



















 Медицина
МедицинаПохожие презентации:










Cystic fibrosis
1.
CYSTIC FIBROSISPRESENTED BY :- HARSHIT SHUKLA
PRESENTED TO :- SVETLANA
SMIRNOVA
SUBJECT :- MEDICAL BIOLOGY
2.
CYSTIC FIBROSIS3.
CYSTIC FIBROSISCystic fibrosis is an inherited life-threatening disorder that
damages the lungs and digestive system.
Cystic fibrosis affects the cells that produce mucus,
sweat and digestive juices. It causes these fluids to
become thick and sticky. They then plug up tubes, ducts
and passageways.
4.
Cystic fibrosis affects the cells that produce sweat, mucus, anddigestive enzymes. Normally, these secreted fluids are thin and
smooth like olive oil. They lubricate various organs and tissues,
preventing them from getting too dry or infected.
In people with cystic fibrosis, however, a faulty gene causes the
fluids to become thick and sticky. Instead of acting as a lubricant,
the fluids clog the ducts, tubes, and passageways in the body.
5.
Symtoms :Symptoms vary and can include cough, repeated lunginfections, inability to gain weight and fatty stools.
6.
Symptoms may appear at infancy, but for otherchildren, symptoms may not begin until after puberty
or even later in life. As time passes, the symptoms
associated with the disease may get better or worse.
One of the first signs of cystic fibrosis is a strong salty
taste to the skin. Parents of children with cystic
fibrosis have mentioned tasting this saltiness when
kissing their children.
Other symptoms of cystic fibrosis result from
complications that affect:
the lungs
the pancreas
the liver
other glandular organs
7.
Respiratory problemsThe thick, sticky mucus associated with cystic fibrosis often blocks
the passageways that carry air into and out of the lungs. This can
cause the following symptoms-
wheezing
a persistent cough that produces thick mucus or phlegm
shortness of breath, especially when exercising
recurrent lung infections
a stuffy nose
stuffy sinuses
8.
Digestiveproblems
The abnormal mucus can also plug up the channels that carry the
enzymes produced by the pancreas to the small intestine. Without
these digestive enzymes, the intestine can’t absorb the necessary
nutrients from food. This can result in greasy, foul-smelling stools
constipation
nausea
a swollen abdomen
loss of appetite
poor weight gain in children
delayed growth in children
9.
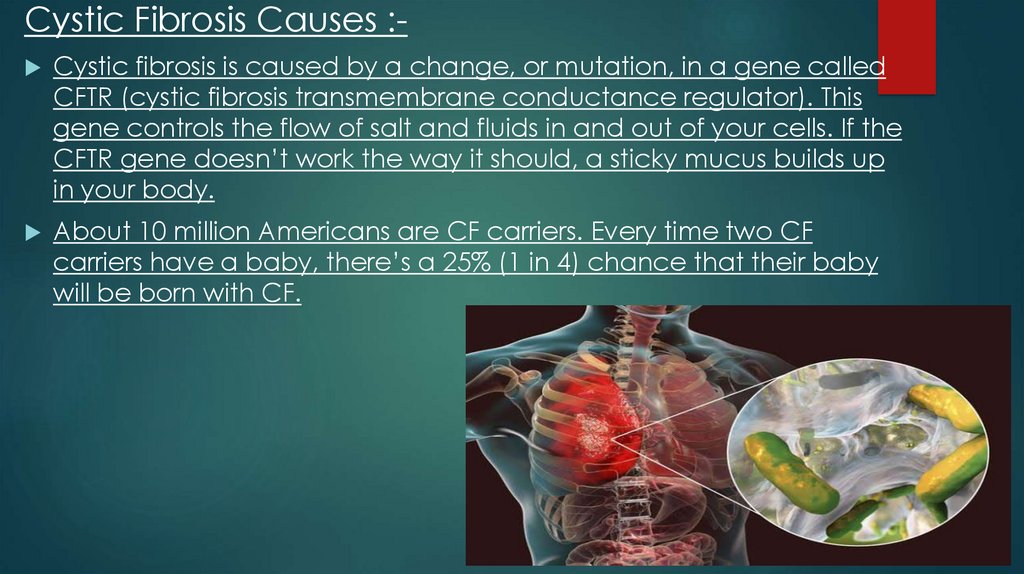
Cystic Fibrosis Causes :Cystic fibrosis is caused by a change, or mutation, in a gene called
CFTR (cystic fibrosis transmembrane conductance regulator). This
gene controls the flow of salt and fluids in and out of your cells. If the
CFTR gene doesn’t work the way it should, a sticky mucus builds up
in your body.
About 10 million Americans are CF carriers. Every time two CF
carriers have a baby, there’s a 25% (1 in 4) chance that their baby
will be born with CF.
10.
Cystic fibrosis occurs as a result of a defect in what’s called the“cystic fibrosis transmembrane conductance regulator” gene, or
CFTR gene. This gene controls the movement of water and salt in
and out of your body’s cells.
A sudden mutation, or change, in the CFTR gene causes your mucus
to become thicker and stickier than it’s supposed to be. This
abnormal mucus builds up in various organs throughout the body,
including the:
intestines
pancreas
liver
lungs
11.
It also increases the amount of salt in your sweat.Many different defects can affect the CFTR gene. The type of
defect is associated with the severity of cystic fibrosis. The damaged
gene is passed on to the child from their parents.
In order to have cystic fibrosis, a child must inherit one copy of the
gene from each parent.
If they only inherit one copy of the gene, they won’t develop the
disease. However, they’ll be a carrier of the defective gene, which
means that they may pass the gene on to their own children.
12.
Cystic Fibrosis Diagnosis :Early diagnosis means early treatment and better healthlater in life. Tests newborns for cystic fibrosis using one or
more of these three tests:
Blood test :- This test checks the levels of
immunoreactive trypsinogen (IRT). People with CF have
higher levels of it in their blood.
DNA test :- This looks for mutations to the CFTR gene.
Sweat test :- It measures the salt in your sweat.
Higher than normal results suggest CF.
13.
Approximately 1,000 people are diagnosed with cystic fibrosisevery year in the United States. Although people with the
condition require daily care, they can still lead a relatively
normal life and work or attend school.
Screening tests and treatment methods have improved in recent
years, so many people with cystic fibrosis can now live into their
40s and 50s.
14.
Other diagnostic tests that may be performed include:1:- Immunoreactive trypsinogen (IRT) test
2:- Sweat chloride test
3:- Sputum test
4:- Chest X-ray
5:- CT scan
6:- Pulmonary function tests (PFTs)
15.
Treatment :Although there’s no cure for cystic fibrosis, there are various treatments available that may
help relieve symptoms and reduce the risk of complications.
Medication :- Antibiotics may be prescribed to get rid of a lung infection and to prevent
another
infection.. In more severe cases, injections or infusions of antibiotics can be given intravenously
(through a vein).
Mucus-thinning medications make the mucus thinner and less sticky. They also help you to
cough up the mucus so it leaves the lungs. This significantly improves lung function
Surgical procedures:-
Bowel surgery :- This is an emergency surgery that involves the removal of a
section of the bowel. It may be performed to relieve a blockage in the
bowels.
16.
Feeding tube :- Cystic fibrosis may interfere with digestion andprevent the absorption of nutrients from food. A feeding tube to
supply nutrition can be passed through the nose or surgically
inserted directly into the stomach.
Double-lung transplant :- When medical management
alone can no longer maintain lung health and physical function, this
procedure can improve the length and quality of life for a person
with cystic fibrosis.
17.
Transplant benefits :Most transplant recipients report improved strength and energy, and freedom from symptoms
such as coughing and shortness of breath
While having a double-lung transplant can’t cure cystic fibrosis, as the defective gene
remains in the body, the donor lungs don’t contain the mutated gene
Home care :
Cystic fibrosis can prevent the intestines from absorbing necessary nutrients from food
If you have cystic fibrosis, you might need more calories per day than people who don’t the
disease. You may also need to take pancreatic enzyme capsules with every meal.
Drink plenty of fluids, because they can help thin the mucus in the lungs.
Exercise regularly to help loosen mucus in the airways. Walking, biking, and swimming are
great options.
Avoid smoke, pollen, and mold whenever possible. These irritants can make symptoms worse.
Get influenza and pneumonia vaccinations regularly.
18.
The long-term outlook for people with cystic fibrosis :The outlook for people with cystic fibrosis has improved dramatically in recent years, largelydue to advances in treatment. Today, many people with the disease live into their 40s and
50s, and even longer in some cases.
However, there’s no cure for cystic fibrosis, so lung function will steadily decline over time.
The resulting damage to the lungs can cause severe breathing problems and other
complications.
How can cystic fibrosis be prevented?
Cystic fibrosis can’t be prevented. However, genetic testing should be performed for
couples who have cystic fibrosis or who have relatives with the disease.
Genetic testing can determine a child’s risk for cystic fibrosis by testing samples of blood or
saliva from each parent. Tests can also be performed on you if you’re pregnant and
concerned about your baby’s risk.
19.
OVERVIEW OF DISEASE20.
Questions :1:- What is cystic fibrosis ?
2:- What is the symroms of cystic fibrosis ?
3:- How is cystic Fibrosis diagnosed ?
4:- What is the treatment of cystic fibrosis