
























































 Медицина
МедицинаПохожие презентации:



")






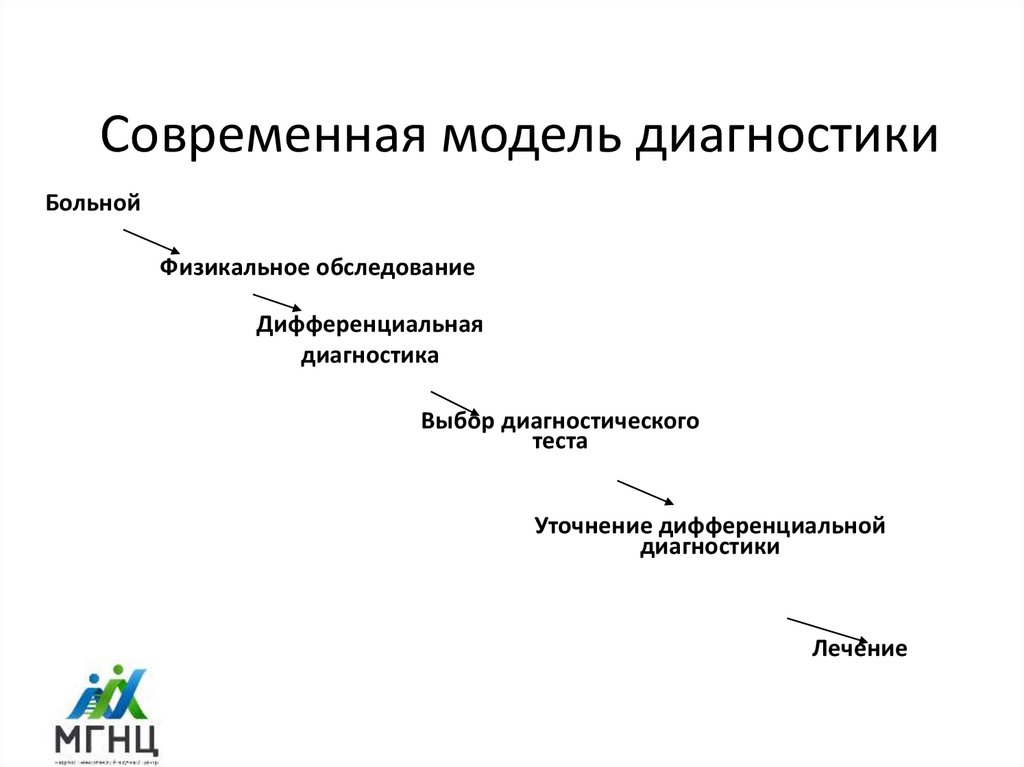
Современная модель диагностики
1.
2.
Современная модель диагностикиБольной
Физикальное обследование
Дифференциальная
диагностика
Выбор диагностического
теста
Уточнение дифференциальной
диагностики
Лечение
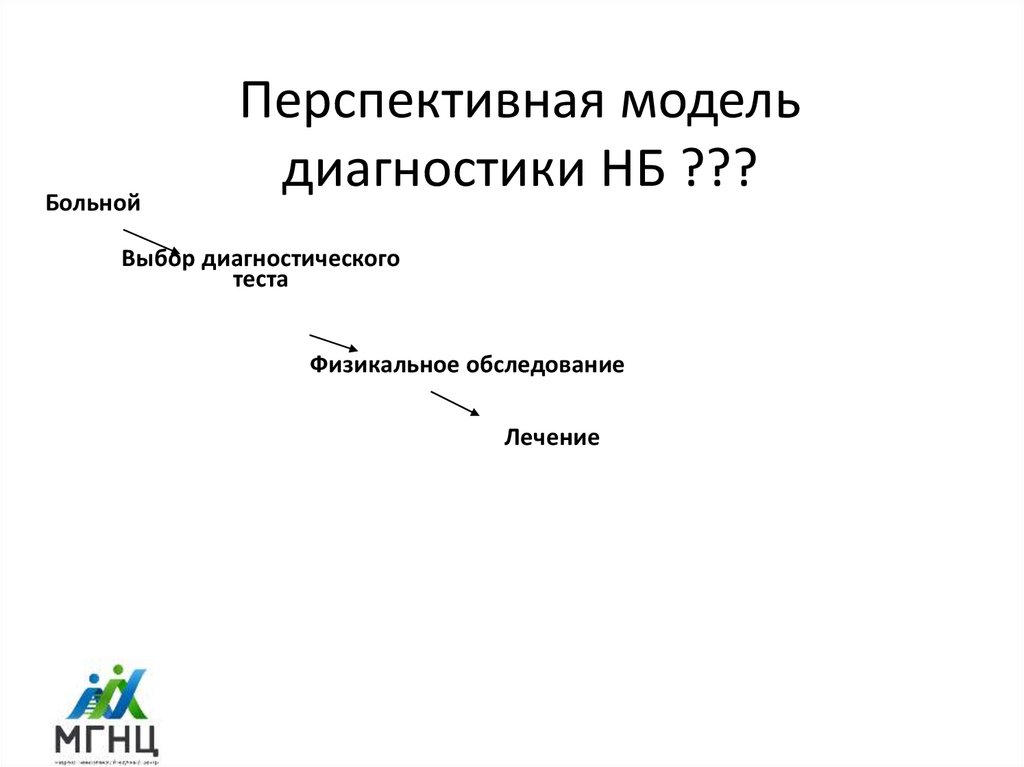
3.
БольнойПерспективная модель
диагностики НБ ???
Выбор диагностического
теста
Физикальное обследование
Лечение
4.
Скрининг – это услуга в области здравоохранения, состоящая в том,что представителям конкретной популяции, которые не
обязательно понимают, что они подвержены риску заболевания или
уже страдают болезнью либо ее осложнениями, предлагается тест
для выявления лиц, которым с большей вероятностью будет
оказана помощь, а не причинен вред, дальнейшими тестами или
лечением с целью снижения риска заболевания или его
осложнений.
Holland W. W., Stewart S., Masseria C.,2008. Screening in Europe. Policy brief. WHO Regional Office for Europe
5.
Критерии скринингаМГНЦ
Holland W. W., Stewart S., Masseria C.,2008. Screening in Europe. Policy brief. WHO Regional Office for Europe
6.
Критерии теста для скринингаCochrane and Holland, 1971
7.
Регламентирующие документыпо неонатальному скринингу и РНС
8.
9.
ДиагностикаПоиск редкого заболевания у
больных с «частыми»
заболеваниями, когда симптомы не
полностью укладываются в клинику ,
в нетипичном возрасте, когда не
удается подобрать лечение, и
заболевание прогрессирует,
несмотря на проводимое лечение
10.
МалоинвазивныйВысокочувствительный
Высокоспецифичный
Быстрый
Один тест = много заболеваний
11.
Масс-спектрометрия. Основы понятия.Масс-спектрометрия (МС)— это физикохимический метод, определения
химической структуры образца или
молекул путем их ионизации с
образованием заряженных молекул или их
фрагментов с последующим измерением
отношения их массы к заряду (m/z)
МС обладает высочайшими
специфичностью и точностью, может
определить структурные компоненты в
концентрации 10-15,10-18, 10-21 грамма
Тандемный масс-спектрометр (ТМС) – это
два масс-спектрометра последовательно
соединенных через «камеру соударений».
12.
Диагностические возможности тандемноймасс-спектрометрии при неонатальном
скрининге
ТМС позволяет количественно определять
большое количество метаболитов,
являющихся маркерами наследственных
болезней обмена (НБО).
ТМС применяется для диагностики трех
основных групп НБО (нарушений обмена
аминокислот, органических кислот и
дефектов митохондриального β-окисления
жирных кислот).
ТМС является первым этапом диагностики
НБО.
ТМС должна использовать в комплексе с
подтверждающими методами диагностики
(ДНК-диагностика , исследование
органических кислот мочи.
13.
№Метаболит Условное обозна- чение
№
Метаболит Условное
обозначение
Аминокислоты
1.
Аланин
Ala
21.
3-метилкротонилкарнитин С5:1
2.
Аргинин
Arg
22.
3-гидроксиизовалерилкарнитин
С50Н
3.
Сукцинилацетон
Sa
23.
Г ексаноилкарнитин
С6
4.
Лейцин
Leu
24.
Октаноилкарнитин
С8
5.
Метионин Met
25.
Октеноилкарнитин
С8:1
6.
Фенилаланин
Phe
26.
Деканоилкарнитин
С10
7.
Тирозин
Туг
27.
Деценоилкарнитин
С10:1
8.
Валин
Val
28.
Додеканоилкарнитин
С12
9.
Пролин
Pro
29.
Миристилкарнитин
С14
10.
Цитруллин Cit
30.
Тетрадеценоилкарнитин С 14:1
11.
Глицин
Gly
31.
Тетрадекадиеноилкарнитин
С14:2
12.
Орнитин
Orn
32.
Гидроксимиристилкарнитин
С140Н
Ацилкарнитины
33.
Пальмитоилкарнитин
С16
13.
Свободный карнитин
CO
34.
Гексадеценоилкарнитин С16:1
14.
Ацетилкарнитин
C2
35.
Гидроксигексадеценоилкарнитин
С16:10Н
15.
Пропионилкарнитин
C3
36.
Г идроксипальмитоилкарнитин
С160Н
16.
17.
18.
Малонилкарнитин
Бутирилкарнитин
Метилмалонилкарнитин
C3DC
C4
C4DC
37.
38.
39.
Стеароилкарнитин
С18
Олеоилкарнитин
С18:1
Г идроксистеароилкарнитин
С180Н
19.
Изовалерилкарнитин
C5
40.
Г идроксиолеоилкарнитин С18:10Н
20.
Глутарикарнитин
C5DC
41.
Г идроксилиноилкарнитин С18:20Н
14.
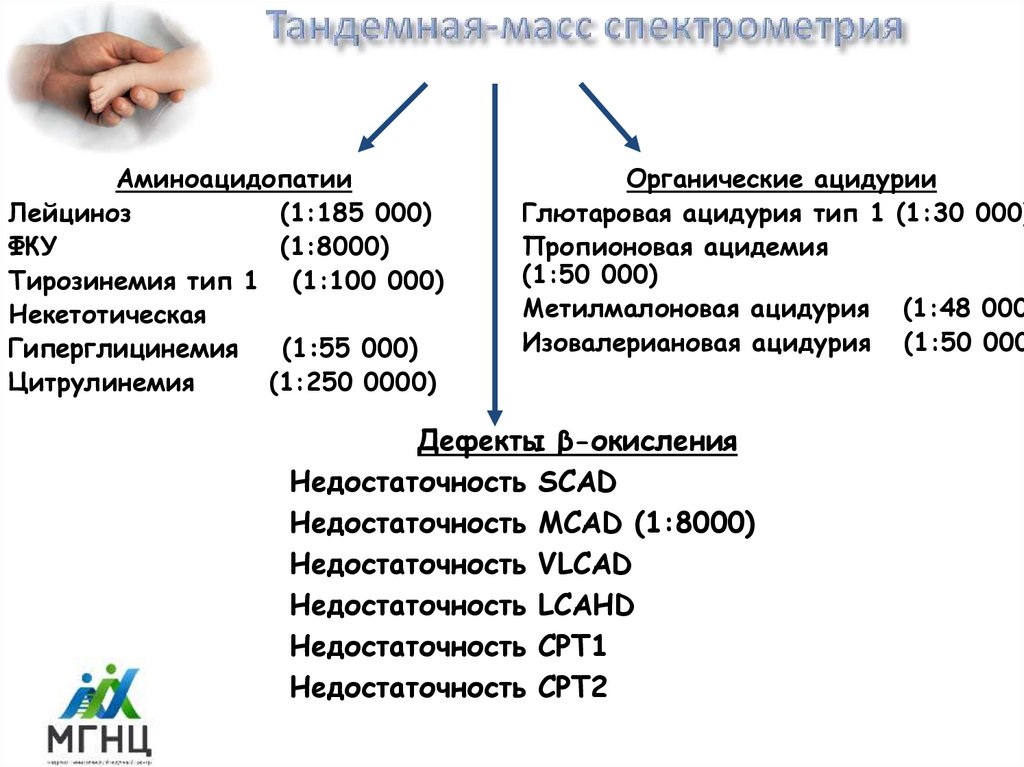
АминоацидопатииЛейциноз
(1:185 000)
ФКУ
(1:8000)
Тирозинемия тип 1 (1:100 000)
Некетотическая
Гиперглицинемия
(1:55 000)
Цитрулинемия
(1:250 0000)
Органические ацидурии
Глютаровая ацидурия тип 1 (1:30 000)
Пропионовая ацидемия
(1:50 000)
Метилмалоновая ацидурия (1:48 000
Изовалериановая ацидурия (1:50 000
Дефекты β-окисления
Недостаточность SCAD
Недостаточность MCAD (1:8000)
Недостаточность VLCAD
Недостаточность LCAHD
Недостаточность CPT1
Недостаточность CPT2
15.
Что такое ацилкарнитины?Это производные L-карнитина, природного
вещества, родственного витаминам группы
В. Карнитин синтезируется в организме,
также его называют витаминоподобным
веществом.
• Ацилкарнитины - это эфиры карнитина и
жирных кислот, которые обладают
способностью проникать внутрь
митохондрии. Сами жирные кислоты
различают по длине углеродной цепи.
Существуют короткоцепочечные,
среднецепочечные и длинноцепочечные
жирные кислоты. Отсюда и ацилкарнитины
делятся на короткоцепочечные
(C2,C3,C4,C5,C5:1) среднецепочечные (C6C12)и длинноцепочечные (C14-C18).
Нарущения обмена органических кислот и
окисления жирных кислот сопровождаются
изменениями концентрации тех или иных
ацилкарнитинов.
16.
Диагностические возможности тандемноймасс-спектрометрии при неонатальном
скрининге
I. Нарушения метаболизма аминокислот:
аминоацидопатии (AAD):
Фенилкетонурия (PKU);
Цитруллинемия (CIT);
Гомоцистинурия (классическая HCY);
Тирозинемия (TYRI);
Недостаточность
орнитинтранскарбамилазы;
Недостаточность аргининосукциназы,
аргиназы
Болезнь c запахом кленового сиропа мочи
(MSUD)
17.
Диагностические возможности тандемноймасс-спектрометрии при неонатальном
скрининге
II. Нарушения митохондриального
β-окисления жирных кислот (FAO):
Дефицит дегидрогеназы Ас-CoA средних
цепей (MCAD);
Дефицит дегидрогеназы Ас-CoA коротких
цепей (SCAD);
Дефицит дегидрогеназы Ас-CoA очень
длинных цепей (VLCAD);
Дефицит карнитинтрансферазы
пальмитиновой кислоты I (CPT I).
•Дефицит карнитинтрансферазы
пальмитиновой кислоты II (CPT II)
18.
Диагностические возможности тандемноймасс-спектрометрии при неонатальном
скрининге
III. Нарушения метаболизма органических
кислот (OAD):
Глутаровая ацидемия I типа (GA I);
Пропионовая ацидемия (PA);
Метилмалоновая ацидемия (MA);
Изовалериановая ацидемия (IVA);
Недостаточность биотинидазы
19.
Критерии отбора пациентов для проведения селективного скринингаметодом ТМС
Прогрессирующее течение заболевания Неврологические симптомы
· Задержка психомоторного развития · Утрата приобретенных
навыков · Судороги, резистентные к базовой терапии · Мышечная
гипотония/гипертония · Снижение зрения · Снижение слуха ·
Умственная отсталость · Коматозные состояния Экстраневральные
симптомы · Задержка физического развития · Срыгивание, рвота ·
Необычный запаха тела и/или мочи («сладкого», «мышиного»,
«вареной капусты», «запаха потных ног» и др.). · Желтуха неясного
генеза · Асцит · Нарушения со стороны других органов (поражение
печени, гепатоспленомегалия, кардиомиопатия, катаракта,
ретинопатия). · Метаболический алкалоз/ацидоз · Нарушения
дыхания (брадипноэ, тахипноэ, апноэ, прерывистое дыхание ,
икота). · Алопеция · Гипергликемии/гипогликемии неясного генеза
20.
Болезнь с запахом кленового сиропа мочи• Болезнь с запахом кленового сиропа мочи
– редкое генетически-гетерогенное
аутосомно-рецессивное заболевание,
обусловленное нарушением метаболизма
разветвлённых аминокислот
• МКБ 10, Е 71.0
• Частота встречаемости 1 :185 000 живых
новорожденных
21.
Этиология и патогенезMSUD - метаболическое заболевание вызванное
недостаточностью дегидрогеназного комплекса альфакетокислот с разветвленной цепью (BCKDC) приводящее к
накоплению аминокислот с разветвленной цепью (лейцин,
изолейцин и валин ) и их токсичных побочных продуктов
(кетокислот) в крови и моче. Ферментный комплекс состоит из
4 субъединиц Е1а, Е1в, Е и Е3. Субъединица Е3 также входит в
состав пируват-дегидрогеназного комплекса и оксоглутарат
дегидрогеназного комплекса.
Тип наследования актосомно-рецессивный.
22.
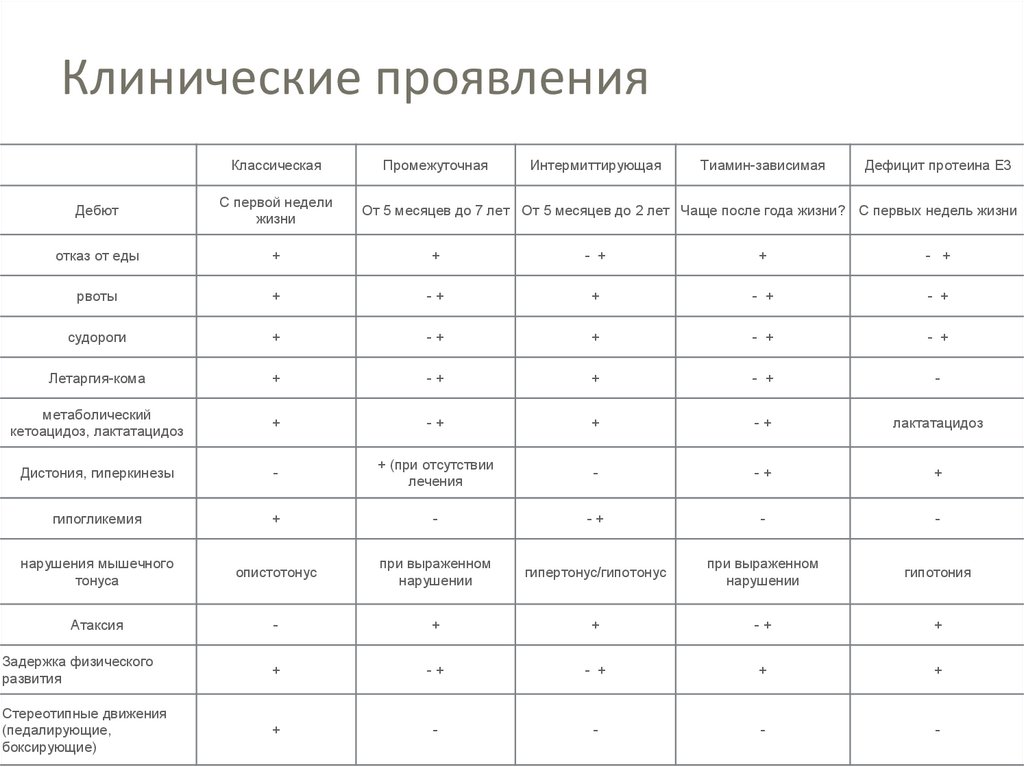
КлассификацияКлассическая, острая или тяжелая форма
Промежуточная
Интермиттирующая
Тиамин-зависимая
Обусловленная дефицитом Е3-протеина и сопровождающаяся
лактат-ацидозом
23.
Клинические проявленияКлассическая
Промежуточная
Интермиттирующая
Тиамин-зависимая
Дефицит протеина Е3
Дебют
С первой недели
жизни
отказ от еды
+
+
- +
+
- +
рвоты
+
-+
+
- +
- +
судороги
+
-+
+
- +
- +
Летаргия-кома
+
-+
+
- +
-
метаболический
кетоацидоз, лактатацидоз
+
-+
+
-+
лактатацидоз
Дистония, гиперкинезы
-
+ (при отсутствии
лечения
-
-+
+
гипогликемия
+
-
-+
-
-
нарушения мышечного
тонуса
опистотонус
при выраженном
нарушении
гипертонус/гипотонус
при выраженном
нарушении
гипотония
Атаксия
-
+
+
-+
+
Задержка физического
развития
+
-+
- +
+
+
Стереотипные движения
(педалирующие,
боксирующие)
+
-
-
-
-
От 5 месяцев до 7 лет От 5 месяцев до 2 лет Чаще после года жизни? С первых недель жизни
24.
ДиагностикаМРТ головного мозга может показать диффузный отек головного мозга: заднее бедро
внутренних капсул, зрительная лучистость, кортикоспинальный путь, иногда ствол мозга и
белое вещество мозжечка представлены диффузным отеком и диффузной миелопатией.
МР-спектроскопия: может показать наличие аминокислот с разветвленной цепью (ВСАА) и
с разветвленной цепью альфа-кетокислоты (BCKA), особенно во время метаболического
кризиса
ТМС и моча на органические кислоты: повышение уровня лейцина, изолейцина, валина,2гидрокси-3-метилвалериановая, 2-гидрокси-изокапроновая, 2-гидрокси-изовалериановая,
2-кето-3-метилвалериановая, 2-кето-изовалериановая.
Генетическая диагностика: BCKDHA (MSUD тип 1A); BCKDHB (MSUD тип 1B); DBT (MSUD тип
2); DLD (MSUD тип 3),
25.
Основы леченияВысококалорийная диета с ограничением лецина, изолейнина, валина
(доза лейцина после 3-4месяцев составляет 40-50мг\кг).
Кофакторная терапия. При установлении диагноза болезни кленового
сиропа для дифференцирования с тиамин-зависимой формой заболевания
рекомендуется осуществлять пробное лечение тиамином, который
назначается в дозе 10 мг/кг/сут сроком на 2 недели.
Терапия левокарнитином. С целью усиления связывания метаболитов
аминокислот с разветвленной цепью больным на длительный срок (напр.,
на 3-6 мес.) назначают карнитин из расчета 50-75 мг/кг/сут (в зависимости
от возраста) за 2-3 приема.
Хирургическая операция - трансплантация печени
26.
Патогенетическая терапия согласностандартам лечения данной
патологии:
1.Диетотерапия – ограничение
белка до 1,5 мг/кг/сутки (MSUD
Анамикс инфант)
2. Тиамина в дозе 85 мг в сутки
3.Левокарнитин 100 мг/кг/сутки
4.Посиндромная терапия
27.
Аминоацидопатии (аминоацидурии) –наследственные патологические состояния,
характеризующиеся повышенной экскрецией
аминокислот и/или их метаболитов с мочой,
а также выраженным аминокислотным
дисбалансом в крови
28.
Наследственная тирозинемия – наследственное нарушениеобмена,
обусловленное
ферментативной
недостаточностью.
Выделяют 3 типа тирозинемии:
I.Наследственная тирозинемия I типа- гепаторенальная
тирозинемия; обусловленная дефицитом
фумарилацетоацетазы, дефицитом
фумарилацетоацетатгидролазы. OMIM 276700)
II.Тирозинемия II типа, с. Ричнера- Ханхарта, дефект
тирозиновой аминотрансферазы; OMIM 276600
III.Тирозинемия III типа-редкое заболевание,
обусловленное дефицитом фермента 4 –
гидрооксифенилпируватдиоксигеназы, OMIM 276710;
29.
Метилмалоновая ацидурия (ММА)• Метилмалоновая
ацидурияредкое
наследственное
генетически гетерогенное заболевание, связанное с нарушение
обмена метилмалоновой кислоты или кобаламина.
• КОД МКБ-10, Е71.1
• Частота тяжелых форм 1:50 000 живых новорожденных, из них
2/3- недостаточность метилмалонил КоА мутазы, 1/3- связаны с
нарушением обмена кобаламина.
• Две клинические формы: В12 чувствительная и В12 резистентная
форма
• Известно несколько генетических вариантов: 1). в зависимости
от остаточной активности фермента метилмалонил КоА мутазы,
2) связанные с нарушением метаболизма кобаламина.
30.
Общие клинические проявления• 80% манифестируют с рождения (классическая форма)
• Провоцирующие факторы ( ОРВИ, вакцинация, хирургические
вмешательства, прием большого количества белка)
• Выраженный метаболический кетоацидоз
• Вторичная гипераммонемия
• Гипогликемия
• Анемия, тромбоцитопения, нейтропения
• Угнетение сознания (сопор, кома)
• Эпилептические приступы
• Гиперкинетические расстройства
• Нарушение психомоторного развития
• Инсультоподобные эпизоды
31.
Диагностика• МРТ головного мозга повышение интенсивности
сигнала в проекции бледных шаров
• Методом тандемной масс спектрометрии
определение концентрации в крови пропионил
(С3) и метилмалонил (С4) карнитинов.
• Методом
хромато-масс-спектрометрии
определение концентрации метилмалоновой,
пропионовой
3
гидрокси-пропионовой,
метиллимонной кислот
• ДНК диагностика
32.
Пренатальная диагностикаПренатальная диагностики возможна путем
определения активности фермента
метилмалонил-КоА- мутазы в культуре
амниоцитов, измерения концентрации
метилмалоновой кислолты в
амниотической жидкости, а также ДНК
диагностика , если генотип пробалда
известен.
33.
Принципы лечения• Низкобелковая диета из расчета белка 1-2
грамм на кг в сутки
• Назначение специализированных смесей
• Коррекция кетоацидоза
• Поддержание нормального нутритивного
статуса
34.
Принципы лечения• Назначение левокарнитина и глицина для
усиления связывания токсичного пропионилрадикала;
• Коррекция вторичной карнитиновой
недостаточности;
• Кофакторная терапия витамином В12
(гидроксикобаламин/цианокобаламин)
• Исключение голодания
• Предотвращение развития ацидоза
• Усиление терапии в период метаболического
криза
35.
Антибактериальная терапияС целью снижения уровня пропионатов в
кишечнике показаны курсы
антибактериальных препаратов:
ампициллин в возрастной дозе в течение 810 дней или метронидазол в дозе 10-15
мг/кг/сут в течение 10-14 дней; через 3-4
недели курс повторяют
36.
Хирургическое лечениеВ ряде случаев (при тяжелом поражении
почек, плохо корректируемых
метаболических нарушениях) возникает
необходимость решать вопрос о
трансплантации печени и почек.
37.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА• Гипоксические поражения нервной системы
• Внутриутробные инфекции
• Поствакцинальные осложнения
• Энцефалит
• В12 зависимая мегалобластная анемия анемия
• Наследственные нарушениями обмена веществ, в
частности с другими формами органических
ацидурий
• Нарушения цикла мочевины.
38.
Диспансеризация• клинический анализ крови
• уровень гемоглобина
• уровень общего белка
• уровень альбумина
• уровень глюкозы
• уровень сывороточного железа
• уровень электролитов, лактата
• уровень аминокислот
• уровень свободного карнитина и
пропионилкарнитина.
• Уровень метилмалоновй кислоты в моче
39.
Специализированные продукты для диетическоголечебного питания для больных аминоацидопатиями и
органическими ацидуриями
• Имеют сбалансированный аминокислотный состав;
• Содержат заменимые и незаменимые аминокислоты,
без патогенетически значимых аминокислот
• По содержанию белкового эквивалента в составе
лечебного продукта, подходят больным разных
возрастных групп, начиная с рождения до взрослого
возраста, оно также показано беременным женщинам,
страдающим указанной патологией.
40.
Гомоцистинурия, обусловленная дефицитомфермента цистатионин - - синтазы, впервые
была описана в 1962 г. N.A. Carson и D.W. Neill
при обследовании группы больных с задержкой
умственного развития.
41.
Частота заболевания среди новорожденных поданным различных авторов колеблется от 1: 50 000 –
1: 250 000 до 1: 311 000.
Патология наследуется по аутосомно-рецессивному
типу.
Фермент цистатионин-β-синтазу кодирует ген CBS,
расположенный в области длинного плеча
хромосомы 21, в локусе q21-q22.3
42.
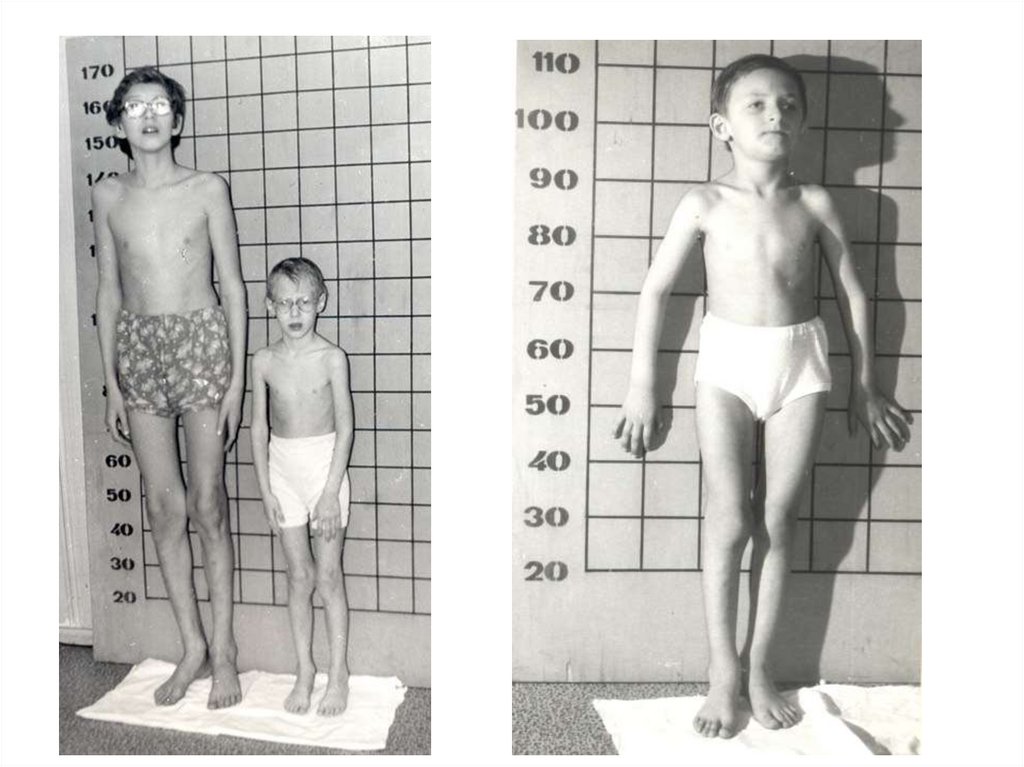
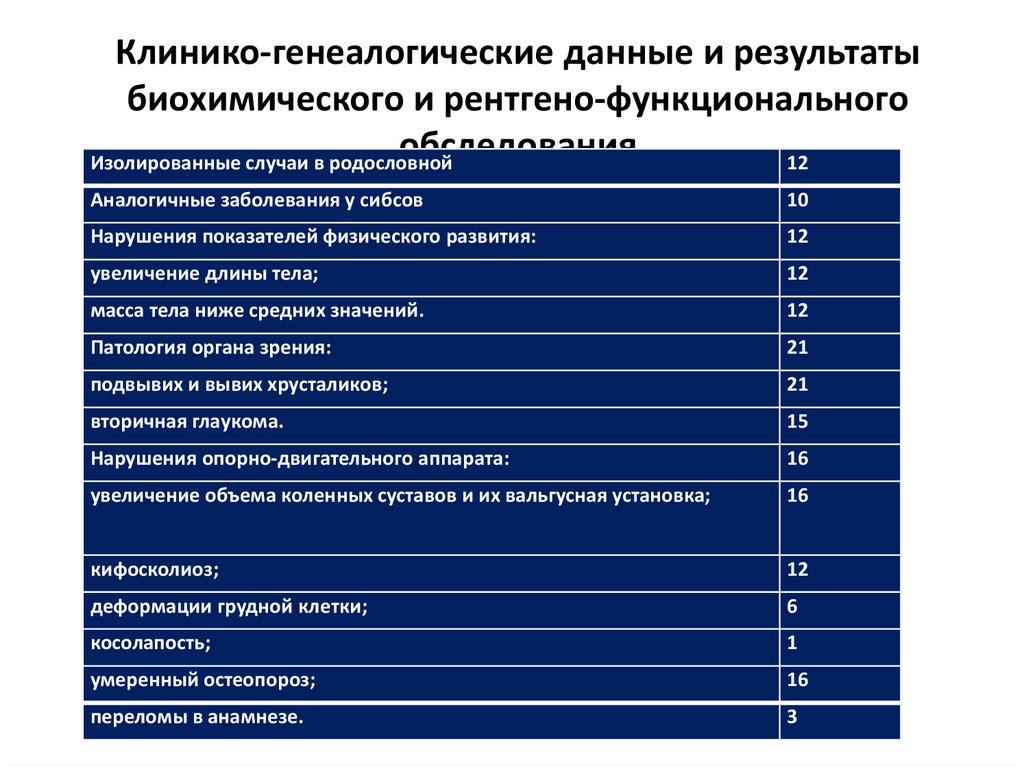
43.
Клинико-генеалогические данные и результатыбиохимического и рентгено-функционального
обследования
Изолированные случаи в родословной
12
Аналогичные заболевания у сибсов
10
Нарушения показателей физического развития:
12
увеличение длины тела;
12
масса тела ниже средних значений.
12
Патология органа зрения:
21
подвывих и вывих хрусталиков;
21
вторичная глаукома.
15
Нарушения опорно-двигательного аппарата:
16
увеличение объема коленных суставов и их вальгусная установка;
16
кифосколиоз;
12
деформации грудной клетки;
6
косолапость;
1
умеренный остеопороз;
16
переломы в анамнезе.
3
44.
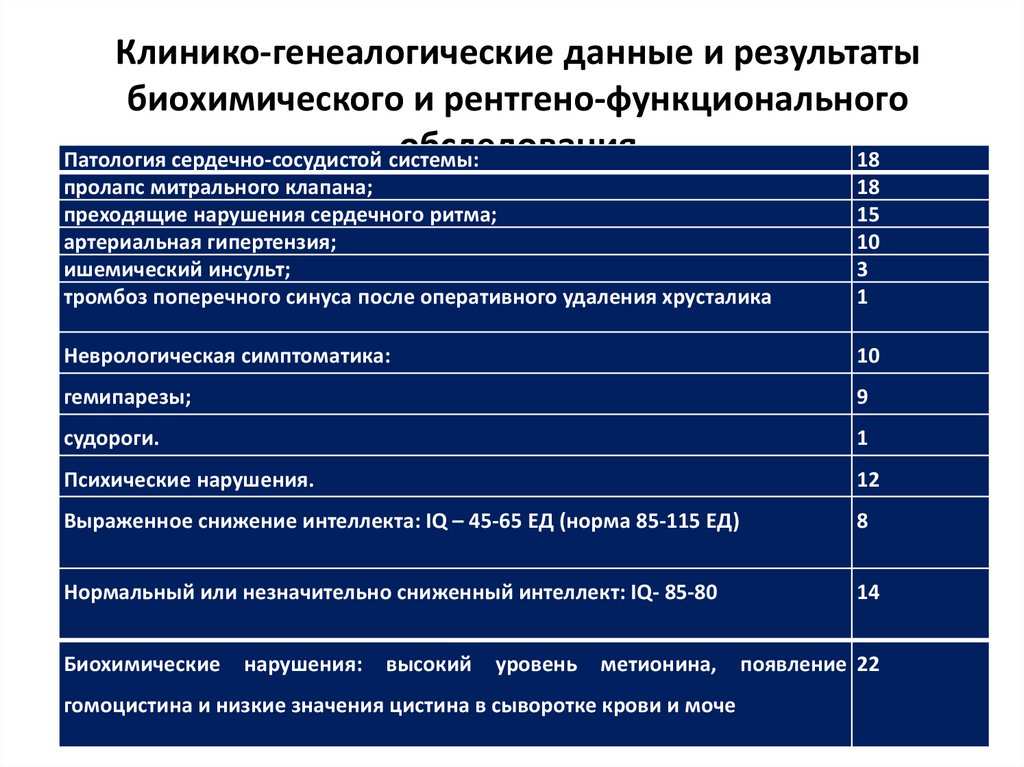
Клинико-генеалогические данные и результатыбиохимического и рентгено-функционального
обследования
Патология сердечно-сосудистой системы:
18
пролапс митрального клапана;
преходящие нарушения сердечного ритма;
артериальная гипертензия;
ишемический инсульт;
тромбоз поперечного синуса после оперативного удаления хрусталика
18
15
10
3
1
Неврологическая симптоматика:
10
гемипарезы;
9
судороги.
1
Психические нарушения.
12
Выраженное снижение интеллекта: IQ – 45-65 ЕД (норма 85-115 ЕД)
8
Нормальный или незначительно сниженный интеллект: IQ- 85-80
14
Биохимические
нарушения:
высокий
уровень
метионина,
гомоцистина и низкие значения цистина в сыворотке крови и моче
появление 22
45.
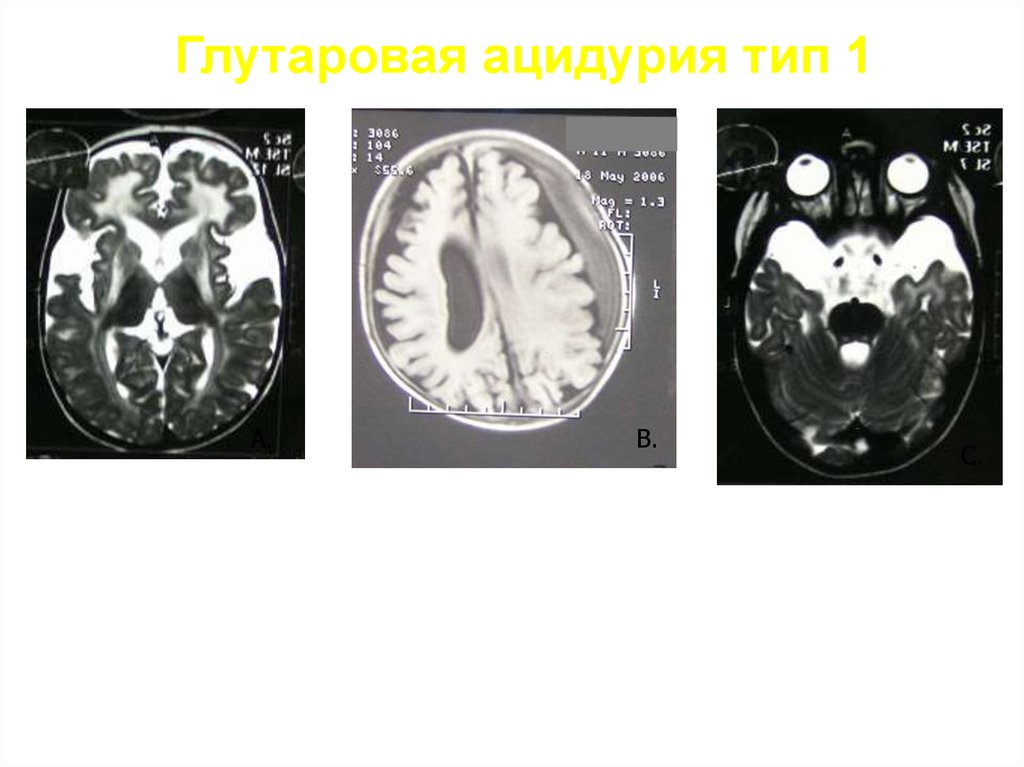
Глутаровая ацидурия тип 1Возраст манифестации 6-18 месяцев
Провоцирующие факторы: черепно-мозговая травма, инфекция,
вакцинация.
- Прогрессирующее течение заболевания
- Макроцефалия 75-80%
1) «Энцефалитоподобное» течение заболевания –
75-80% (судороги, неукротимая рвота, срыгивания, диффузная
мышечная гипотония, различные виды гиперкинезов, угнетение
сознания до сопора и комы, метаболический ацидоз)
Подострое течение заболевания - 20-25%: задержка двигательного
развития с первых месяцев жизни, различные виды гиперкинезов,
эпизоды немотивированной лихорадки, повышенная потливость.
Редко по типу синдрома Рейе- «энцефалитоподобные» кризы в
сочетании с поражением печени
46.
Глутаровая апцидурия тип 126 пациентов
Манифестация заболевания от 3-9 месяцев
У всех пациентов макроцефалия с рождения (97 перцентелей)
- 25 перенесли острые метаболические кризы с
последующей утратой двигательных навыков, формированием
тяжелых дистонических нарушений и орофациальой дискинезии
- У одного пациента клиника спастико-гиперкинетической
формы детского церебрального паралича.
Энцефалитоподобные кризы обычно возникали на фоне
ОРВИ, после вакцинации или легкой ЧМТ
Биохимические изменения
Повышенная экскреция глутаровой и 3 –гидроксиглутаровой
кислот в моче, повышение уровня C5DC в крови
47.
Глутаровая ацидурия тип 148.
Энцефалит?49.
Типичные клинические проявления глутаровой ацидурии тип 1Выступающий
лоб,
относительная
макрокрания,
орофацильные
дискинезии,
дистонияеская
поза.
50.
Черепно-мозговая травма?51.
Глутаровая ацидурия тип1Дебют после
перенесенной ЧМТ
Общемозговая
симптоматика
Утрата
приобретенных
навыков
Тетрапарез
Мышечная дистония
Гиперкинезы
52.
Детский церебральный паралич?53.
Медленно прогрессирующее течениеглутаровой ацидурии тип 1, пациент С.С. 9 лет
От физиологически протекавшей беременности, роды 39
неделя
При рождении макрокрания ( окр головы 40 см) и задержка
двигательных навыков на первом году жизни
К 10 месяцам появилось повышение мышечного тонуса и
атетоидные гиперкинезы конечностей.
Постепенно сформировался спастический тетрапарез
К 1,5 годам устанвоили диагноз ДЦП, спастикогиперкинетическая форма
Диагноз глутаровой ацидурии был установлен к 7 годам по
семейному диагнозу
54.
Лечение глутаровой ацидурии тип 1улучшение двигательных функций
До лечения
Через 6 месяцев после лечения
Низкобелковая диета ( снижение триптофана и
лизина )
Левокарнитин 100 мг/кг/день
Баклофен 2 мг/кг в сутки
Рибофлавин 100 мг в сутки
55.
Глутаровая ацидурия тип 1A.
B.
C.
А.
T2W изображения- билатеральное расширение
Сильвиевых щелей (лобно-височная гипоплазия),
повышение интенсивности сигнала в области бледных
шаров и хвостатых ядрах
В.
Субдуральные гигромы
С.
Битемпоральные арахноидальные кисты
Собственные наблюдения
56.
Лечение глутаровой ацидурии тип 1:
У
большинства пациентов на фоне лечения (
диетотерапия («Глутаридон, « GA1 ANAMIX infant»,
левокарнитин в дозе 100 мг/кг/сутки) отмечается
стабилизация неврологических расстройств:
- уменьшение выраженности гиперкинетических
нарушений
- отсутствуют метаболические кризы
Исход терапии зависит от времени
установления диагноза
57.
Трудности диагностики• Разный возраст манифестации
• Выраженный клинический полиморфизм
• Низкая «орфанная» настороженность
• Плохая оснащенность лабораторным
оборудованием
• Проблемы доставки биологического
материала до референсных лабораторий
• Дороговизна генетических исследований
58.
Тесное сотрудничествоуспех лечения!!!59.
Патогенетическая терапия НБ- Диетотерапия
-Фермент-заместительная терапия
-Ингибиторы ферментов или БАВ
-Субстрат-редуцирующая терапия
-Терапия шаперонами
- Генотерапия
60.
— Куда мне отсюда идти?— А куда ты хочешь попасть?
— А мне все равно, только бы попасть куда-нибудь
— Тогда все равно куда идти. Куда-нибудь ты
обязательно попадешь.
Льюис Кэррол
МГНЦ
61.
Терапия НБОСубстрат-редуцирующая терапия
-Болезнь Нимана-Пика тип С (Миглустат)
Терапия шаперонами
- Фенилкетонурия (Сапропетрин)
Генотерапия
- Дефицит липазы липопротеинов (Глибера)
62.
Терапия НБОСпециализированные продукты лечебного питания для
лечения:
1. ФКУ
2. Тирозинемия
3. Болезнь мочи кленового сиропа
4. Метилмалоновая и
5.Пропионовая ацидемия
6. Гиперлизинемия
7. Некетотическая гиперглицинемия
8. Гомоцистинурия
9. Изовалериановаяацидемия
10. Глутароваяацидемия
11. Галактоземия
63.
64.
Медикаментозное лечениегомоцистинурии
Средства, влияющие на ЦНС
Психостимуляторы и ноотропные препараты:
пантогам, пирацетам, церебролизин
Корректоры мозгового кровообращения:
циннарезин, винпоцетин, инстенон
Антиоксиданты и антигипоксанты:
левокарнитин, актовегин, цитохром С,
Витамины:
тиамин, пиридоксин, фолиевая кислота,
никотинамид, рибофлавин, биотин, витамин С
Средства для лечения остеопороза:
холекальциферол, остеогенон
Антикоагулянты:
тромбо АСС,
Гепатопротекторы:
эссенциале-форте
65.
СПАСИБО ЗАВНИМАНИЕ !