












































































 Медицина
МедицинаПохожие презентации:









")
Дистрофии. Паренхиматозные и ССД дистрофии
1.
Дистрофии.Паренхиматозные и
ССД дистрофии.
2.
Обратимые поврежденияклеток и внеклеточных
структур – дистрофии.
Необратимые – некроз.
3.
ДИСТРОФИИэто количественные и
качественные структурные
изменения в клетках и/или
межклеточном веществе
органов и тканей,
обусловленные нарушением
обменных процессов.
4.
Основные причины дистрофий:Гипоксия
Физические агенты
Химические агенты и лекарства
Инфекционные агенты
Иммунные реакции
Генетические нарушения
Дисбаланс питания
Факторы окружающей среды (пыль, уголь,
асбест, алкоголь, наркотики, гербициды,
консерванты и т.д.)
5.
Непосредственные причиныдистрофий:
1.
2.
3.
Нарушение ауторегуляции клетки
Нарушение энергетических и
транспортных систем клетки
Нарушение эндокринной и нервной
регуляции клетки
6.
Механизмы дистрофий:Инфильтрация
Декомпозиция (фанероз)
Трансформация
Извращенный синтез
7.
Морфологическая сущностьдистрофий:
Увеличение или уменьшение
количества каких-либо веществ.
Изменение качества (физикохимических свойств) веществ.
Появление обычных веществ в
необычном месте.
Появление и накопление новых
(необычных) веществ.
8.
Классификация дистрофийВ зависимости от локализации патологических изменений:
Паренхиматозные
Стромально-сосудистые (мезенхимальные)
Смешанные
2. По виду нарушенного обмена:
Белковые
Жировые
Углеводные
Минеральные
3. По влиянию наследственных факторов
Приобретенные
Наследственные
4. По распространенности процесса
Общие
Местные
1.
9.
Паренхиматозные дистрофии –это структурные изменения в
высокоспециализированных в
функциональном отношении
клетках, связанные с нарушением
обмена веществ.
10.
Паренхиматозные дистрофии:1. Белковые (диспротеинозы)
2. Жировые (липидозы)
3. Углеводные
11.
Приобретенные паренхиматозныебелковые дистрофии:
1.
Гиалиново-капельная дистрофия
2.
Гидропическая дистрофия
3.
Роговая дистрофия
12.
Гиалиново-капельная дистрофия:Локализация: гепатоциты, кардиомиоциты, эпителий
извитых канальцев почки;
Причины: инфекции, интоксикации, аллергические
реакции;
Патогенез: декомпозиция, инфильтрация,
извращенный синтез;
Макро: без изменений;
Микро: гиалиноподобные розовые капли в цитоплазме
клеток. Например, алкогольный гиалин;
Исход: неблагоприятный - необратимый процесс,
коагуляционный некроз клетки.
13.
Гиалиново-капельная дистрофия почек14.
Алкогольный гиалин15.
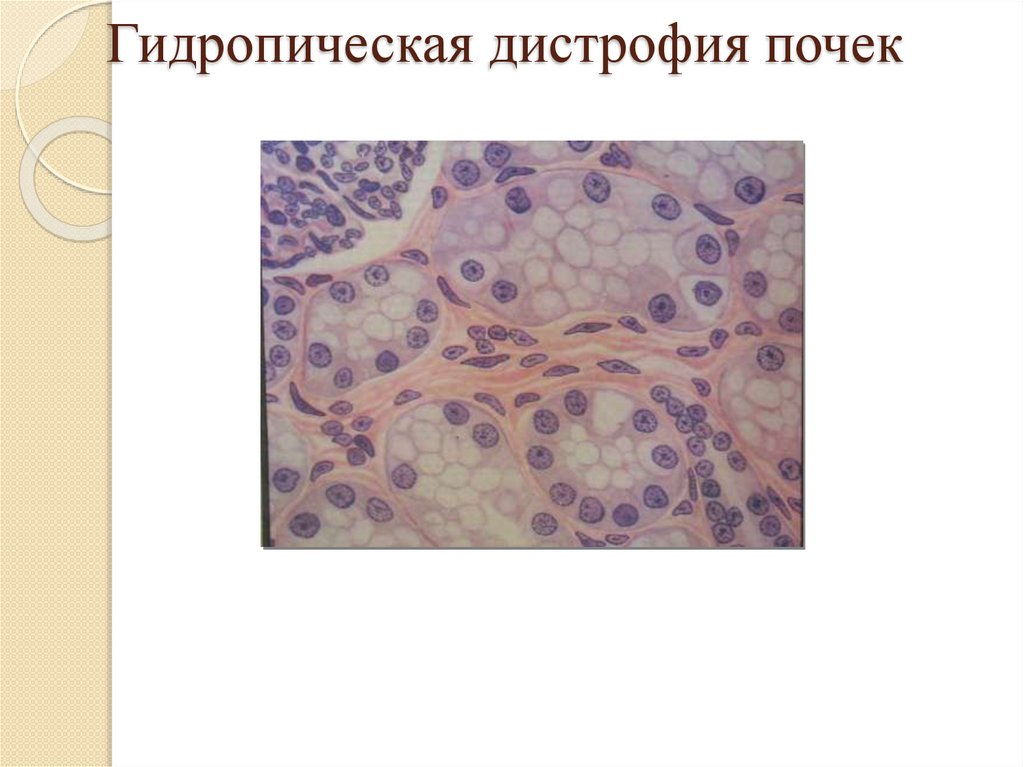
Гидропическая дистрофия:Локализация: гепатоциты, кардиомиоциты, эпителий
кожи и извитых канальцев почки, клетки коры
надпочечников;
Причины: инфекции, воспаление, воздействие
физических и химических веществ;
Патогенез: инфильтрация;
Макро: без изменений, пузыри с жидкостью на коже;
Микро: вакуоли заполненные жидкостью в цитоплазме
клеток;
Исход: неблагоприятный - необратимый процесс,
колликвационный некроз клетки.
16.
Гидропическая дистрофия почек17.
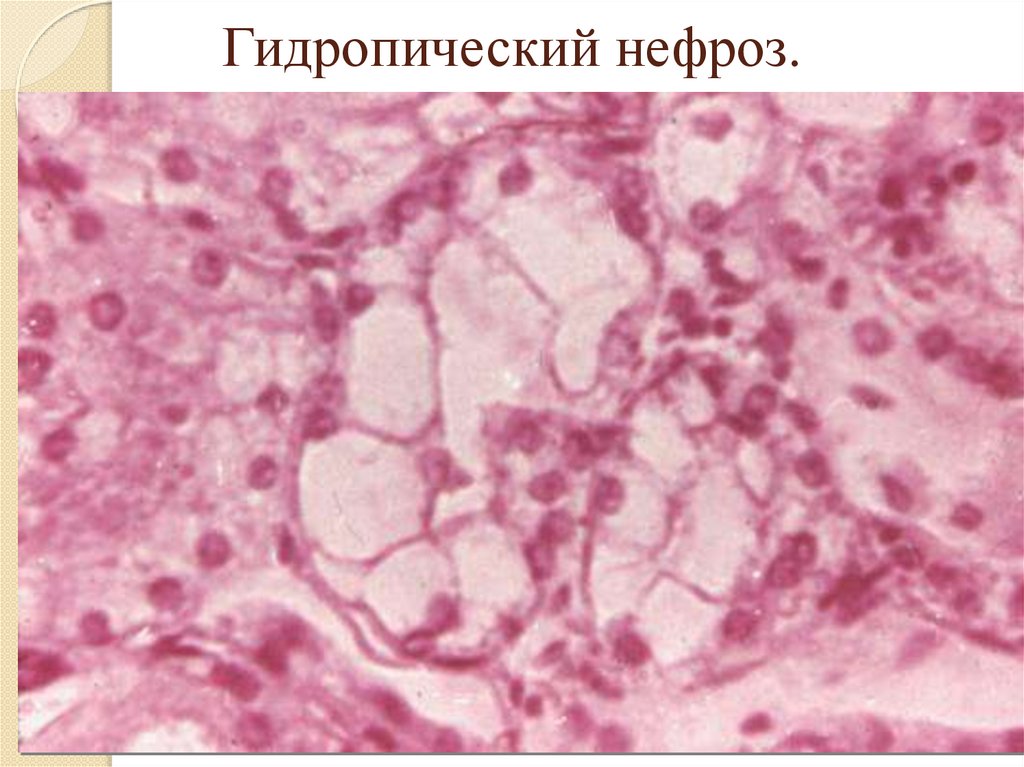
Гидропический нефроз.18.
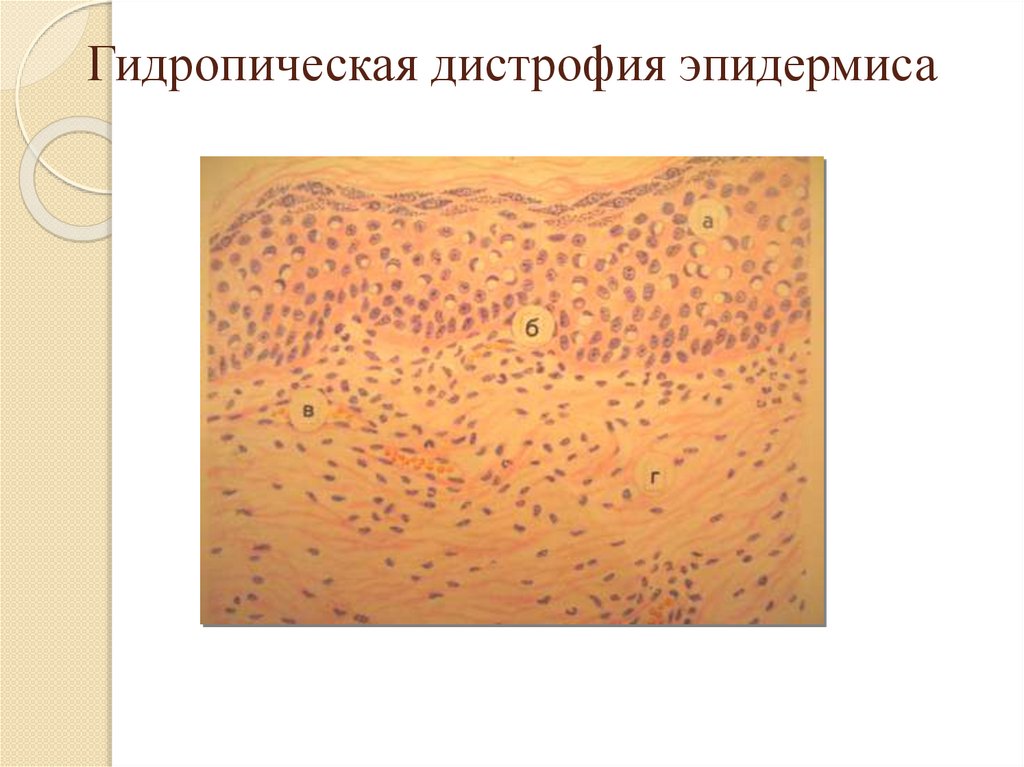
Гидропическая дистрофия эпидермиса19.
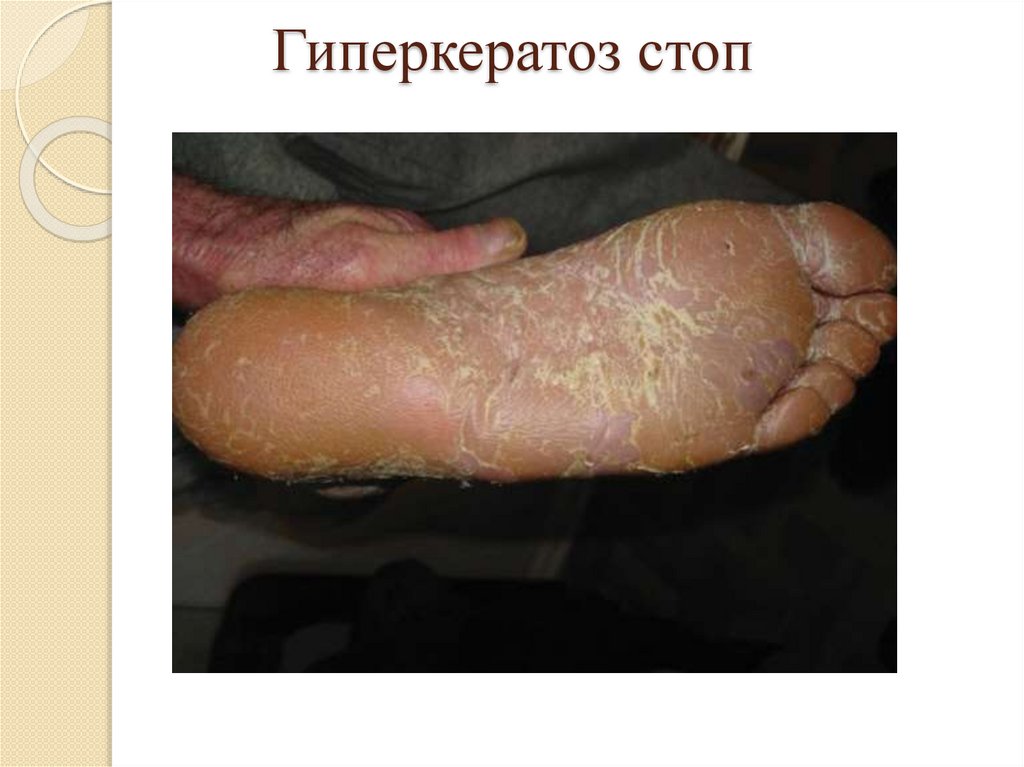
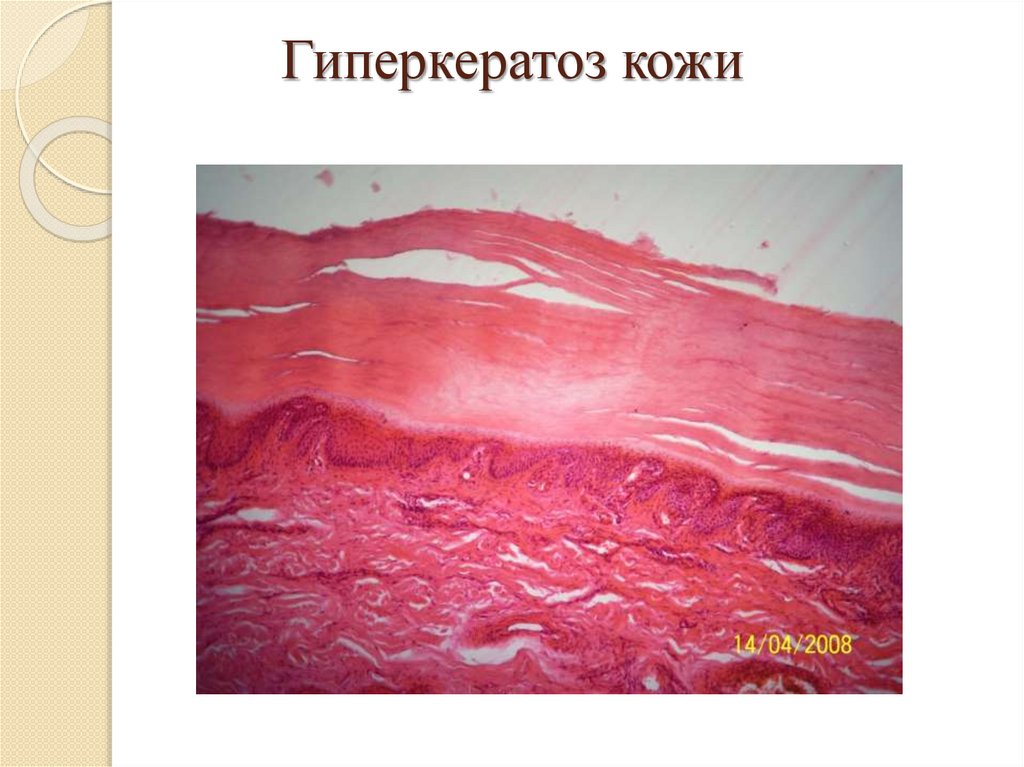
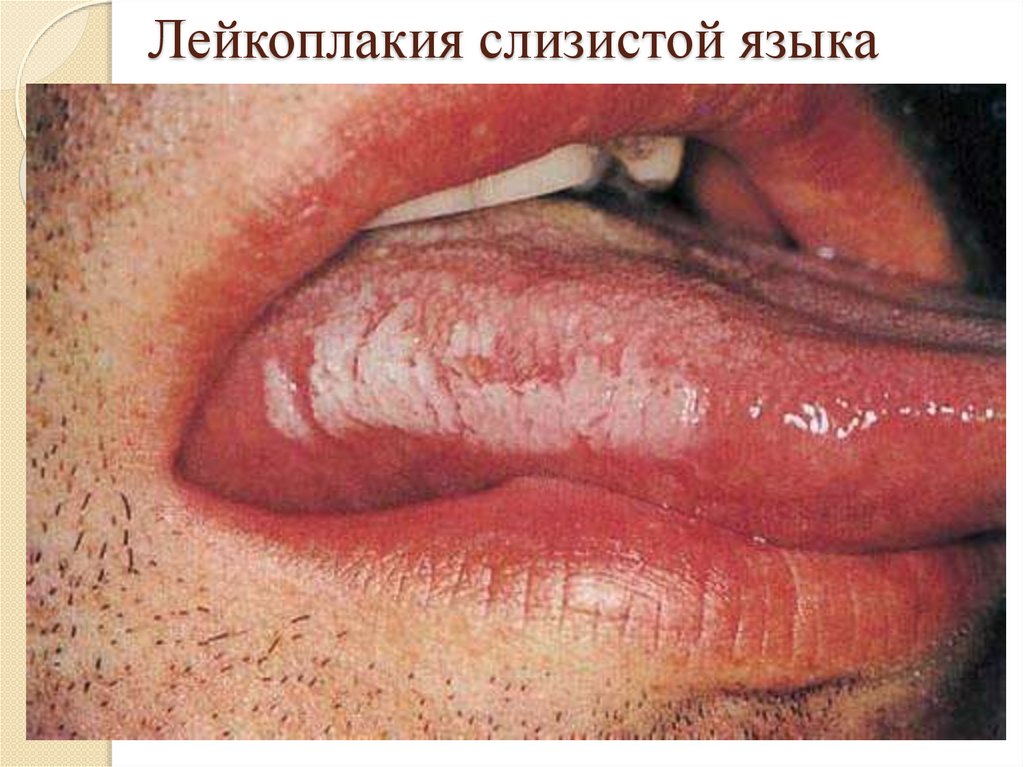
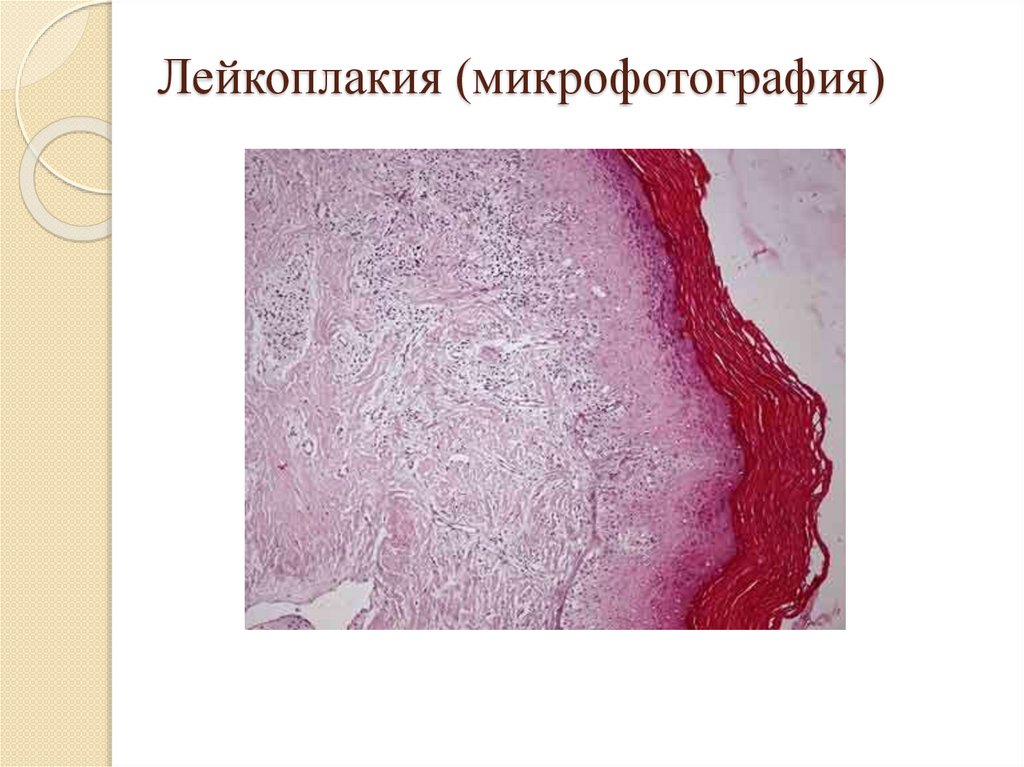
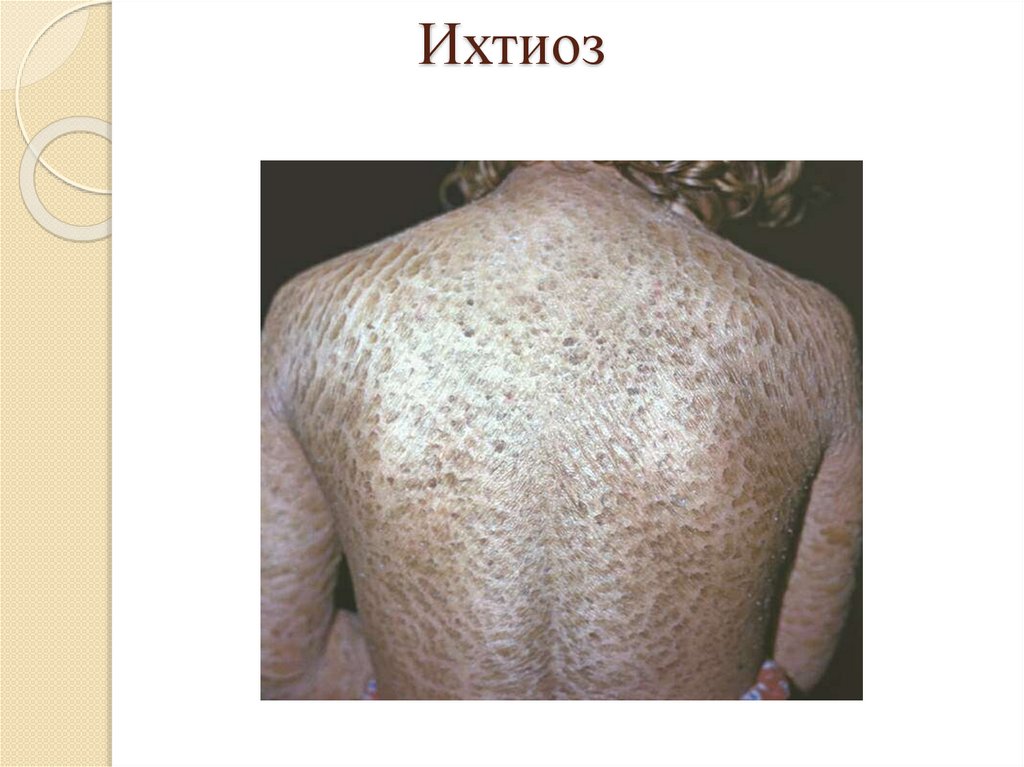
Роговая дистрофия:Локализация: кожа и слизистые оболочки;
Причины: инфекции, воспаление, воздействие
физических и химических веществ, авитаминозы,
наследственные болезни (ихтиоз);
Патогенез: избыточный нарушенный синтез кератина;
Макро: утолщение и огрубление кожи, на слизистых –
белые пятна – «лейкоплакия»;
Микро: утолщение в несколько раз рогового слоя кожи
или появление рогового слоя на слизистых;
Исход: возможно восстановление при устранении
причины, при лейкоплакии – возникновение раковой
опухоли.
20.
Гиперкератоз стоп21.
Гиперкератоз кожи22.
Лейкоплакия слизистой языка23.
Лейкоплакия (микрофотография)24.
Ихтиоз25.
Наследственныепаренхиматозные
белковые дистрофии
26.
Цистиноз (аминокислота накапливается впечени, почках, селезёнке, глазах, костном
мозге, л/у, коже).
Тирозиноз (недостаток
тирозинаминотрансферазы), аминокислота
накапливается в печени, почках, костях.
Фенилпировиноградная олигофрения
(недостаток фенилаланин-4-гидроксилазы),
накопление аминокислоты наблюдается в
нервной системе, мышцах, коже, крови,
моче.
27.
Приобретенныепаренхиматозные жировые
дистрофии (липидозы):
Морфологически проявляются:
увеличением количества жиров,
появлением там, где их не бывает в норме,
образование необычного химического
состава.
28.
Причины жировой дистрофии:Кислородное голодание (гипоксия);
Тяжелые и длительно протекающие
инфекции (дифтерия, туберкулез, сепсис);
Интоксикации (фосфор, мышьяк,
хлороформ, алкоголь);
Авитаминозы и одностороннее ( с
недостаточным содержанием белка)
питание.
29.
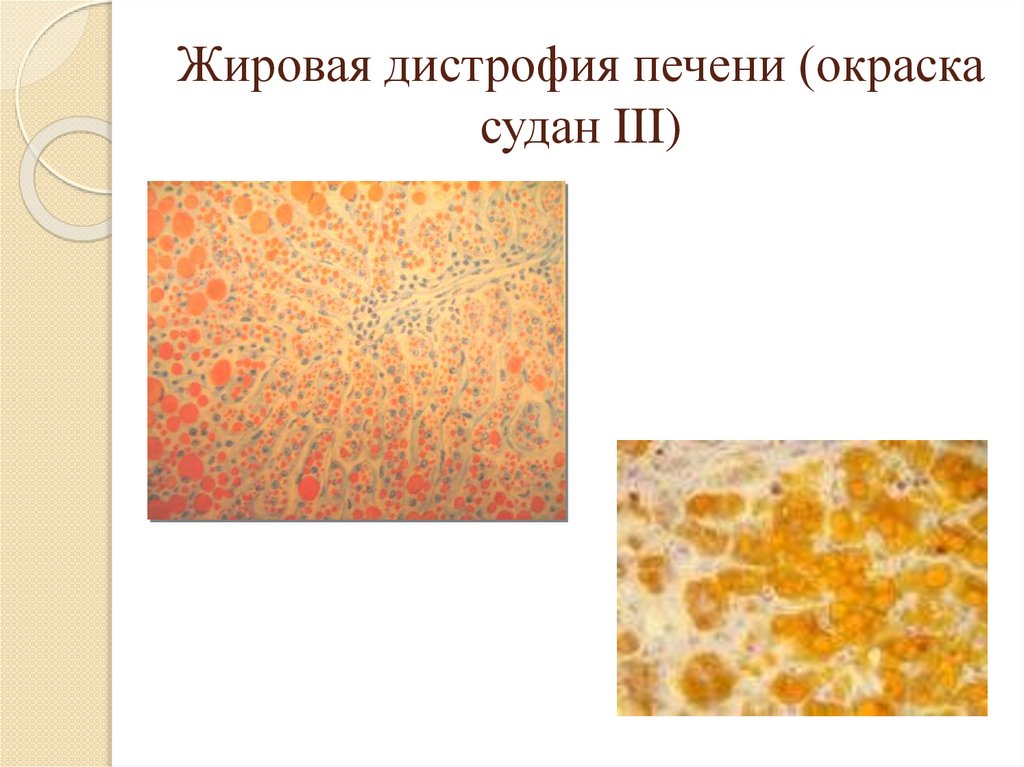
Патогенез паренхимотозных жировыхдистрофий – декомпозиция;
Микроскопически жир в клетках и тканях
можно обнаружить при помощи
специфических гистохимических реакций:
- Судан IV окрашивает жиры в красный цвет,
- Судан III – в оранжевый,
- Судан черный В и осмиевая кислота – в
черный цвет,
- Сульфат нильского голубого окрашивает
жирные кислоты в темно-синий цвет, а
нейтральные жиры – в красный.
30.
Локализация паренхиматозныхлипидозов:
Печень
Сердце
Почки
31.
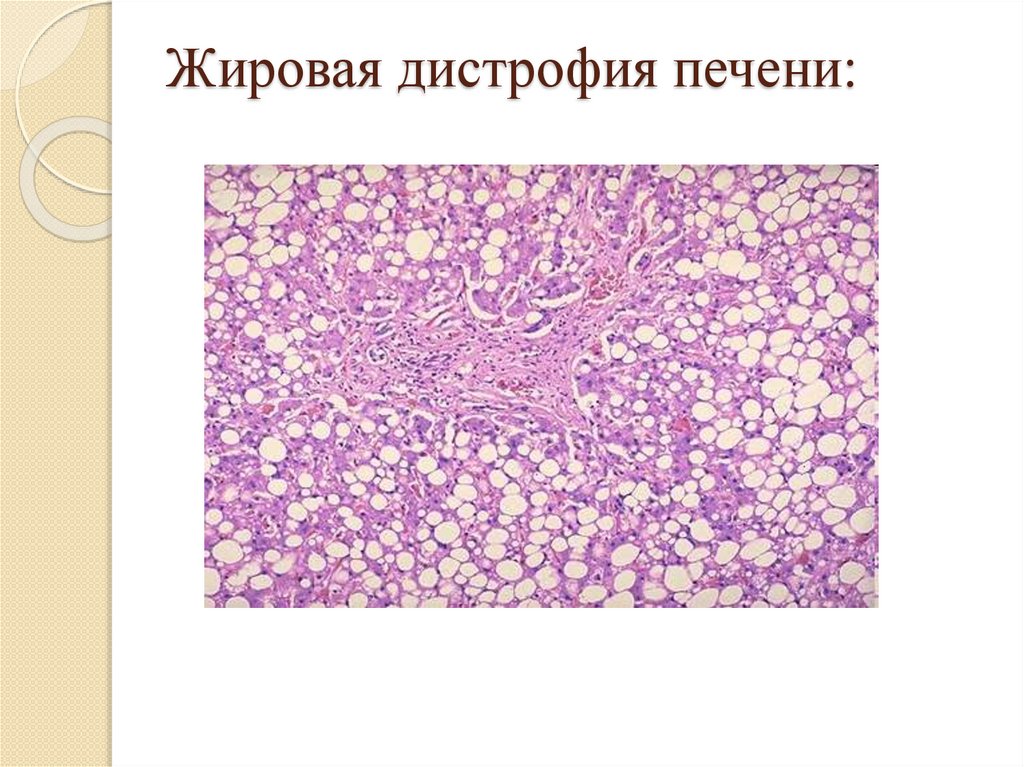
Жировая дистрофия печени:Причины: острые и хронические
интоксикации;
Макро: печень увеличена, дряблая,
желтого цвета – «гусиная печень»;
Микро: появление пылинок, мелких или
крупных капель жира в гепатоцитах, в
тяжелых случаях гепатоциты
превращаются в липоциты;
Исход: возможно восстановление при
устранении причины, иначе – печеночная
недостаточность.
32.
Жировая дистрофия печени:33.
Жировая дистрофия печени (окраскасудан III)
34.
Жировая дистрофия миокарда:Причины: хронические гипоксические
состояния, интоксикации – при дифтерии;
Макро: сердце увеличено, дряблое, под
эндокардом желтые полоски – «тигровое
сердце»;
Микро: появление пылинок, мелких или
крупных капель жира в кардиомиоцитах,
исчерченность мышечных волокон исчезает;
Исход: возможно восстановление при
устранении причины, либо сердечная
недостаточность.
35.
Жировая дистрофия миокарда (окраскасудан III)
36.
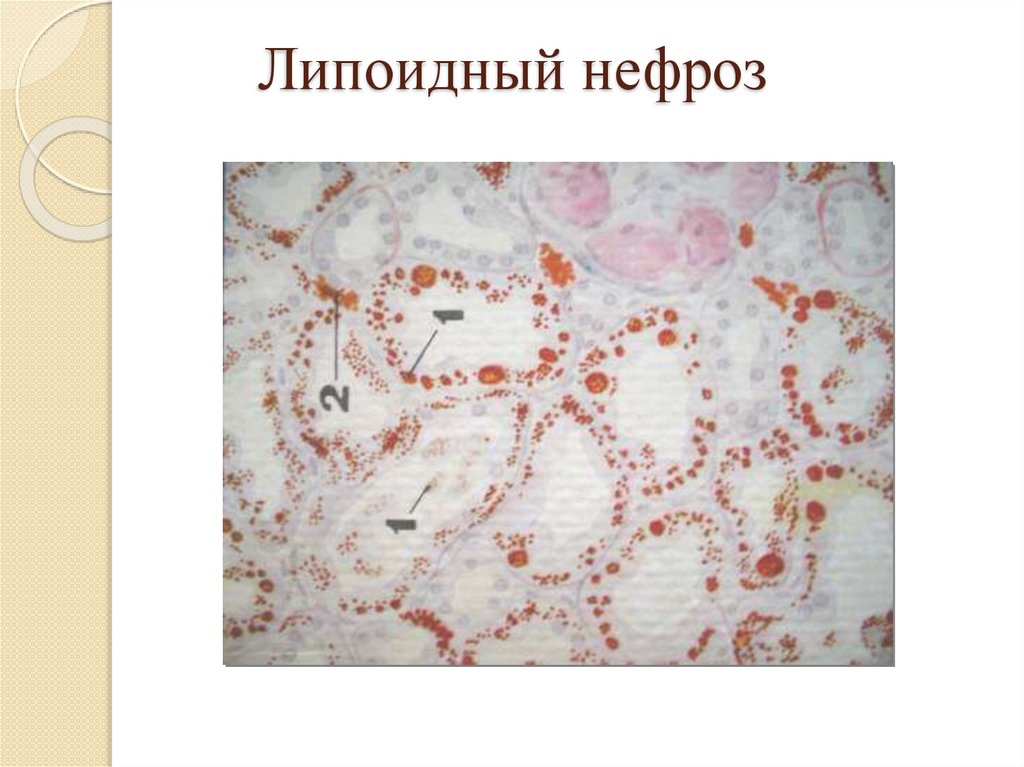
Жировая дистрофия эпителия канальцевпочек:
Причины: нефротический синдром;
Патогенез: инфильтрация;
Макро: почки увеличены, дряблые, корковое
вещество серое с желтым крапом;
Микро: появление пылинок, мелких или
крупных капель жира в эпителии извитых
канальцев почек;
Исход: возможно восстановление при
устранении причины, глубокое нарушение
обмена жиров ведет к гибели клетки.
37.
Липоидный нефроз38.
Врожденныепаренхиматозные
жировые дистрофии
39.
Болезнь Гоше (дефицит ферментаглюкоцереброзидазы), накопление жиров в
печени, селезенке, ЦНС, костном мозге.
Болезнь Ниманна-Пика (дефицит
сфингомиелиназы).
Амавротическая идиотия (Б. Тея-Сакса) дефицит
гексоаминидазы.
Б. Нормана-Ландинга (дефицит β-галактозидазы).
40.
Паренхиматозныеуглеводные
дистрофии
41.
Углеводы, которые определяются в клеткахи тканях и могут быть идентифицированы
гистохимически, делят на:
1. Полисахариды:
Гликоген
2. Гликозаминогликаны (мукополисахариды):
Нейтральные
Кислые
3. Гликопротеиды:
Муцины
Мукоиды
42.
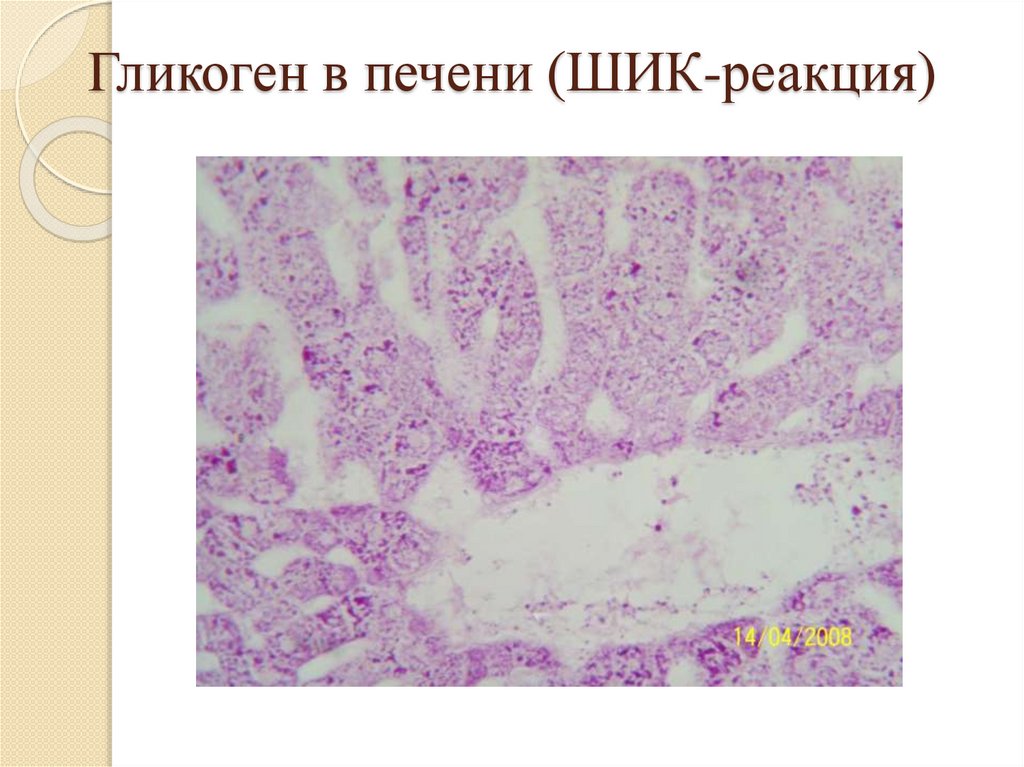
Гистохимические методы выявленияуглеводов:
Гликоген, гликозаминогликаны и гликопротеиды
выявляются с помощью ШИК-реакции (краснофиолетовое окрашивание);
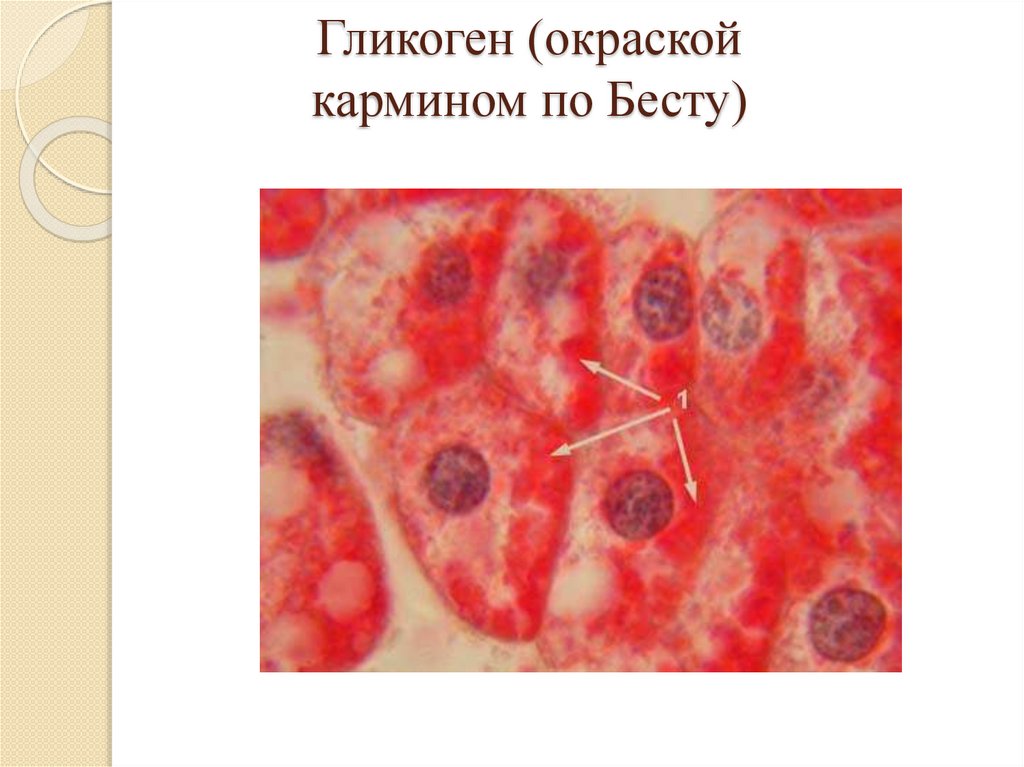
Гликоген окрашивается кармином по Бесту в
красный цвет;
Гликозаминогликаны и гликопротеиды
определяют с помощью реакции метахромазии
(толуидиновый синий, метиленовый синий).
43.
Гликоген в печени (ШИК-реакция)44.
Гликоген (окраскойкармином по Бесту)
45.
Паренхиматозные углеводныедистрофии могут быть
связаны с нарушением:
1. ГЛИКОГЕНА
2. ГЛИКОПРОТЕИДОВ
46.
Нарушение обмена гликогенаможет быть:
Приобретенным (при СД)
Врожденным (при
гликогенозах)
47.
Сахарный диабет характеризуется:Патологией β-клеток островков поджелудочной
железы, что ведет к недостаточной выработке
инсулина,
Происходит недостаточное использование
глюкозы тканями и накопление глюкозы в крови
(гипергликемия) и выведение глюкозы с мочой
(глюкозурия)
Тканевые запасы гликогена резко уменьшаются.
48.
Морфологические проявления сахарногодиабета в тканях:
В печени уменьшается синтез гликогена, что
ведет к жировой инфильтрации гепатоцитов и
жировой дистрофии печени.
С глюкозурией связана гликогенная
инфильтрация эпителия канальцев почек, однако
страдают не только канальцы, но и клубочки, что
ведет к диабетическому гломерулосклерозу.
49.
Наследственные углеводные дистрофии(гликогенозы)
Болезнь Гирке (I тип),
Болезнь Помпе (II тип),
Болезнь Мак-Ардля (V тип),
Болезнь Герса (VI тип), при них структура
накапливаемого в тканях гликогена не нарушена;
Болезнь Форбса-Кори (III тип),
Болезнь Андерсена (IV тип), при них структура
гликогена резко изменена.
50.
Углеводные дистрофии, связанные снарушением обмена гликопротеидов:
Происходит накопление слизистых и
слизеподобных веществ.
Причины: воспаление слизистых оболочек.
Происходит не только увеличение
количества образования слизи, но и
изменение химического состава слизи.
Слизистая дистрофия лежит в основе
заболевания – муковисцидоз.
Исход: атрофия, склероз слизистых,
возможно и восстановление.
51.
Стромально-сосудистыедистрофии
52.
Стромально-сосудистые (мезенхимальные)дистрофии развиваются на территории
гистиона в результате нарушений обмена в
соединительной ткани и выявляются в
строме органов и стенках сосудов.
Гистион - отрезок микроциркуляторного
русла с окружающими его элементами
соединительной ткани и нервными
волокнами.
53.
Белковые стромально-сосудистыедистрофии
Мукоидное набухание
Фибриноидное набухание
Гиалиноз
Амилоидоз
54.
Мукоидное набуханиеповерхностная и обратимая дезорганизация
соединительной ткани (син. слизеподобное
набухание, хромотропный отек)
диагностируется при получении метахроматического
окрашивания с красителем – толуидиновым синим
развивается при повышении сосудистой
проницаемости и высоком содержании жидкости в
основном веществе соединительной ткани
55.
Мукоидное набухание56.
Причины мукоидного набухания:гипоксия
инфекция
реакции гиперчувствительности
57.
Звенья морфогенеза мукоидногонабухания:
Накопление и перераспределение
гликозаминогликанов в основном веществе
соединительной ткани за счёт увеличения
содержания гиалуроновой кислоты.
Повышение сосудисто-тканевой проницаемости.
Гидратация и набухание основного вещества и
коллагеновых волокон соединительной ткани.
58.
Исходы мукоидного набухания:полное восстановление ткани
переход в фибриноидное набухание
59.
Фибриноидное набухание–
накопление в основном веществе
соединительной ткани белковых масс
(плазменных
белков,
содержащих
фибриноген) - фибриноида. При образовании
комплексов с коллагеном процесс становится
необратимым
может перейти в фибриноидный некроз
Исходы – склероз, гиалиноз
60.
Звенья морфогенеза фибриноидногонабухания:
Резкое повышение сосудисто-тканевой
проницаемости.
Плазморрагия.
Выход грубодисперсных плазменных белков, в
первую очередь фибриногена.
Превращение фибриногена в фибрин.
Деструкция коллагена и основного вещества
соединительной ткани.
Образование фибриноида.
61.
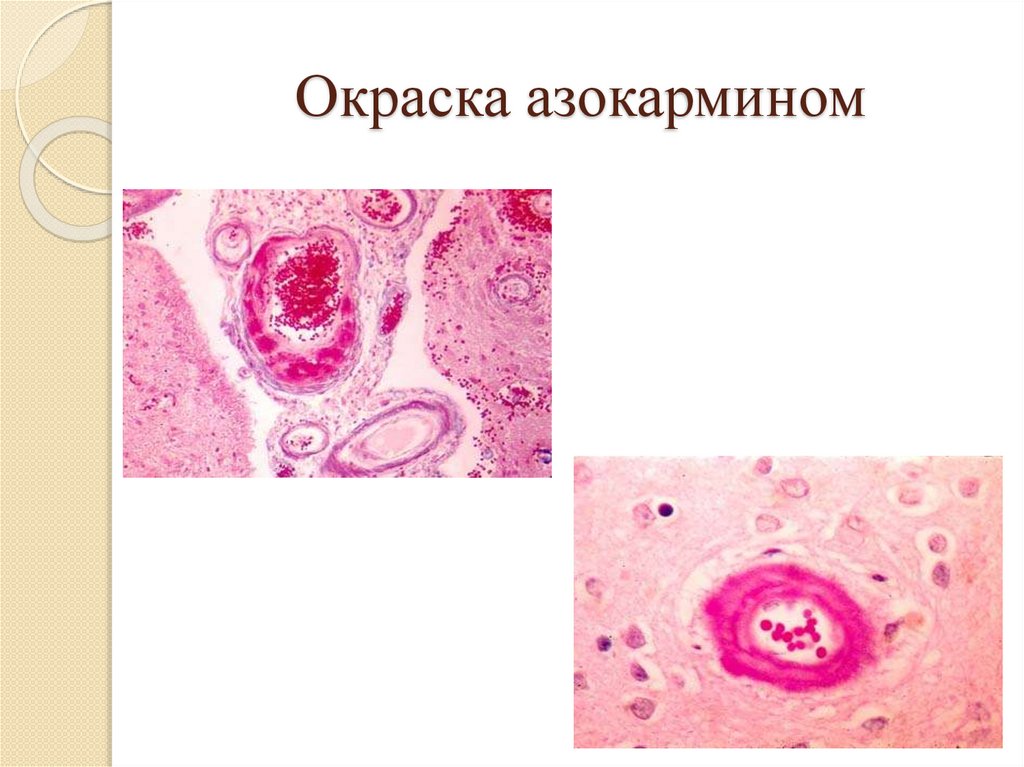
Окраска азокармином62.
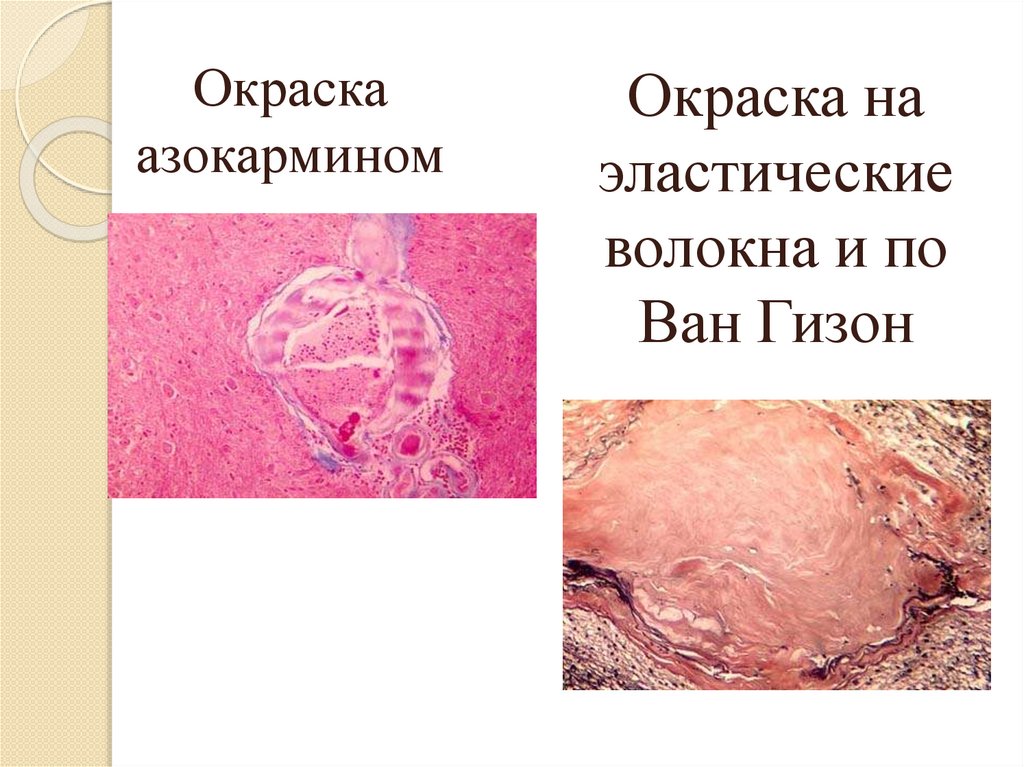
Окраскаазокармином
Окраска на
эластические
волокна и по
Ван Гизон
63.
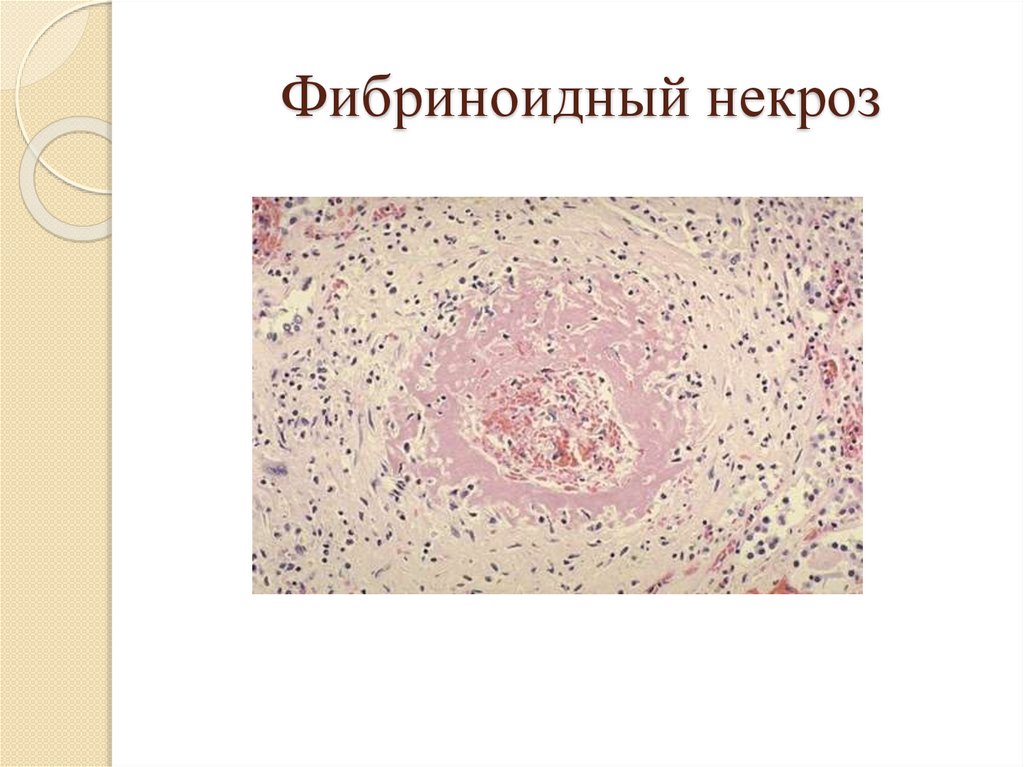
Фибриноидный некроз64.
Гиалинозотложение белковых масс, которое
при макроскопическом
исследовании напоминает
гиалиновый хрящ
65.
Классификация:В зависимости от преимущественной
локализации выделяют:
– гиалиноз сосудов
– гиалиноз стромы
По составу выделяют:
– простой гиалин
– сложный гиалин
– липогиалин
66.
Простой гиалин возникает в результатепропитывания стенки сосуда белками
плазмы при ГБ и атеросклерозе
Сложный
гиалин
состоит
из
иммунных комплексов, фибрина и
компонентов
сосудистой
стенки,
образуется при иммунопатологических
процессах
67.
Исходы гиалиноза:ослизнение ткани;
липоидоз;
обызвествление;
рассасывание гиалиновых масс.
68.
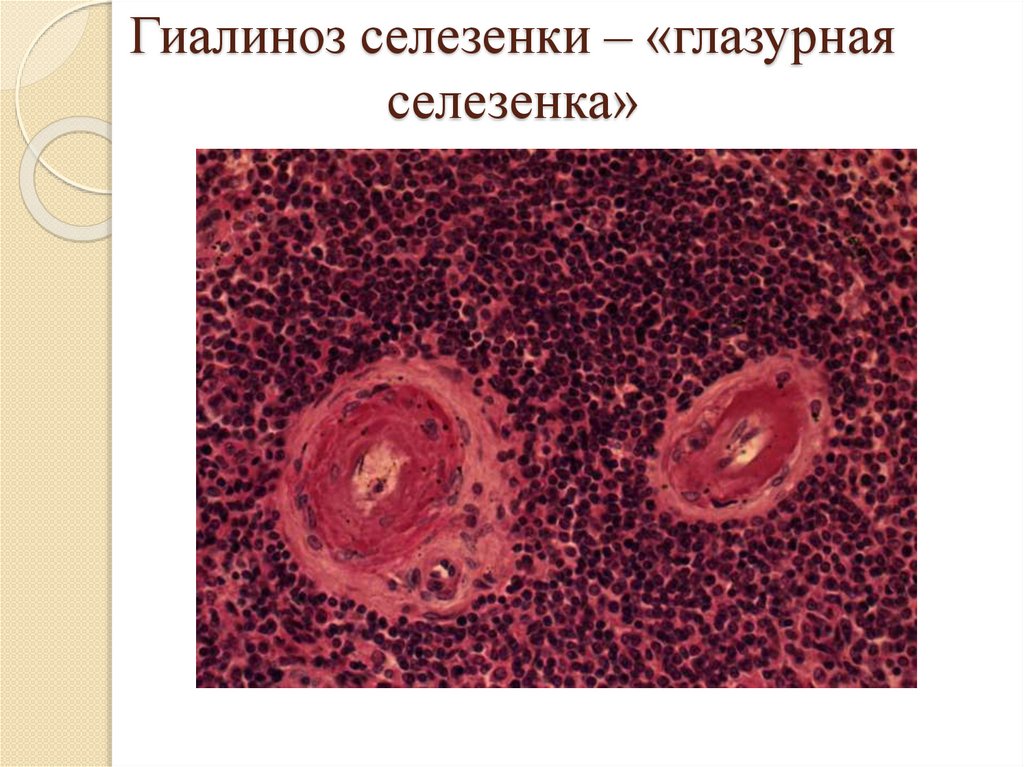
Гиалиноз селезенки – «глазурнаяселезенка»
69.
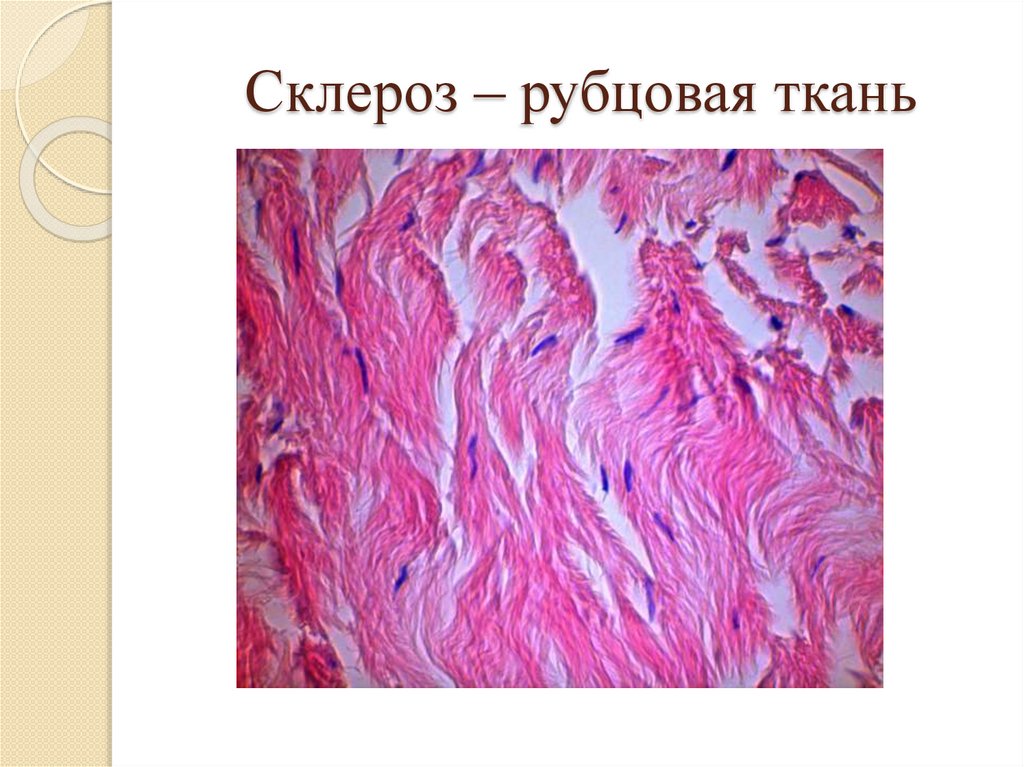
Склероз – рубцовая ткань70.
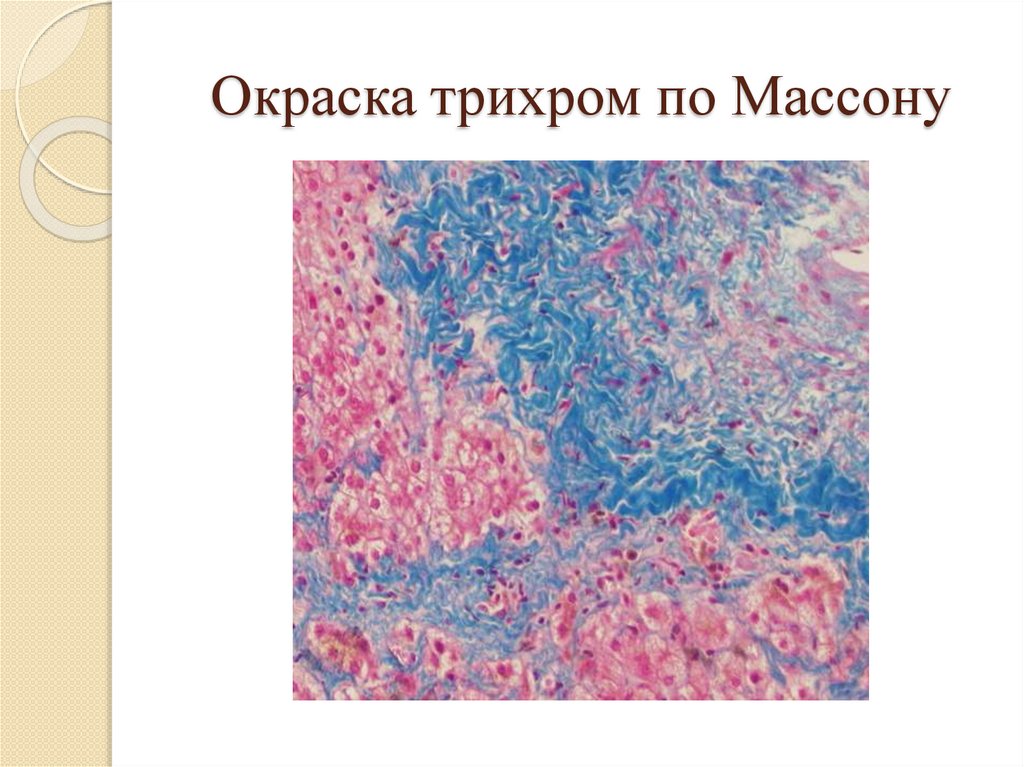
Окраска трихром по Массону71.
АмилоидозГруппа заболеваний, характеризующихся
отложением гомогенных эозинофильных
нерастворимых белковых масс – амилоида.
Амилоид окрашивается красителем Конго
красным в красный цвет и дает двойное
лучепреломление и зеленое свечение в
поляризационном свете.
72.
Амилоидоз«Большая сальная почка» - отложение
амилоида обнаруживается в клубочках,
стенках сосудов и строме
«Сальная селезенка» («ветчинная
селезенка») – диффузно в строме и
капсуле
«Саговая селезенка» - преимущественно в
проекции лимфоидных фолликулов
73.
АмилоидозПатогенез:
- Стимул
- Растворимый белок предшественник
- Образование нерастворимых фибрилл
амилоида
Причины отложения амилоида:
- Нарушения в структурной организации белков
- Замены (мутации) в аминокислотной
последовательности белков
74.
КЛАССИФИКАЦИЯАМИЛОИДОЗА:
• ПО ВХОДЯЩЕМУ В СОСТАВ АМИЛОИДА БЕЛКУ
– на сегодняшний день выделено более 25 белков
• ПО РАСПРОСТРАНЕННОСТИ
– системный
– местный
• ПО ПРОИСХОЖДЕНИЮ
– наследственный
– приобретенный
• ПО КЛИНИЧЕСКИМ ПРОЯВЛЕНИЯМ
– с преимущественным поражением тех или иных органов и
систем (почек, печени, ЖКТ и т.д.)
75.
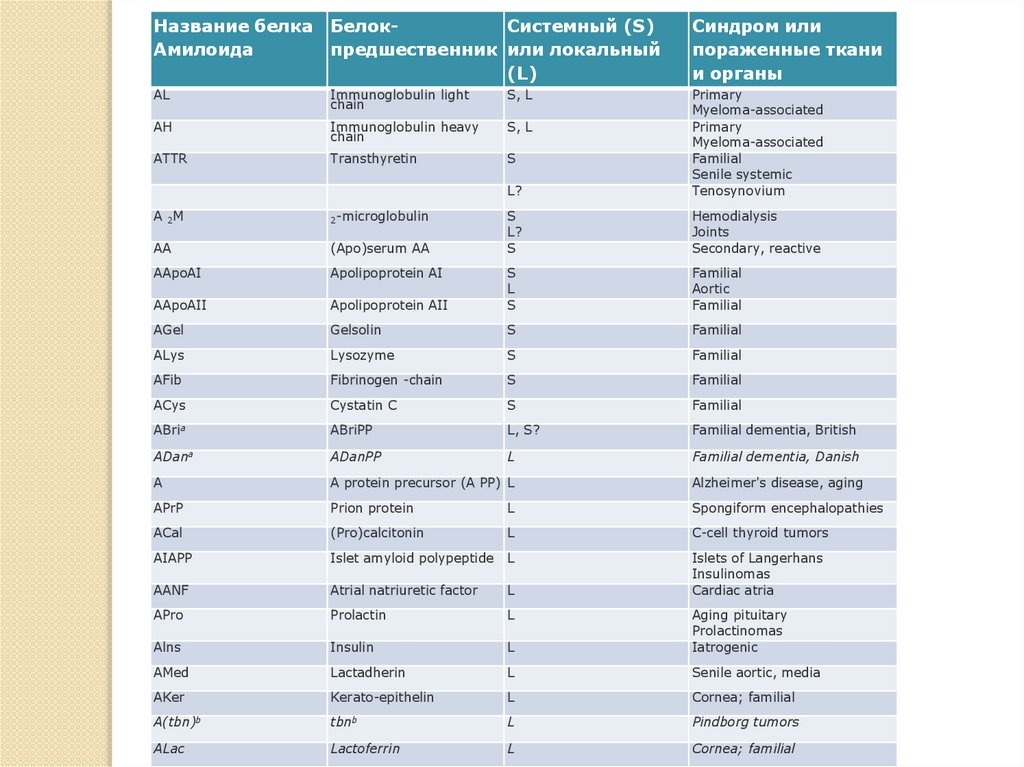
Название белкаАмилоида
БелокСистемный (S)
предшественник или локальный
(L)
Синдром или
пораженные ткани
и органы
AL
Immunoglobulin light
chain
S, L
AH
Immunoglobulin heavy
chain
S, L
ATTR
Transthyretin
S
L?
Primary
Myeloma-associated
Primary
Myeloma-associated
Familial
Senile systemic
Tenosynovium
S
L?
S
Hemodialysis
Joints
Secondary, reactive
Familial
Aortic
Familial
A 2M
2-microglobulin
AA
(Apo)serum AA
AApoAI
Apolipoprotein AI
AApoAII
Apolipoprotein AII
S
L
S
AGel
Gelsolin
S
Familial
ALys
Lysozyme
S
Familial
AFib
Fibrinogen -chain
S
Familial
ACys
Cystatin C
S
Familial
ABria
ABriPP
L, S?
Familial dementia, British
ADana
ADanPP
L
Familial dementia, Danish
A
A protein precursor (A PP) L
Alzheimer's disease, aging
APrP
Prion protein
L
Spongiform encephalopathies
ACal
(Pro)calcitonin
L
C-cell thyroid tumors
AIAPP
Islet amyloid polypeptide
L
AANF
Atrial natriuretic factor
L
Islets of Langerhans
Insulinomas
Cardiac atria
APro
Prolactin
L
Alns
Insulin
L
Aging pituitary
Prolactinomas
Iatrogenic
AMed
Lactadherin
L
Senile aortic, media
AKer
Kerato-epithelin
L
Cornea; familial
A(tbn)b
tbnb
L
Pindborg tumors
ALac
Lactoferrin
L
Cornea; familial
76.
Наиболее клинически значимыесистемные амилоидозы:
- Первичный
- Вторичный
- Наследственный
- Диализ-ассоциированный
77.
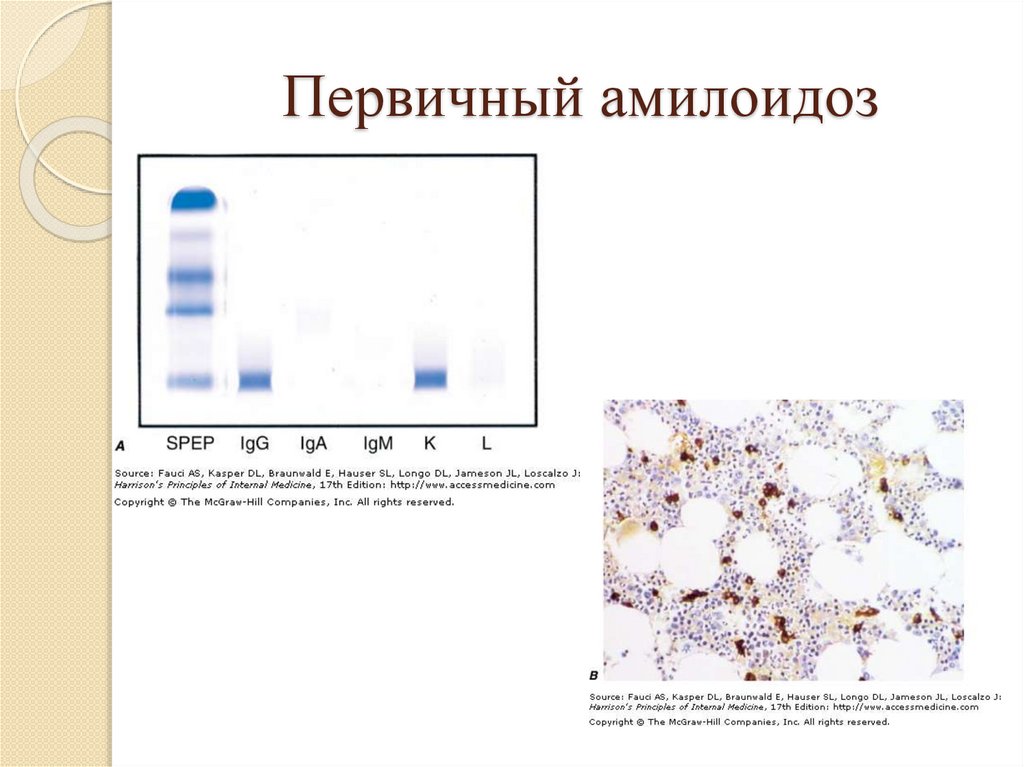
AL амилоидозПервичный
амилоидоз - (в состав
амилоида входят легкие цепи
иммуноглобулинов)
Патогенез:
- Неизвестный стимул (канцероген?)
- Моноклональная пролиферация Влимфоцитов
- Секреция плазматическими клетками
моноклональных L-цепей
78.
Первичный амилоидоз:- Наблюдается при миеломной болезни
- При заболеваниях, сопровождающихся
избыточной продукцией
моноклональных иммуноглобулинов
- Идиопатический
79.
Первичный амилоидоз80.
AА амилоидозВторичный (реактивный) амилоидоз - (в
состав амилоида входит SAA-белок –
сывороточный белок предшественник)
Патогенез:
- Хроническое воспаление
- Активация макрофагов
- Продукция интерлейкинов (ИЛ1 и ИЛ6)
- Продукция гепатоцитами белка SAA
- Образование АА амилоида
81.
Вторичный (реактивный) амилоидознаблюдается при:
- Туберкулез, лепра (до 17% больных)
- Анкилозирующий спондилит,
ревматоидный артрит, болезнь Крона
(0.5-13%)
- Хронический остеомиелит, хронический
абсцесс легкого, бронхоэктазы
82.
АмилоидозНаследственный амилоидоз:
- ATTR амилоидоз (транстиретин,
преальбумин – плазменный белок
связывающий и переносящий
тироксин и ретинол) обнаруживается
при семейных амилоидных
полинейропатиях и старческом
семейном амилоидозе
83.
АмилоидозДиализ-ассоциированный
амилоидоз:
- Aβm амилоидоз (β2-микроглобулин
входит в состав главного комплекса
гистосовместимости 1 класса) и
определяется при длительном
гемодиализе
84.
Наиболее клинически значимыелокальные амилоидозы:
- Болезнь Альцгеймера
- Сердечный
- Инсулярный
- Опухолевый
85.
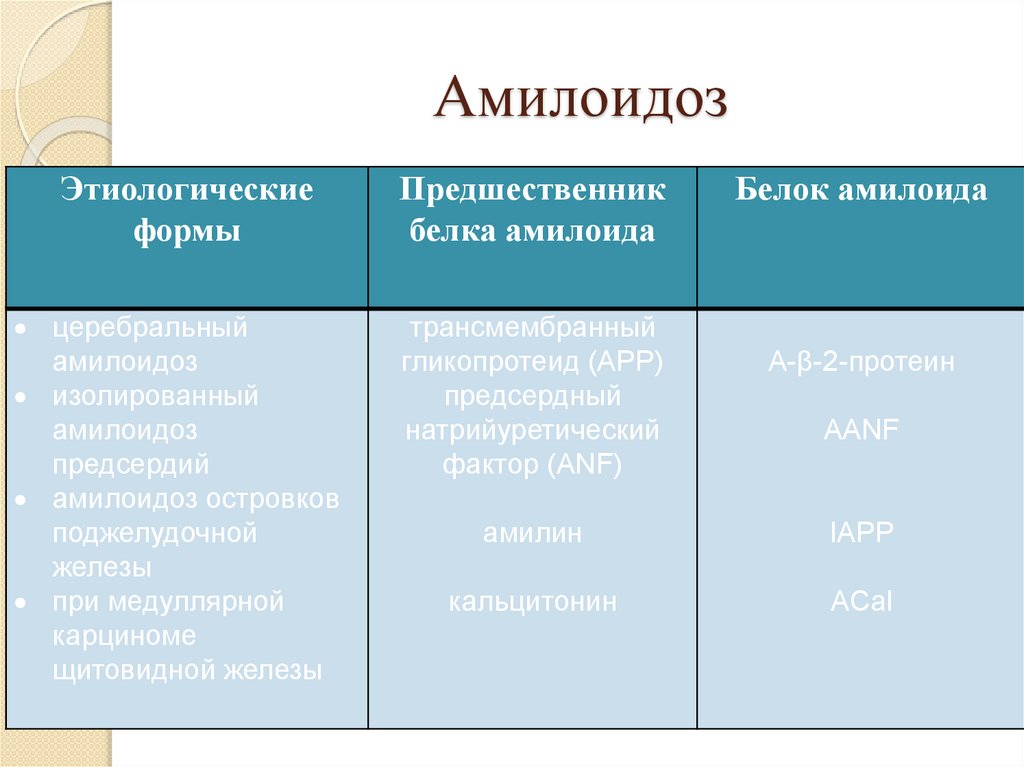
АмилоидозЭтиологические
формы
Предшественник
белка амилоида
церебральный
амилоидоз
изолированный
амилоидоз
предсердий
амилоидоз островков
поджелудочной
железы
при медуллярной
карциноме
щитовидной железы
трансмембранный
гликопротеид (АРР)
предсердный
натрийуретический
фактор (ANF)
Белок амилоида
A-β-2-протеин
AANF
амилин
IAPP
кальцитонин
ACal
86.
Амилоидоз87.
Амилоидоз88.
Амилоидоз89.
Жировые стомально-сосудистыедистрофии (ожирение)
Нарушения
обмена нейтральных
жиров проявляются в увеличении
или уменьшении их запасов в
жировой ткани, могут иметь общий
или местный характер.
90.
ОжирениеОжирение (тучность) –
увеличение количества
нейтральных жиров в жировых
депо, имеющее общий характер
91.
Локализация отложения жира:подкожная жировая клетчатка
сальник
брыжейка
средостение
эпикард
92.
Классификация:По механизму:
Наследственное
- Алиментарное
- Эндокринное
- Церебральное
- Смешанное
-
По внешним проявлениям:
симметричное
- верхнее
- среднее
- нижнее
-
93.
По превышению массы тела (ИМТ):1 степени (избыточная масса составляет 20-29%)
- 2 степени (30-49%)
- 3 степени (50-99%)
- 4 степени (100% и более)
-
По локализации:
общее
- местное
-
По морфологии:
Гипертрофическое – увеличение размеров жировых
клеток (липоцитов)
- Гиперпластическое – увеличение количества клеток
-
94.
Ожирение миокарда95.
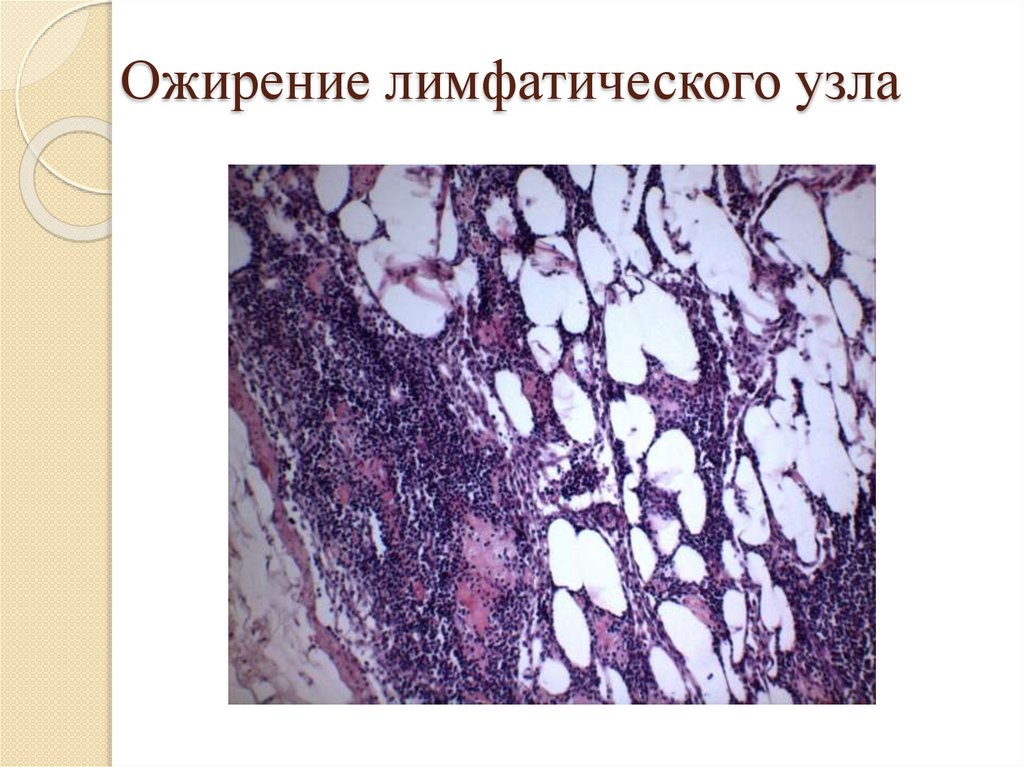
Ожирение лимфатического узла96.
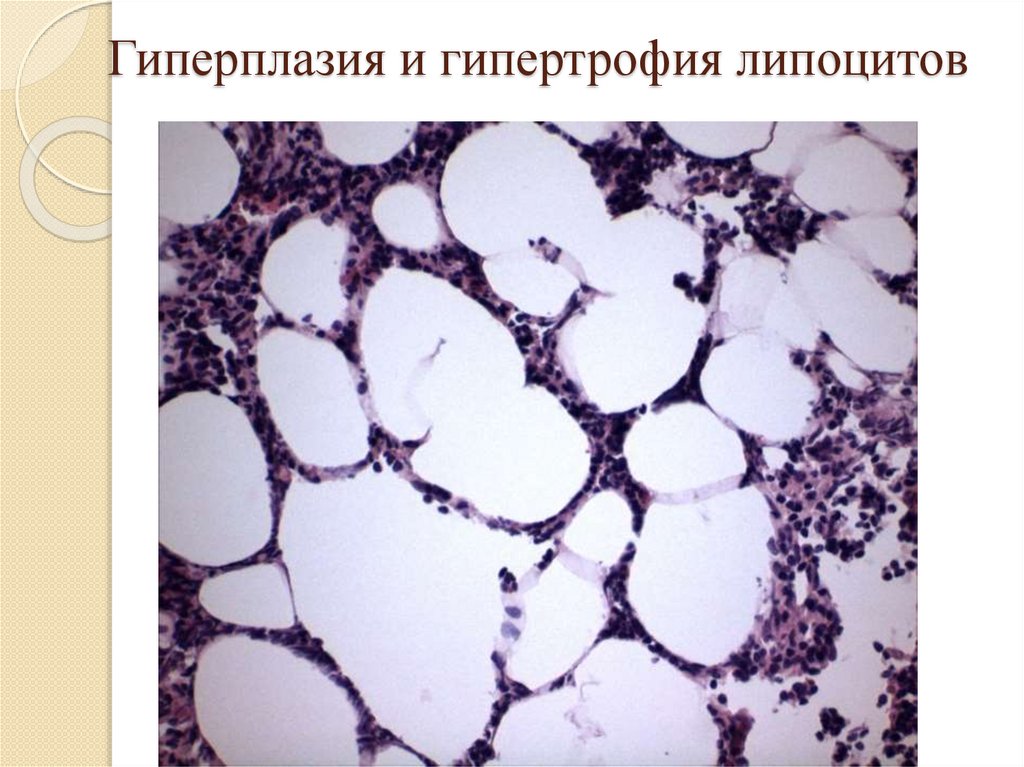
Гиперплазия и гипертрофия липоцитов97.
Жировые сосудисто-стромальныедистрофии (ожирение)
Липоматоз – диффузное или
множественное опухолевидное
разрастание жировой ткани в
организме.
Вакатное ожирение – местное
увеличение количества жировой
ткани при атрофии органа (жировое
замещение).
98.
Жировые сосудисто-стромальныедистрофии (ожирение)
Истощение – снижение веса,
сопровождающееся сокращением
количества нейтральных жиров в
жировых депо, уменьшением объема и
массы органов с утратой способности к
выполнению физиологических функций.
Кахексия – резко выраженное
истощение организма.
99.
Жировые сосудисто-стромальныедистрофии (ожирение)
Регионарные липодистрофии
характеризуются очаговой
деструкцией жировой ткани и
распадом жиров, часто с
воспалительной реакцией и
образованием липогранулём.
100.
Углеводные сосудистостромальные дистрофииСтромально-сосудистые
углеводные дистрофии могут
быть связаны с нарушением
баланса
гликопротеидов
и
гликозаминогликанов
101.
Углеводные сосудисто-стромальныедистрофии
Ослизнение тканей – стромально-
сосудистая дистрофия, связанная с
нарушением обмена гликопротеидов,
при которой хромотропные вещества
высвобождаются из связей с белками
и накапливаются в межуточном
веществе соединительной ткани.
102.
Углеводные сосудисто-стромальныедистрофии
Основные причины ослизнения
тканей:
истощение, кахексия любого
генеза;
дисфункция эндокринных желез.
103.
Углеводные сосудисто-стромальныедистрофии
Наследственные нарушения обмена
гликозаминогликанов
(мукополисахаридов) представлены
большой группой болезней
накопления – мукополисахаридозами.