

















 Химия
ХимияПохожие презентации:






")



Органические реакции. Кислоты и основания. Классификация органических реакций. Одноэлектронные реакции. Теория Бренстеда
1.
Северо-Казахстанский университет им. М. КозыбаеваКафедра химии и химических технологий
Тема лекции:
Органические реакции. Кислоты и основания.
Классификация органических реакций. Одноэлектронные реакции. Теория Бренстеда.
Кислотно-основные реакции Льюиса.
Лектор: Луценко А.А.
Доктор PhD, доцент.
Казахстан
Петропавловск, 2024 г.
2.
КЛАССИФИКАЦИЯ ОРГАНИЧЕСКИХ РЕАКЦИЙОрганические реакции классифицируют по различным признакам:
— по типу превращения субстрата;
— по типу активирования;
— по характеру разрыва связей.
Особую группу органических реакций составляют одноэлектронные реакции.
Классификация по типу превращения субстрата
Реакции замещения.
Замещение — реакция, в ходе которой атом водорода (или функциональная группа) в органической молекуле
замещается на какую-либо функциональную группу (или атом водорода). В общем виде реакцию замещения X—>Y
можно записать в виде:
Исходные соединения в органических реакциях называют реагентами, а образующиеся соединения — продуктами. В
уравнении (1) R–X и Y — реагенты, а R–Y и X — продукты.
Для удобства один из реагентов принято называть субстратом, а другой — атакующим реагентом. Как правило,
субстрат имеет более сложное строение, атакующий реагент часто имеет неорганическую природу.
3.
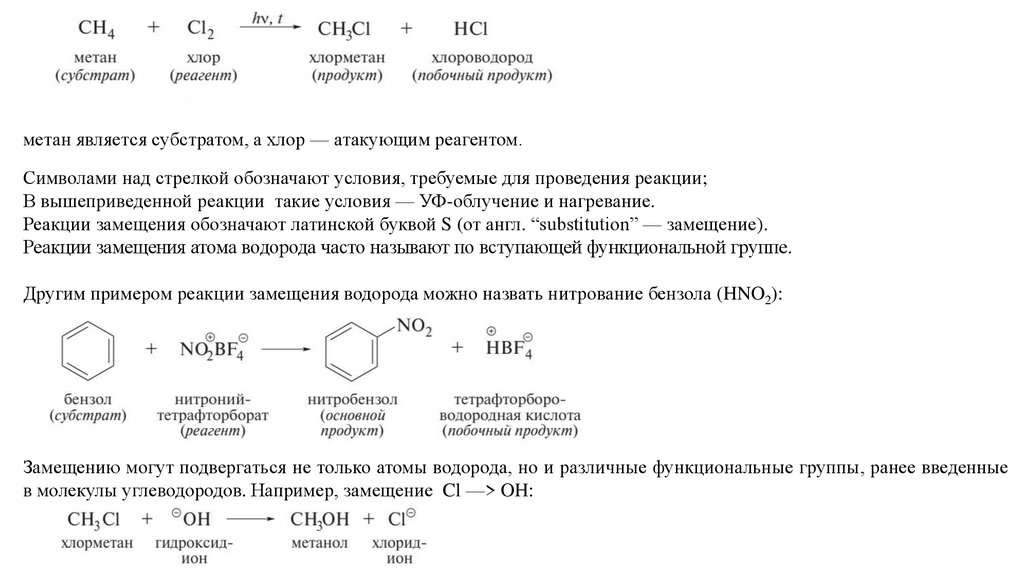
метан является субстратом, а хлор — атакующим реагентом.Символами над стрелкой обозначают условия, требуемые для проведения реакции;
В вышеприведенной реакции такие условия — УФ-облучение и нагревание.
Реакции замещения обозначают латинской буквой S (от англ. “substitution” — замещение).
Реакции замещения атома водорода часто называют по вступающей функциональной группе.
Другим примером реакции замещения водорода можно назвать нитрование бензола (HNO2):
Замещению могут подвергаться не только атомы водорода, но и различные функциональные группы, ранее введенные
в молекулы углеводородов. Например, замещение Cl —> OH:
4.
Реакции присоединения.Присоединение — реакция, в ходе которой реагент присоединяется по кратной связи (С=С, С=О, С=N) молекулы
субстрата. Это, например, гидробромирование этилена:
Реакции присоединения обозначают латинским символом Аd (от англ. “addition” — присоединение). Продукт такой
реакции обычно называют аддуктом. Другие примеры реакций присоединения: — образование циангидрина
ацетальдегида:
— присоединение HCl к ацетилен
Реакции элиминирования.
Элиминирование — реакция, в ходе которой от субстрата отщепляется молекула или частица (вода, галогеноводород
и т.д.). Этот тип превращения обозначают латинской буквой Е (от англ. “elimination” — элиминирование,
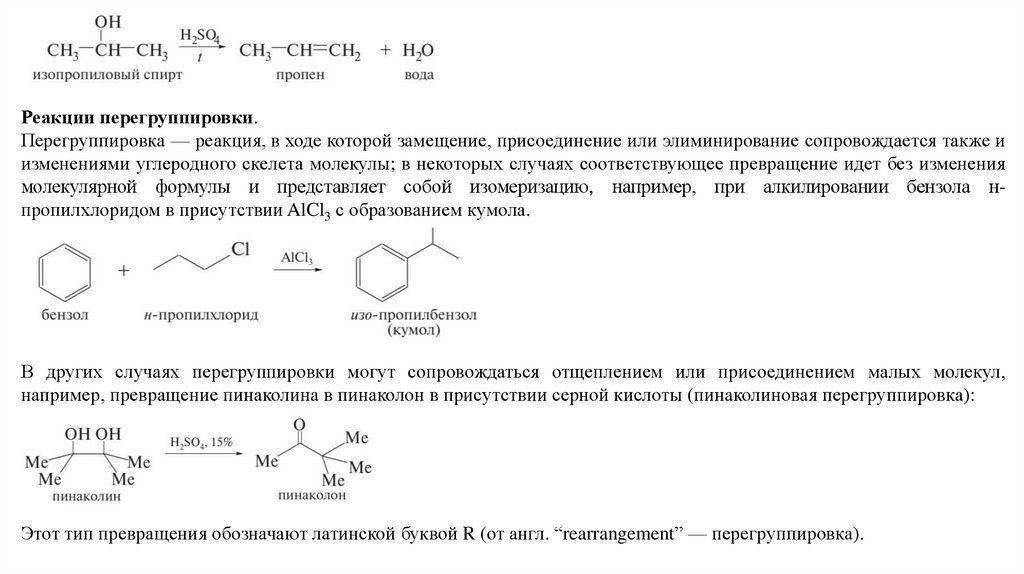
отщепление). Например, дегидратация изопропилового спирта представляет собой реакцию элиминирования.
5.
Реакции перегруппировки.Перегруппировка — реакция, в ходе которой замещение, присоединение или элиминирование сопровождается также и
изменениями углеродного скелета молекулы; в некоторых случаях соответствующее превращение идет без изменения
молекулярной формулы и представляет собой изомеризацию, например, при алкилировании бензола нпропилхлоридом в присутствии AlCl3 с образованием кумола.
В других случаях перегруппировки могут сопровождаться отщеплением или присоединением малых молекул,
например, превращение пинаколина в пинаколон в присутствии серной кислоты (пинаколиновая перегруппировка):
Этот тип превращения обозначают латинской буквой R (от англ. “rearrangement” — перегруппировка).
6.
Еще пример — перегруппировка 1-хлорпропана в 2-хлорпропан в присутствии хлорида алюминия:Некаталитическими являются реакции, которые не требуют присутствия катализатора. Эти реакции ускоряются
только при повышении температуры, их иногда называют термическими. Такой способ активирования обозначают
значком t.
Исходными реагентами в некаталитических реакциях служат высокополярные или заряженные частицы.
Каталитическими называют реакции, протекание которых требует присутствия катализатора.
Если в качестве катализатора выступает кислота, речь идет о кислотном катализе.
Если в качестве катализатора выступает основание, речь идет об основном катализе.
Фотохимические реакции — реакции, которые активируют облучением; такой способ активирования обозначают hν.
Фотохимически активируют также и реакцию димеризации этилена:
Важно отметить, что эта реакция не протекает в темноте даже при значительном нагревании.
7.
КЛАССИФИКАЦИЯ ПО ХАРАКТЕРУ РАЗРЫВА СВЯЗЕЙРадикальные реакции
Радикальные реакции сопровождаются гомолитическим разрывом связей и образованием радикалов — нейтральных
частиц, содержащих один или несколько неспаренных электронов:
Радикальные реакции особенно распространены в превращениях алканов.
SR
Ионные реакции
Ионные реакции протекают с участием ионов и сопровождаются гетеролитическим разрывом связей в субстрате:
Заряженную частицу, имеющую вакантную р-орбиталь на атоме углерода, называют карбокатионом.
Заряженную частицу, содержащую НЭП на атоме углерода, называют карбанионом.
8.
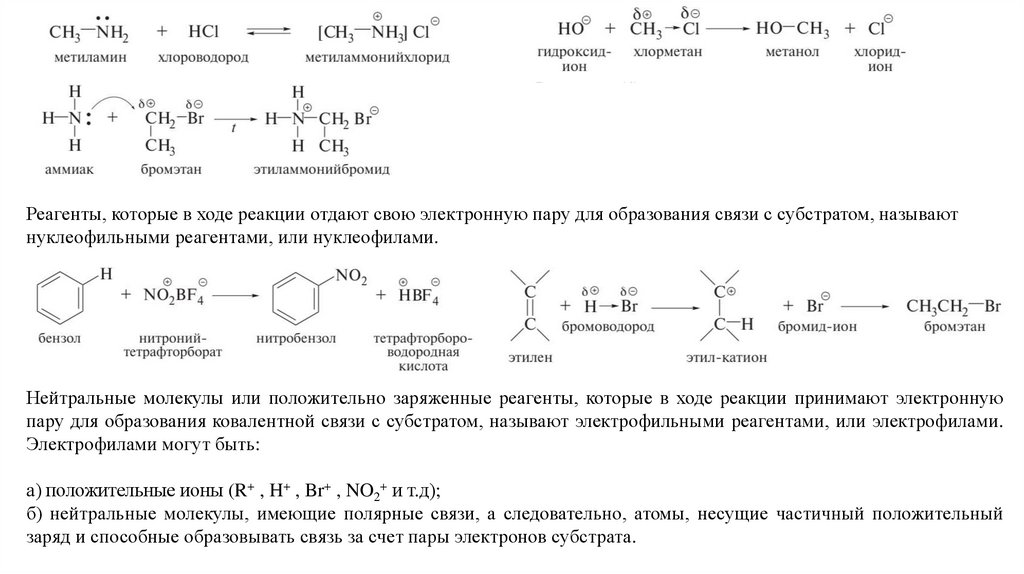
Реагенты, которые в ходе реакции отдают свою электронную пару для образования связи с субстратом, называютнуклеофильными реагентами, или нуклеофилами.
Нейтральные молекулы или положительно заряженные реагенты, которые в ходе реакции принимают электронную
пару для образования ковалентной связи с субстратом, называют электрофильными реагентами, или электрофилами.
Электрофилами могут быть:
а) положительные ионы (R+ , H+ , Br+ , NO2+ и т.д);
б) нейтральные молекулы, имеющие полярные связи, а следовательно, атомы, несущие частичный положительный
заряд и способные образовывать связь за счет пары электронов субстрата.
9.
Определение характера реагента — радикальный, нуклеофильный или электрофильный — позволяет уточнитьклассификацию органических реакций по типу превращения.
Определение характера реагента — радикальный, нуклеофильный или электрофильный — позволяет уточнить
классификацию органических реакций по типу превращения.
Синхронные реакции.
Синхронные реакции протекают без промежуточного образования ионов и радикалов: разрыв старых и образование
новых связей происходят одновременно (синхронно).
Примером синхронной реакции является диеновый синтез — реакция Дильса–Альдера:
10.
Отдельную группу органических реакций составляют одноэлектронные реакции. Их своеобразие заключается в том,что они не сопровождаются разрывом старых и образованием новых связей. В ходе этих реакций органическая
молекула не подвергается заметной трансформации, поскольку она лишь отдает или присоединяет один электрон.
Одноэлектронные реакции являются промежуточными стадиями многих процессов, в которых участвуют органические
соединения. В частности, отрыв электрона от органической молекулы часто сопровождает реакции электрофильного
замещения (SE) и окисления.
Отрыв электрона от органической молекулы ведет к образованию катион-радикала:
Присоединение электрона к молекуле субстрата наблюдается в реакциях восстановления.
Присоединение электрона к органической молекуле ведет к образованию анион-радикала:
11.
КИСЛОТЫ И ОСНОВАНИЯ. ТЕОРИЯ БРЁНСТЕДАВ органической химии известно несколько теорий, которые объясняют кислотно-основные свойства органических
соединений. Прежде всего это теории Брёнстеда и Льюиса.
Согласно теории Брёнстеда (Й. Брёнстед, 1923 г.), кислота — это любое вещество, способное диссоциировать с
отщеплением протона, а основание — любое соединение, способное присоединять протон:
Кислоты Брёнстеда.
Мерой силы кислоты Брёнстеда является константа ее диссоциации Kа, определяемая по отношению к воде как
основанию, или соответствующее значение pKа .
12.
Наиболее известные примеры органических кислот — алифатические и ароматические карбоновые кислоты.Отщеплять протон могут органические соединения, относящиеся и к другим классам. Среди органических соединений
различают OH-, SH-, NH- и CH-кислоты. OH-кислоты > NH-кислоты > CH-кислоты
Отдельно следует отметить наиболее сильную из органических кислот — трифторметансульфоновую кислоту
CF3SO3H, которая относится к группе так называемых суперкислот.
Сила этой кислоты превышает кислотность сильных минеральных кислот, в том числе серной и соляной кислот
(значение рКа достигает –15).
Чем устойчивее сопряженное основание, тем ниже свободная энергия диссоциации ΔG°, тем более равновесие смещено
вправо, тем сильнее кислота. Зависимость между значениями ΔG° и lgKa выражается уравнением
ΔG° = –2,303 RT lgKa ,
где R = 8,31 ⋅ 10–3 кДж/(К⋅моль), T — температура в градусах Кельвина.
1. Чем выше номер группы периодической системы, к которой относится элемент, тем сильнее он удерживает
электроны в сопряженном основании, тем сильнее соответствующая кислота.
13.
2. Более высокая кислотность карбоновых кислот по сравнению со спиртами объясняется большейустойчивостью карбоксилат-иона по сравнению с алкоксид-ионом. Отрицательный заряд в карбоксилат-ионе,
например в ацетат-ионе, резонансно делокализован на двух атомах кислорода:
В алкоксид-ионе, например в метоксид-ионе, такая делокализация невозможна.
14.
3. Введение электронодонорных заместителей дестабилизирует сопряженное основание и снижает силу кислоты.4. Введение электроноакцепторных заместителей стабилизирует сопряженное основание и повышает силу
кислоты. Ниже на примере замещенных уксусных кислот показана стабилизация карбоксилат-ионов за счет
индуктивного эффекта заместителя Х.
5. Удаление по цепи электроноакцепторного заместителя от карбоксильной группы ведет к снижению силы
кислоты.
15.
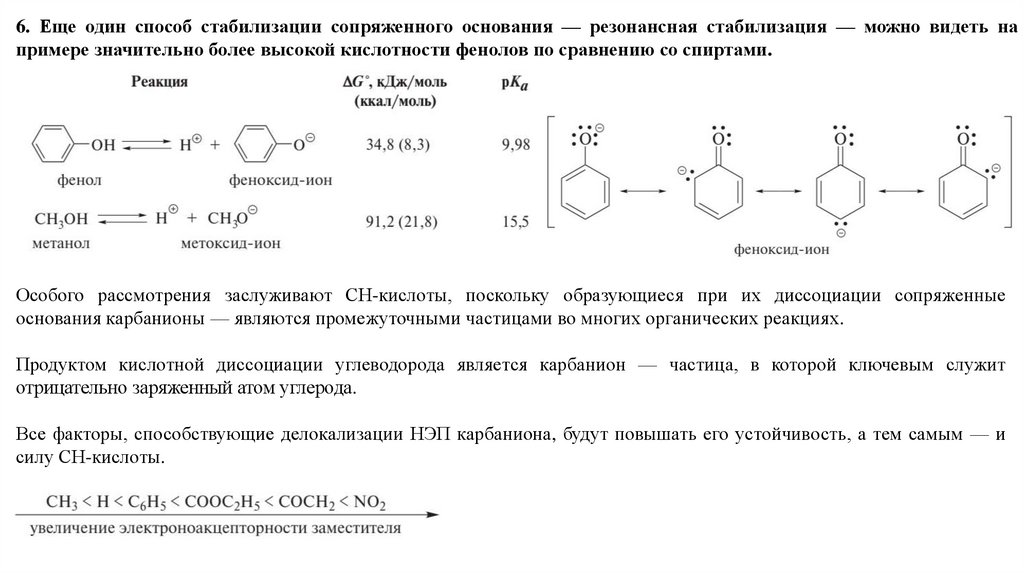
6. Еще один способ стабилизации сопряженного основания — резонансная стабилизация — можно видеть напримере значительно более высокой кислотности фенолов по сравнению со спиртами.
Особого рассмотрения заслуживают СН-кислоты, поскольку образующиеся при их диссоциации сопряженные
основания карбанионы — являются промежуточными частицами во многих органических реакциях.
Продуктом кислотной диссоциации углеводорода является карбанион — частица, в которой ключевым служит
отрицательно заряженный атом углерода.
Все факторы, способствующие делокализации НЭП карбаниона, будут повышать его устойчивость, а тем самым — и
силу СН-кислоты.
16.
Эффект делокализации отрицательного заряда в бензил-анионе столь велик, что его геометрия приближается кплоской, при этом углеродный атом карбанионного центра меняет свою гибридизацию с sp3 на sp2.
Электронодонорные заместители, повышающие электронную плотность на карбанионном центре, дестабилизируют
карбанион и тем самым снижают кислотность соответствующего углеводорода.
Основания Брёнстеда
Сравнение устойчивости исходного соединения и конечного продукта — объясняет закономерности в изменении силы
органических оснований, в том числе оснований Брёнстеда.
17.
Константу соответствующего равновесия обозначают как Kb:Чем больше значение Kb, тем выше сила основания. Более универсальной оценкой силы органического основания,
однако, является величина pKa его сопряженной кислоты — pKa(ВН⊕).
Слабым основаниям соответствуют сильные сопряженные кислоты и наоборот: сильным основаниям
соответствуют слабые сопряженные кислоты.
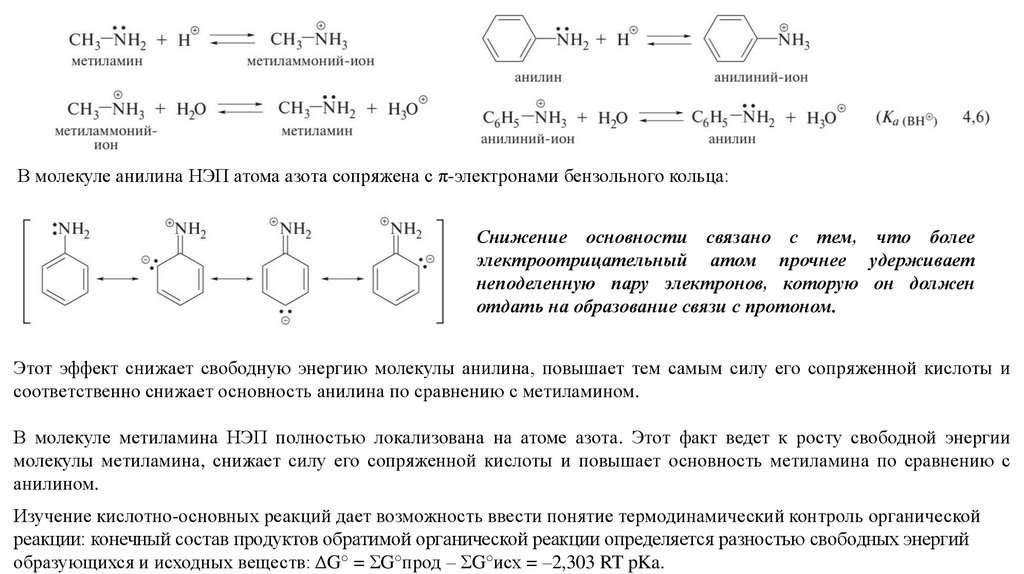
Типичными основаниями по Брёнстеду являются органические амины.
При этом алифатические амины — значительно более сильные основания, нежели ароматические амины.
аммониевые основания > оксониевые основания
18.
В молекуле анилина НЭП атома азота сопряжена с π-электронами бензольного кольца:Снижение основности связано с тем, что более
электроотрицательный атом прочнее удерживает
неподеленную пару электронов, которую он должен
отдать на образование связи с протоном.
Этот эффект снижает свободную энергию молекулы анилина, повышает тем самым силу его сопряженной кислоты и
соответственно снижает основность анилина по сравнению с метиламином.
В молекуле метиламина НЭП полностью локализована на атоме азота. Этот факт ведет к росту свободной энергии
молекулы метиламина, снижает силу его сопряженной кислоты и повышает основность метиламина по сравнению с
анилином.
Изучение кислотно-основных реакций дает возможность ввести понятие термодинамический контроль органической
реакции: конечный состав продуктов обратимой органической реакции определяется разностью свободных энергий
образующихся и исходных веществ: ΔG° = ΣG°прод – ΣG°исх = –2,303 RT pKa.
19.
Чем выше ΔG°, тем менее диссоциирована (т. е. тем слабее) кислота.Чем ниже ΔG°, тем более диссоциирована (т. е. тем сильнее) кислота.
ОБОБЩЕННАЯ ТЕОРИЯ КИСЛОТ И ОСНОВАНИЙ. КИСЛОТНО-ОСНОВНЫЕ РЕАКЦИИ ЛЬЮИСА
Кислота Льюиса — любая молекула или частица, способная принимать (акцептировать) электроны на вакантную
орбиталь. В качестве вакантной орбитали выступает, как правило, НСМО. Она может иметь как σ-, так и π-симметрию.
Соответственно этому среди кислот Льюиса можно различать σ- и π-кислоты.
незаряженные (нейтральные) – молекулы и частицы: BF3, AlCl3, FeBr3, ZnCl2, :CCl2;
положительно заряженные – частицы (катионы): NO2+, Br+ , CH3+ , R3C+, NO+, RCO+, H+.
Типичной σ-кислотой является протон. Большинство кислот Льюиса представляют собой π-кислоты.
К π-кислотам следует отнести также алкены и арены, содержащие электроноакцепторные заместители:
Основание Льюиса — любая частица, способная выступать донором пары электронов. Различают:
1) π-основания (π-доноры),
2) p-основания (p-доноры),
3) σ-основания (σ-доноры).
В π-основаниях высшей занятой молекулярной орбиталью является π-орбиталь. Ими являются алкены R—СH=СH2,
бензол и его производные, другие арены. В p-основаниях донорной является орбиталь, занимаемая НЭП: R-OH, R3N,
R2S, R3C-, RO-, R2N-, σ-основаниях высшей занятой молекулярной орбиталью является σ-орбиталь. Алканы и
циклоалканы, в которых ВЗМО локализованы в области C—C- и С—Н-связей, могут выступать в качестве доноров
электронов при взаимодействии со сверхсильными кислотами и комплексами переходных металлов.
20.
Таким образом, основания Бренстеда и Льюиса – это одни и те же частицы и молекулы.Однако основность по Бренстеду есть способность присоединять только протон, в то время как основность по Льюису –
cпособность к взаимодействию с любой частицей, имеющей низколежащую свободную орбиталь.
Кислотно-основное взаимодействие по Льюису есть донорно-акцепторное взаимодействие, и любую гетеролитическую
реакцию можно представить как взаимодействие кислоты и основания Льюиса:
Для оценки легкости протекания кислотно-основного взаимодействия по Льюису Р. Пирсоном была предложена
качественная теория «жестких» и «мягких» кислот и оснований.
Жесткие основания обладают высокой электроотрицательностью и низкой поляризуемостью. Они трудно
окисляются. Их высшие занятые молекулярные орбитали имеют низкую энергию.
Мягкие основания имеют низкую электроотрицательность и высокую поляризуемость. Они легко окисляются.
Их высшие занятые молекулярные орбитали (ВЗМО) имеют высокую энергию.
21.
Жесткие кислоты имеют высокую электроотрицательность и низкую поляризуемость. Они трудновосстанавливаются. Их низшие свободные молекулярные орбитали имеют низкую энергию.
Мягкие кислоты обладают низкой электроотрицательностью и высокой поляризуемостью. Они легко
восстанавливаются. Их низшие свободные молекулярные орбитали имеют высокую энергию.
Самая жесткая кислота – Н+, самая мягкая – СН3Hg+.
Наиболее жесткие основания – F– и OH–, наиболее мягкие – I– и Н–.
Кислотно-основные реакции Льюиса
Карбокатионы
В кислотно-основной реакции Льюиса основание (D) является донором электронной пары и образует ковалентную
связь с кислотой (А), предоставляющей для этого вакантную (акцепторную) орбиталь.
На первой стадии кислотно-основной реакции Льюиса, как правило, образуется донорно-акцепторный комплекс
(ДАК):
Донорно-акцепторный комплекс — продукт кислотно-основной реакции Льюиса, в котором основание (донор) и
кислота (акцептор) связаны координационной связью.
22.
Взаимодействие бромметана (основание Льюиса) с алюминийтрибромидом (кислота Льюиса) протекает через рядстадий:
Появление полного положительного заряда на атоме брома в ДАК ведет к еще большей поляризации связи С–Br (по
сравнению с исходной молекуой бромметана) вплоть до ее полного гетеролитического разрыва (гетеролиз).
Причиной нестабильности метил-катиона является отсутствие каких-либо факторов, способствующих делокализации
положительного заряда в этой частице. Поскольку атом углерода метил-катиона sp2-гибридизован, все четыре атома
находятся в од- ной плоскости, а вакантная орбиталь перпендикулярна этой плоскости, т. е. является π-орбиталью.
Вследствие электронодонорного влияния метильных групп по индуктивному механизму, а также по механизму
сверхсопряжения (гиперконъюгация) устойчивость карбокатионов увеличивается в ряду: CH3 < CH2CH3 < CH(CH3)2 <
C(CH3)3. К примеру делокализация положительного заряда в трет-бутил-катионе за счет +I- и +Мh-эффектов трех
метильных групп:
23.
Способность фенильной и винильной групп к резонансной стабилизации положительного заряда на соседнемС-атоме резко увеличивает устойчивость бензильного и аллильного катионов.
Кроме винильной и фенильной групп стабилизировать соседний катионный центр могут и заместители, обладающие
НЭП: