9.5
Аутосомно-рецессивные
2.0 – 3.5
Х-сцепленные
0.5 – 15.0
2. ХРОМОСОМНЫЕ
5.0 – 7.0
3. МУЛЬТИФАКТОРИАЛЬНЫЕ
7.0 – 10.0
4. ВРОЖДЕННЫЕ ПОРОКИ РАЗВИТИЯ 19.0 – 22.0
45.
Уродства – дефекты морфогенеза, наиболеетяжелые проявления ВПР
Дисплазии – деформации, морфологические
врожденные изменения, выходящие за рамки
общепринятых норм
Малые аномалии развития – стигмы
дизэмбриогенеза: синдактилия, ямочки на
щеках, искривление мизинца, дефекты, не
требующие медицинского вмешательства
Клинически значимые пороки развития –
врожденные аномалии требующие
медицинского вмешательства
46.
ЛС, алкоголь, марихуана, героин, кокаин, гипертемия,вещества, загрязняющие окружающую среду,
инфекционные заболевания:
Вирус краснухи
1 мес – 50% ВПР
2 мес – 22%
3-4 мес – 6-10%
ЦМВ
1-2% случаев всех беременностей
ВПГ II типа
< чем в 0,02% случаев, ВПР очень редко в
I триместре- смерть плода.
Токсоплазма
30% всех инфицированных беременных