













 Химия
ХимияПохожие презентации:










Кислородсодержащие органические соединения
1.
ЛекцияКислородсодержащие органические соединения
2.
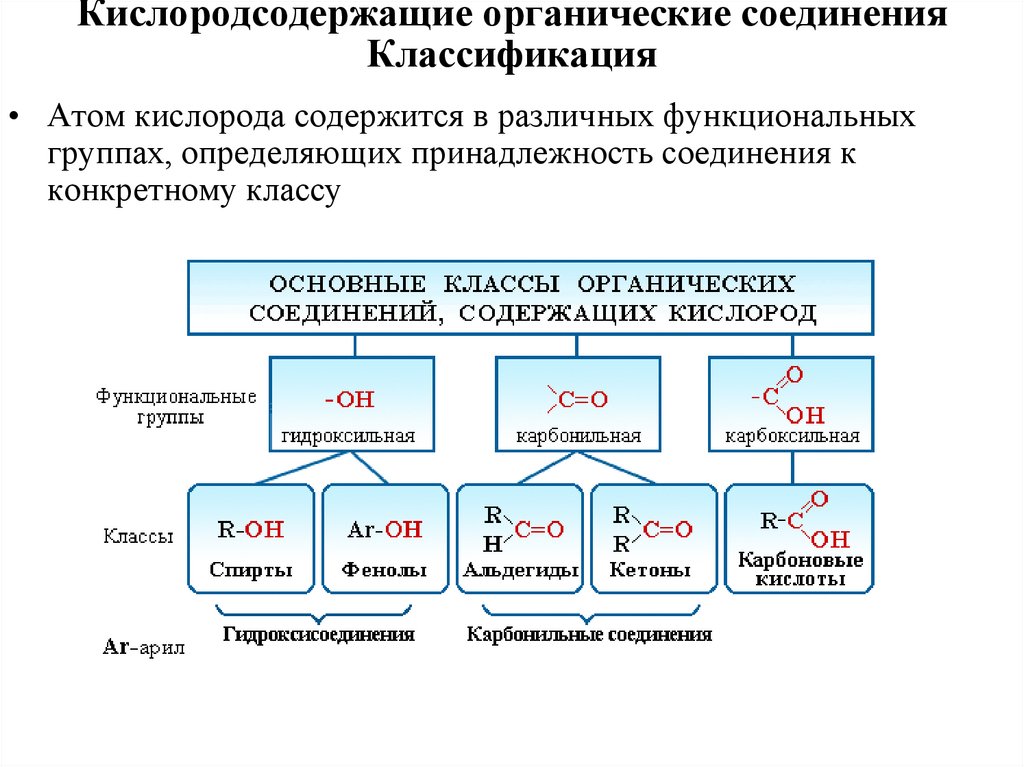
Кислородсодержащие органические соединенияКлассификация
• Атом кислорода содержится в различных функциональных
группах, определяющих принадлежность соединения к
конкретному классу
3.
Кислородсодержащие органические соединенияКлассификация
• Coединения каждого класса образуют различные производные. Например
к производным спиртов относятся простые эфиры R-O-R’, к производным
карбоновых кислот – сложные эфиры RC(O)-OR’, амиды RC(O)-NH2,
ангидриды (RCO)2O, хлорангидриды RCOCl и т.д.
• Кроме того, большую группу составляют гетерофункциональные
соединения, содержащие различные функциональные группы:
• гидроксиальдегиды HO-R-CHO; гидроксикетоны HO-R-C(O)-R’;
гидроксикислоты HO-R-COOH и т.д.
• К важнейшим гетерофункциональным кислородсодержащим относятся
углеводы, молекулы которых включают гидроксильные, карбонильные и
производные от них группы.
4.
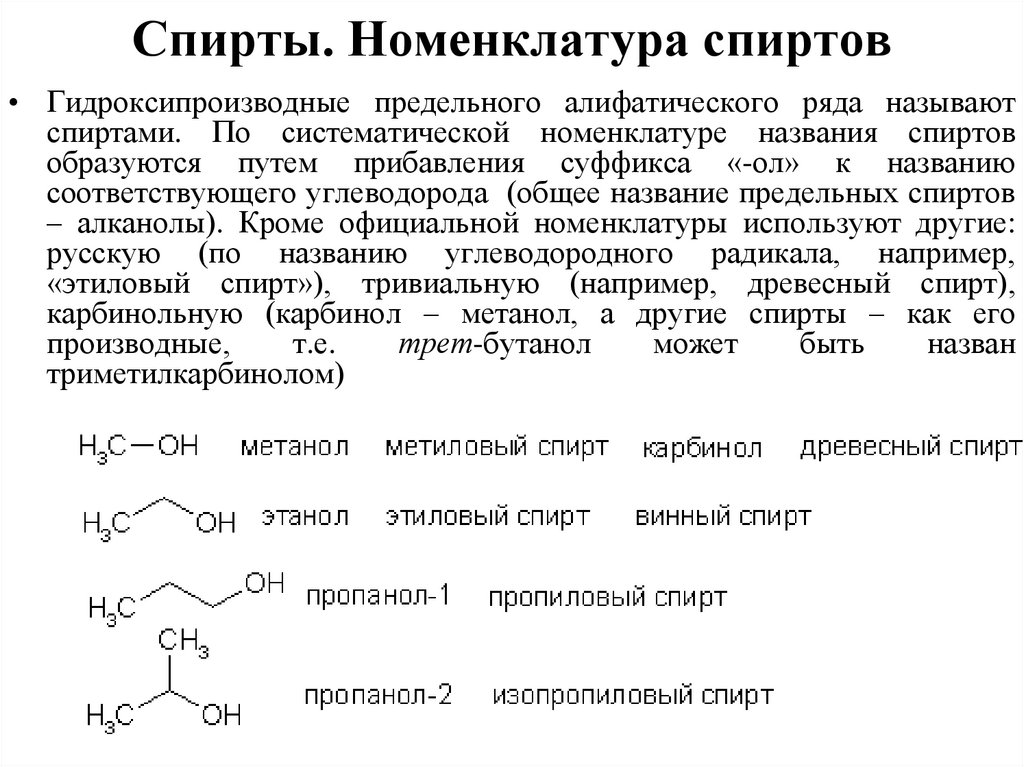
Спирты. Номенклатура спиртов• Гидроксипроизводные предельного алифатического ряда называют
спиртами. По систематической номенклатуре названия спиртов
образуются путем прибавления суффикса «-ол» к названию
соответствующего углеводорода (общее название предельных спиртов
– алканолы). Кроме официальной номенклатуры используют другие:
русскую (по названию углеводородного радикала, например,
«этиловый спирт»), тривиальную (например, древесный спирт),
карбинольную (карбинол – метанол, а другие спирты – как его
производные,
т.е.
трет-бутанол
может
быть
назван
триметилкарбинолом)
5.
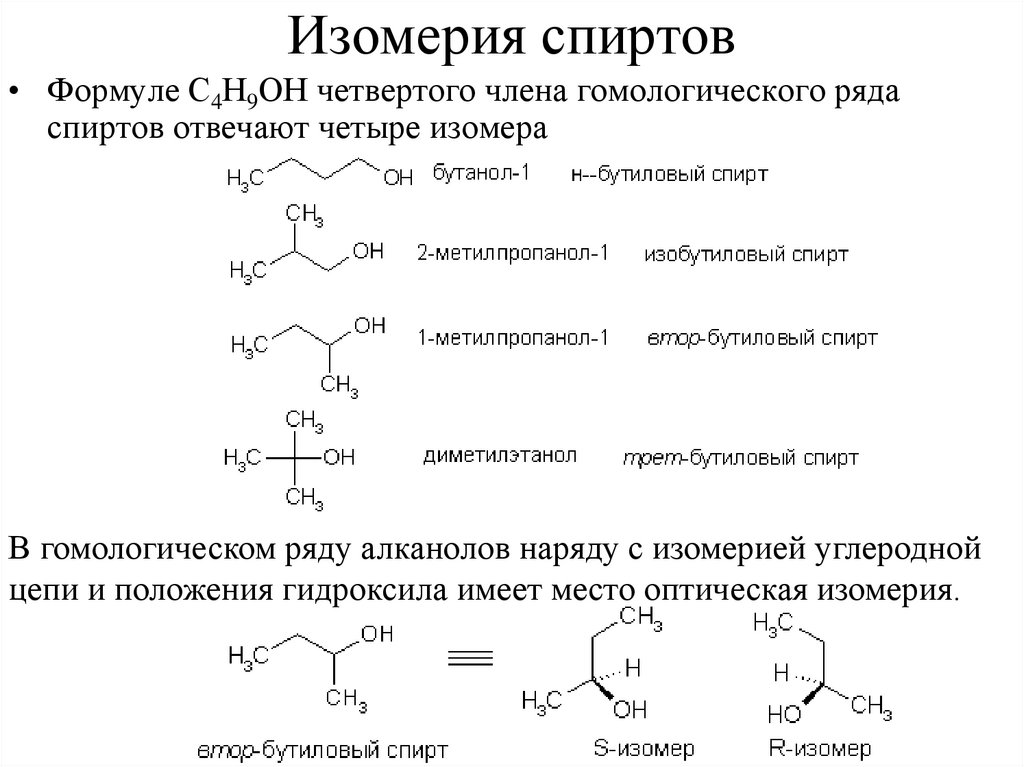
Изомерия спиртов• Формуле С4Н9ОН четвертого члена гомологического ряда
спиртов отвечают четыре изомера
В гомологическом ряду алканолов наряду с изомерией углеродной
цепи и положения гидроксила имеет место оптическая изомерия.
6.
Непредельные спирты• Непредельные спирты бывают двух типов: виниловые спирты,
содержащие гидроксил при этиленовом углероде, и аллильные спирты, в
которых гидроксил отделен от этиленовой связи группой СН2.
Ароматические спирты рассматривают как алканолы, в молекулах
которых присутствуют ароматические кольца. Те спирты, в которых
кратные связи отделены от ОН-группы более чем одним sp3-гибридным
атомом углерода, по свойствам не отличаются от обычных алканолов
Виниловые спирты, не содержащие сильных акцепторов в молекуле, крайне
неустойчивы и переходят в более стабильную кето-форму. Они, являются
таутомерной формой карбонильных соединений, которая, как правило,
присутствует в веществе в незначительной концентрации:
7.
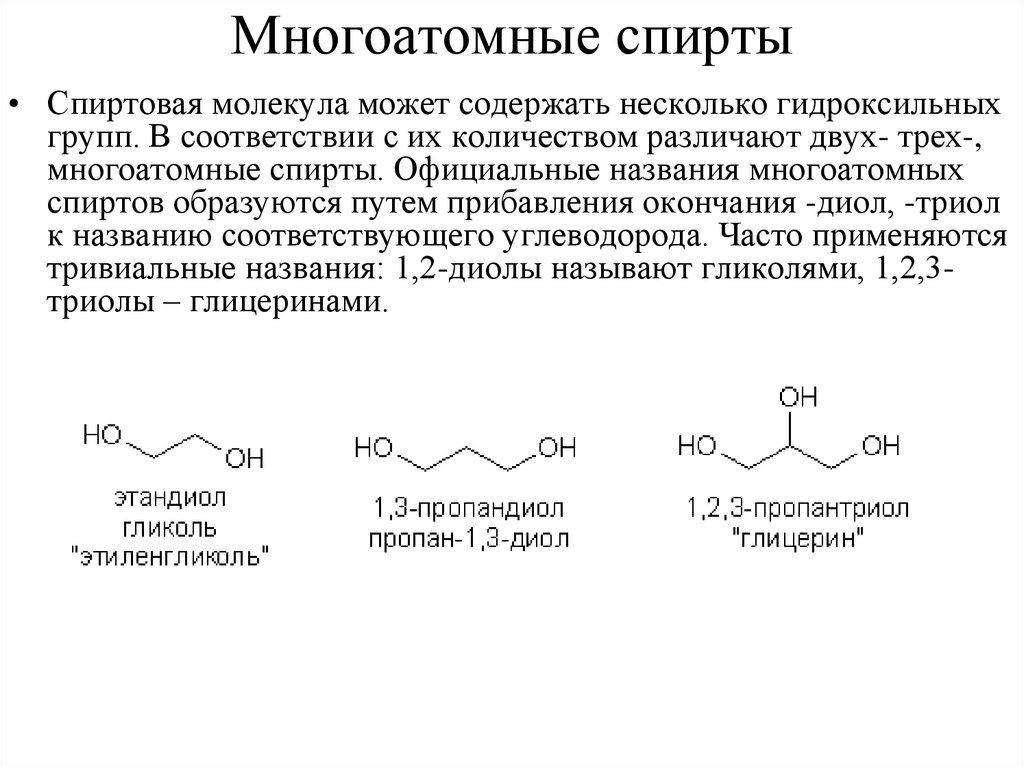
Многоатомные спирты• Спиртовая молекула может содержать несколько гидроксильных
групп. В соответствии с их количеством различают двух- трех-,
многоатомные спирты. Официальные названия многоатомных
спиртов образуются путем прибавления окончания -диол, -триол
к названию соответствующего углеводорода. Часто применяются
тривиальные названия: 1,2-диолы называют гликолями, 1,2,3триолы – глицеринами.
8.
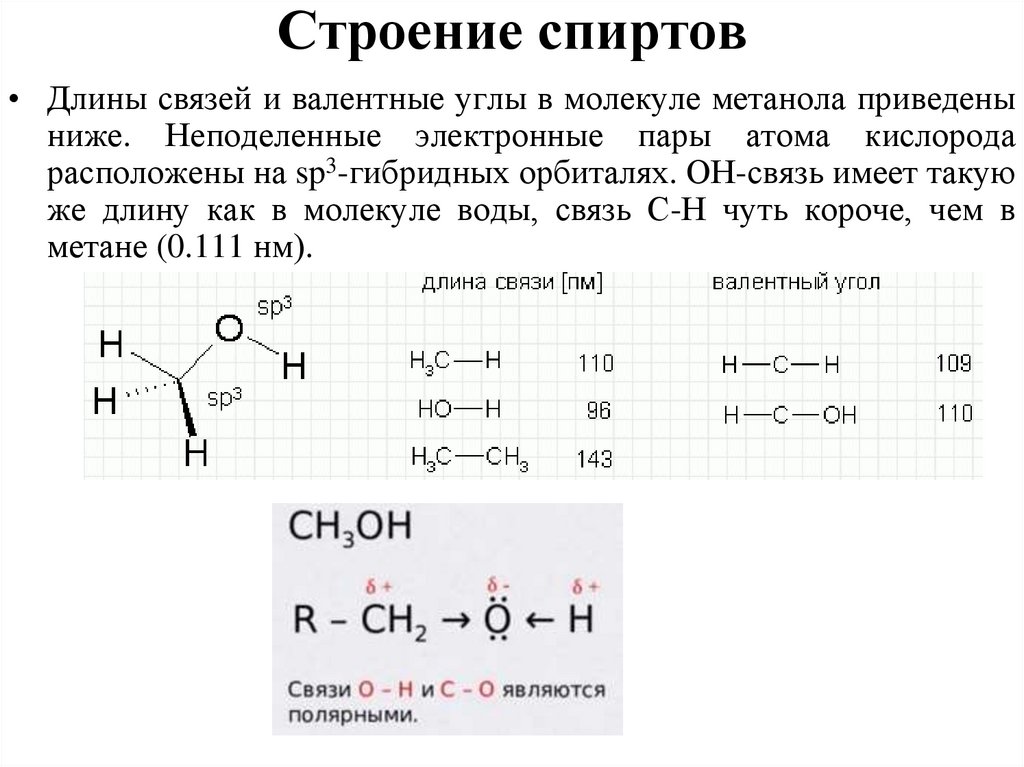
Строение спиртов• Длины связей и валентные углы в молекуле метанола приведены
ниже. Неподеленные электронные пары атома кислорода
расположены на sp3-гибридных орбиталях. ОН-связь имеет такую
же длину как в молекуле воды, связь С-Н чуть короче, чем в
метане (0.111 нм).
9.
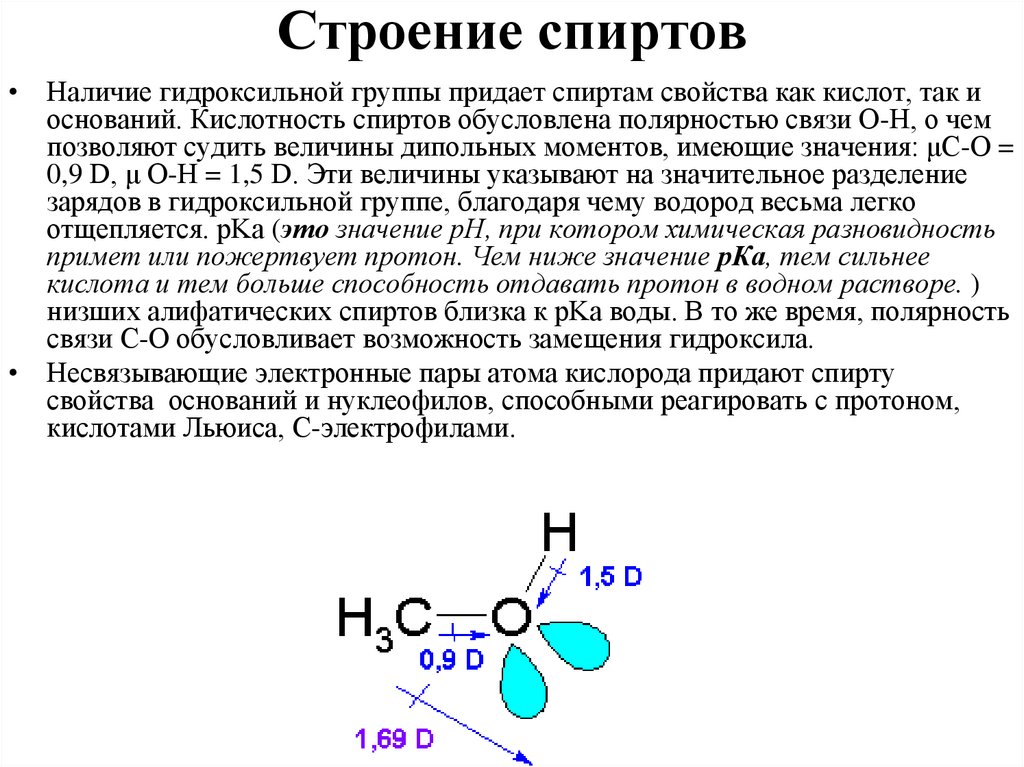
Строение спиртов• Наличие гидроксильной группы придает спиртам свойства как кислот, так и
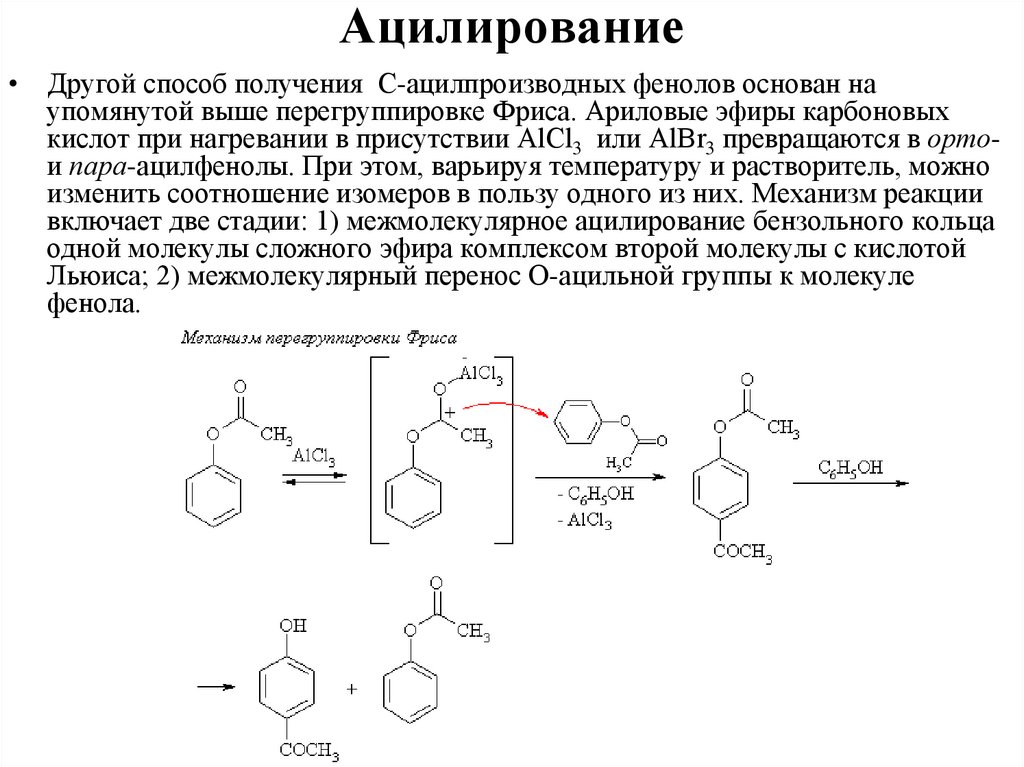
оснований. Кислотность спиртов обусловлена полярностью связи О-Н, о чем
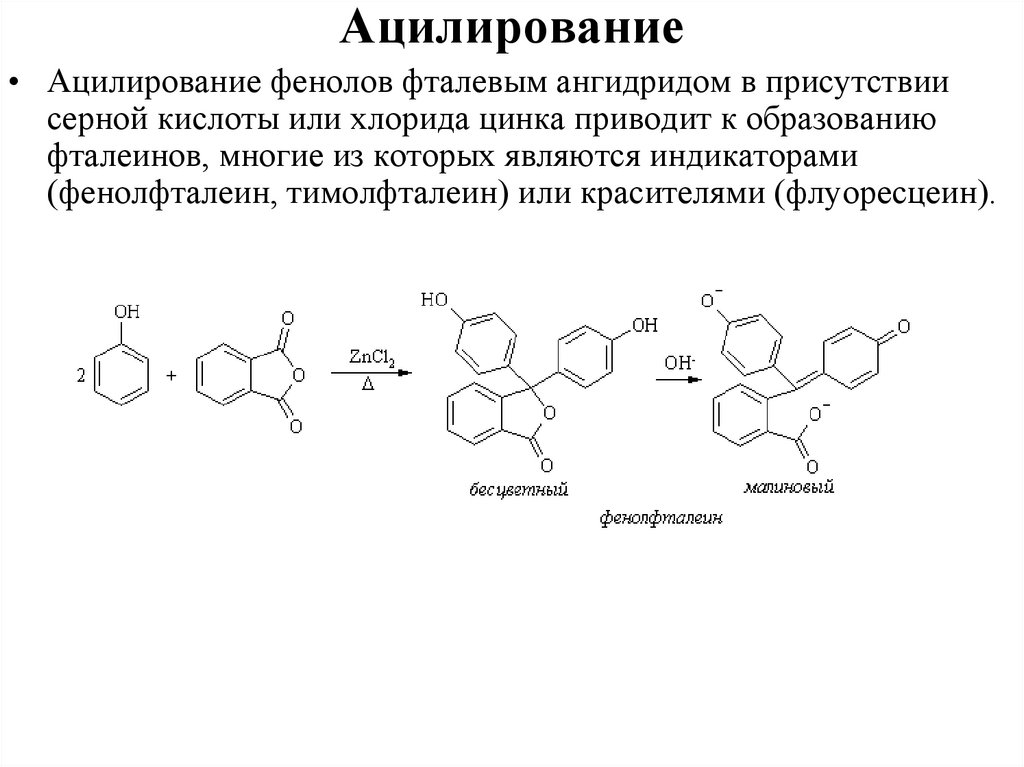
позволяют судить величины дипольных моментов, имеющие значения: μС-О =
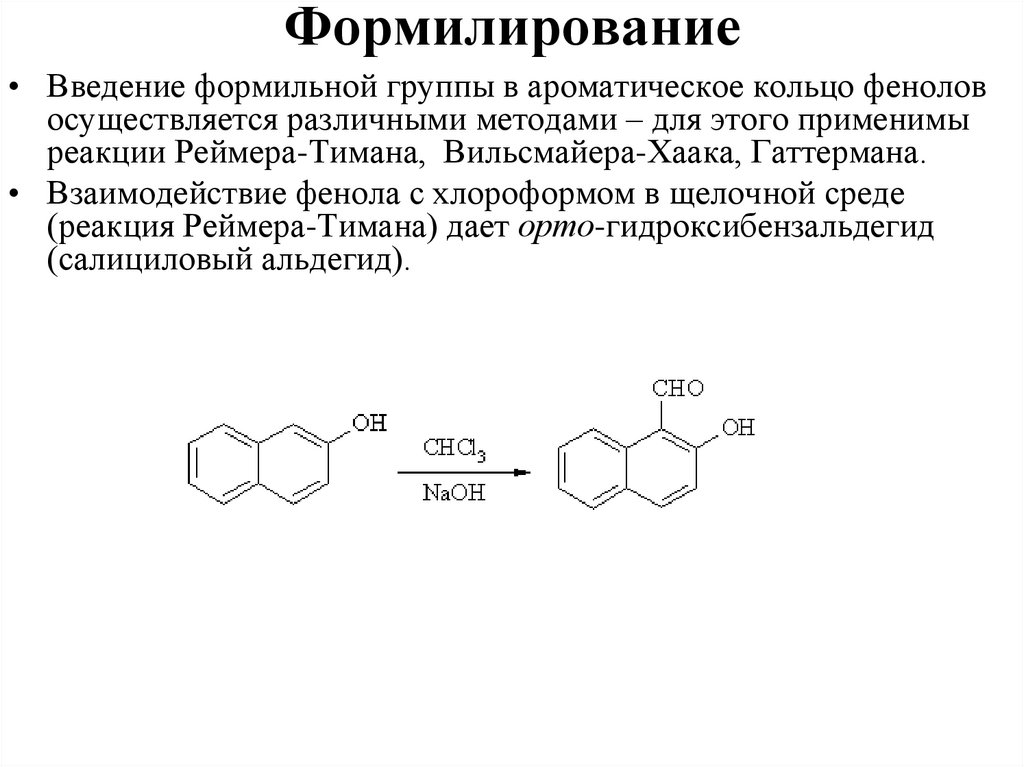
0,9 D, μ О-Н = 1,5 D. Эти величины указывают на значительное разделение
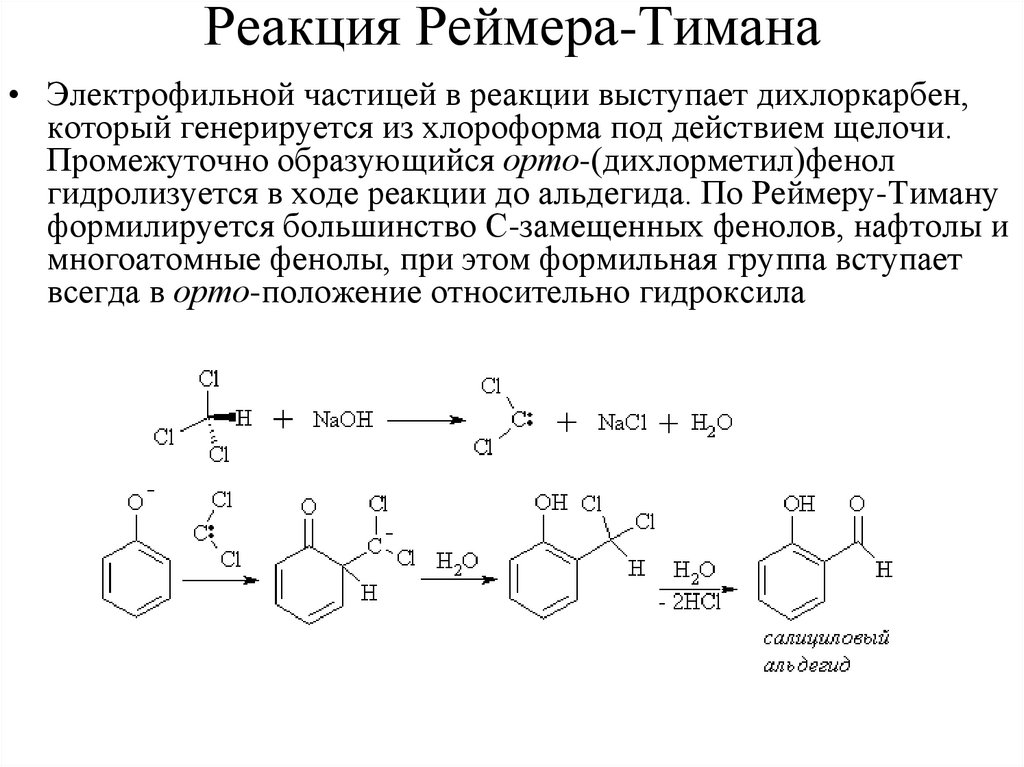
зарядов в гидроксильной группе, благодаря чему водород весьма легко
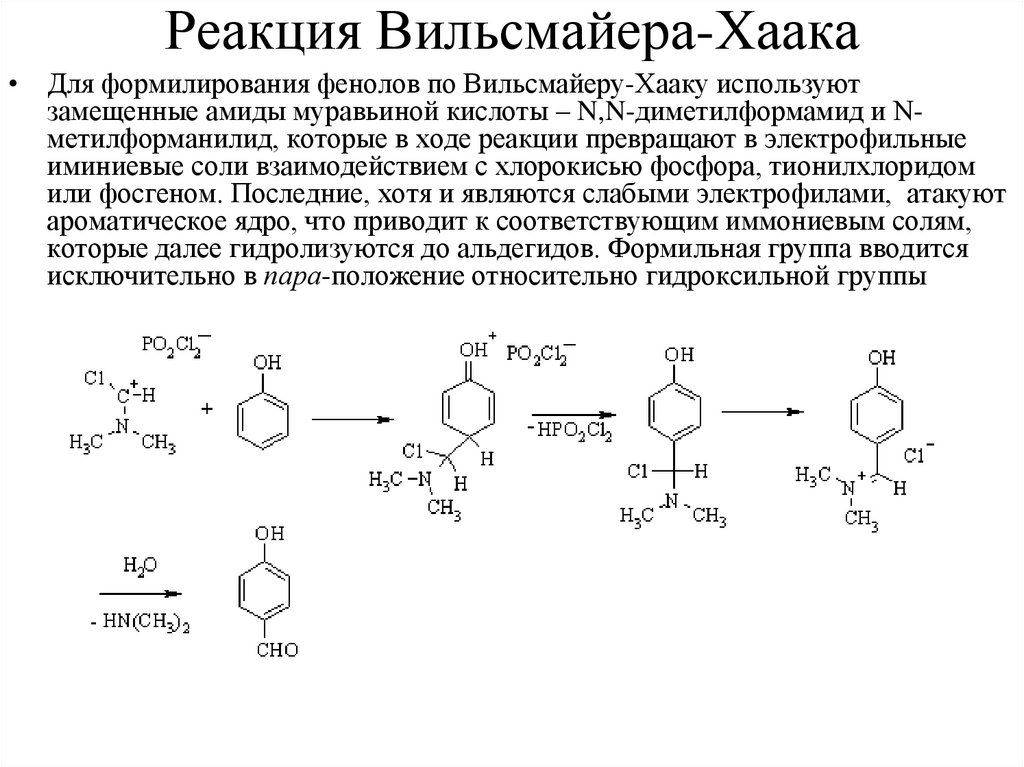
отщепляется. pKa (это значение pH, при котором химическая разновидность
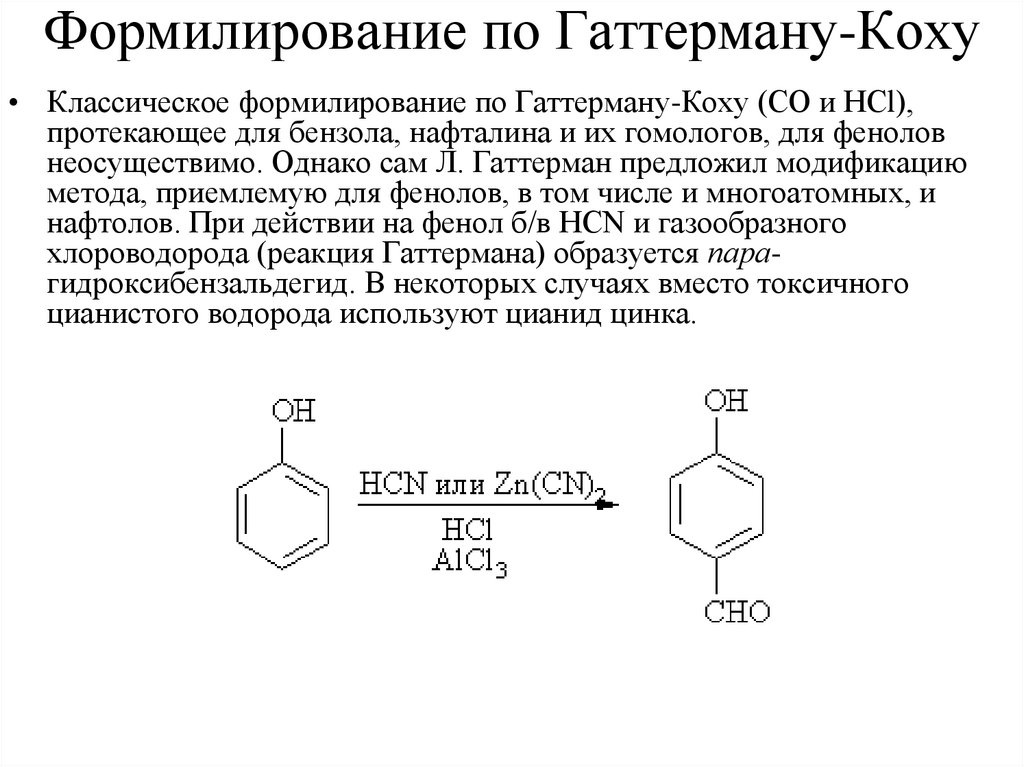
примет или пожертвует протон. Чем ниже значение рКа, тем сильнее
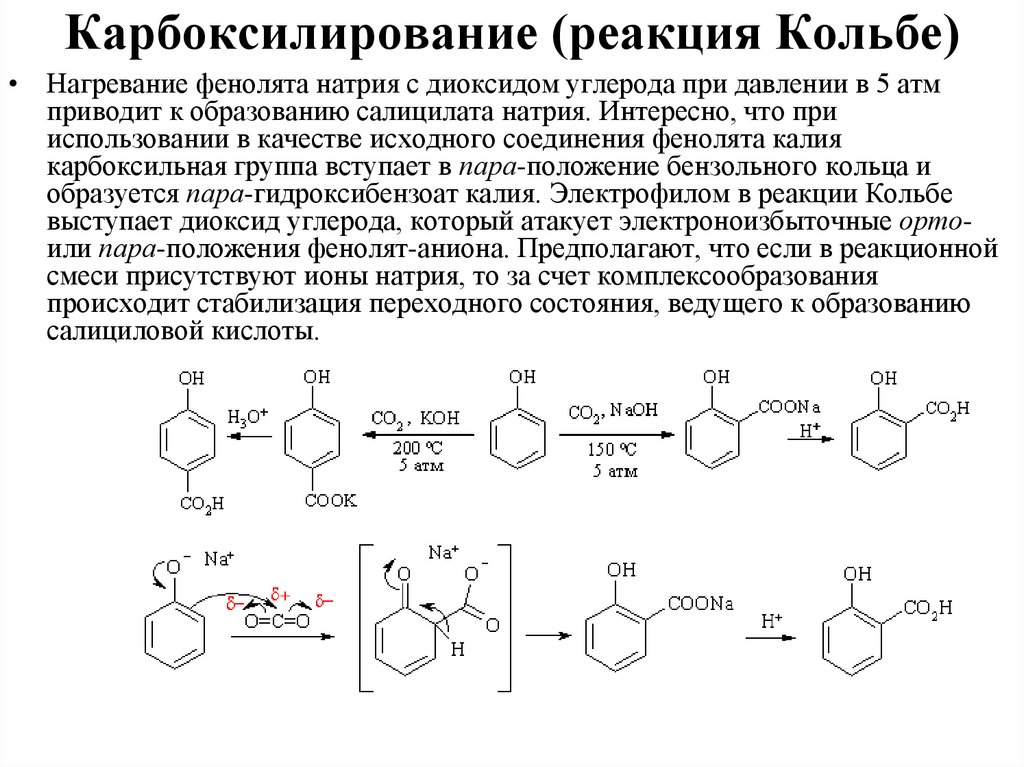
кислота и тем больше способность отдавать протон в водном растворе. )
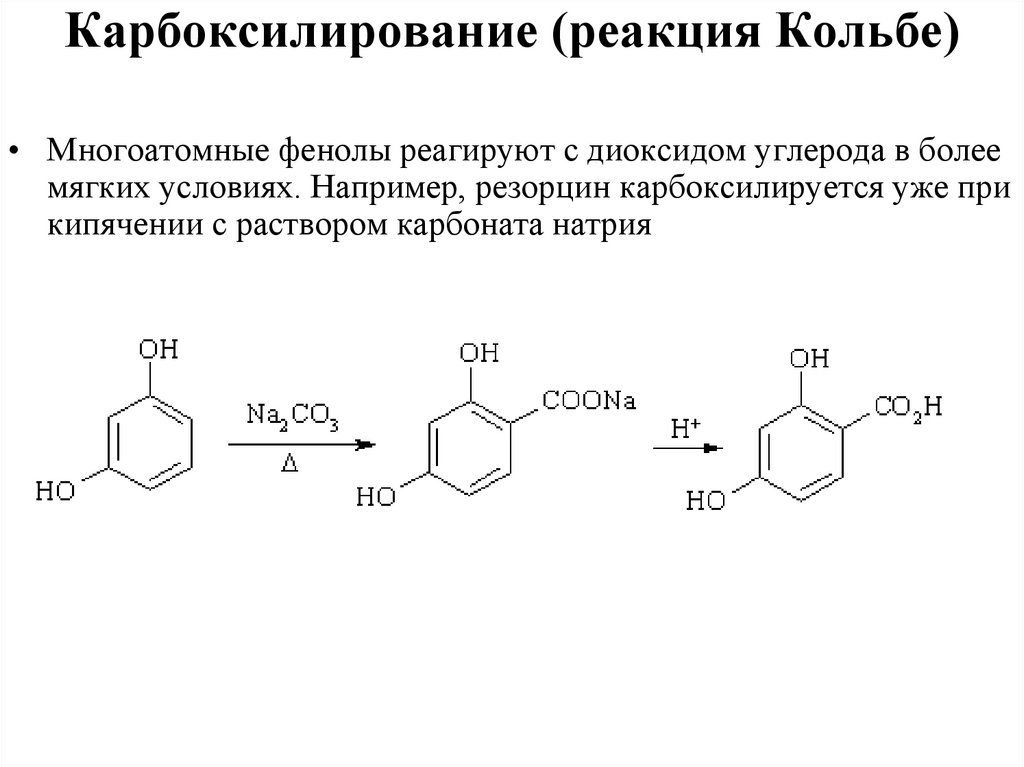
низших алифатических спиртов близка к pKa воды. В то же время, полярность
связи С-О обусловливает возможность замещения гидроксила.
• Несвязывающие электронные пары атома кислорода придают спирту
свойства оснований и нуклеофилов, способными реагировать с протоном,
кислотами Льюиса, С-электрофилами.
10.
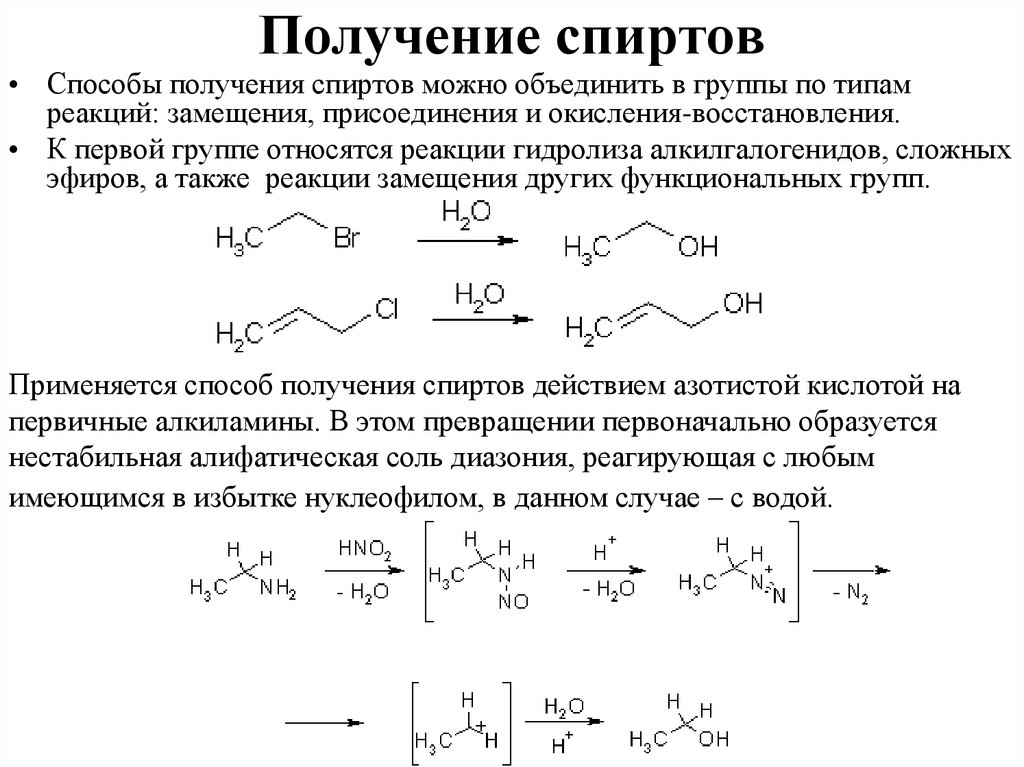
Получение спиртов• Способы получения спиртов можно объединить в группы по типам
реакций: замещения, присоединения и окисления-восстановления.
• К первой группе относятся реакции гидролиза алкилгалогенидов, сложных
эфиров, а также реакции замещения других функциональных групп.
Применяется способ получения спиртов действием азотистой кислотой на
первичные алкиламины. В этом превращении первоначально образуется
нестабильная алифатическая соль диазония, реагирующая с любым
имеющимся в избытке нуклеофилом, в данном случае – с водой.
11.
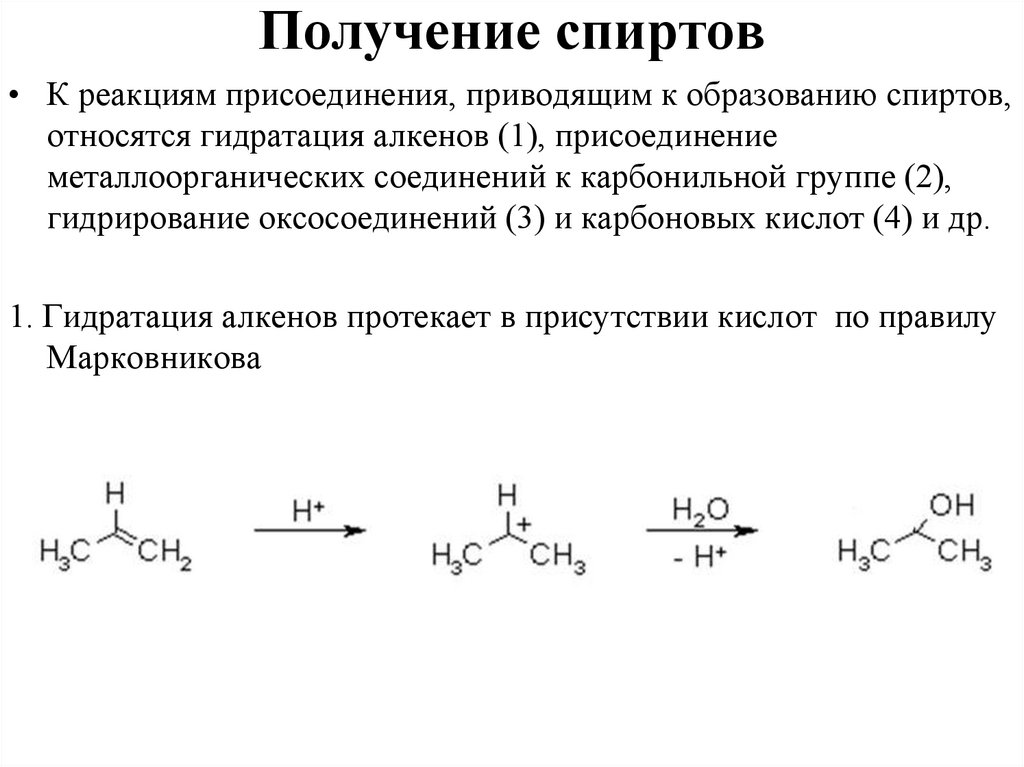
Получение спиртов• К реакциям присоединения, приводящим к образованию спиртов,
относятся гидратация алкенов (1), присоединение
металлоорганических соединений к карбонильной группе (2),
гидрирование оксосоединений (3) и карбоновых кислот (4) и др.
1. Гидратация алкенов протекает в присутствии кислот по правилу
Марковникова
12.
Получение спиртов• 2. Взаимодействие реактивов Гриньяра и других металлоорганических
соединений с альдегидами позволяет получать спирты различного
строения и имеет важное препаративное значение. В случае использования
формальдегида образуются первичные спирты, из других альдегидов
образуются вторичные, а из кетонов – третичные спирты, например:
13.
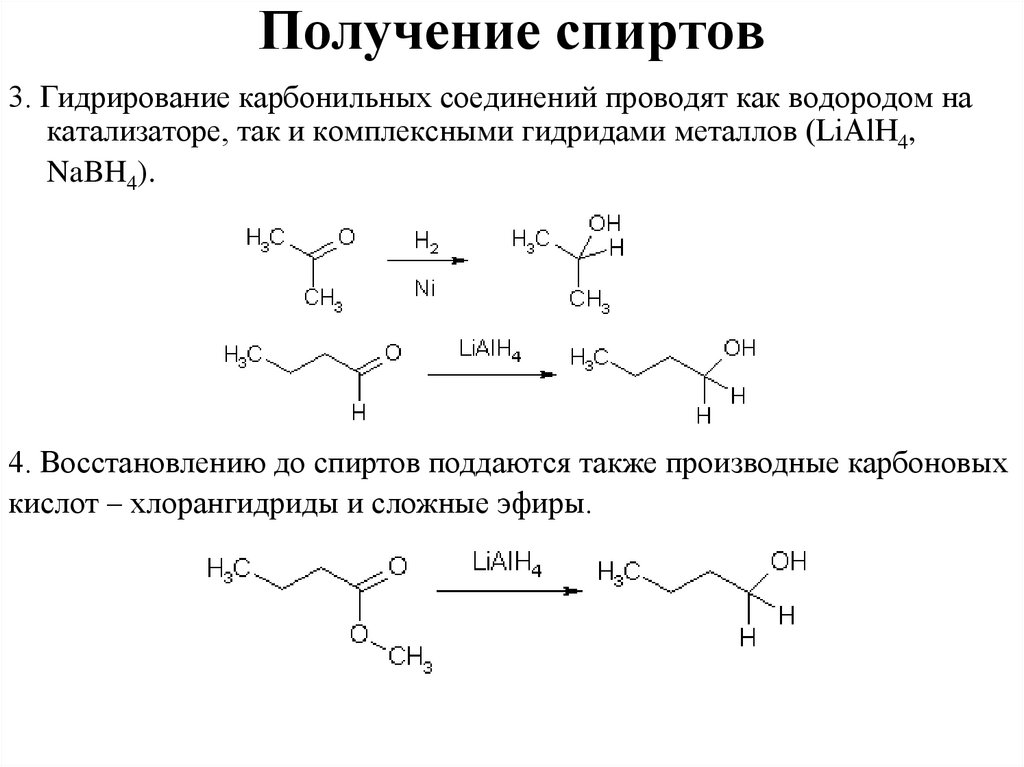
Получение спиртов3. Гидрирование карбонильных соединений проводят как водородом на
катализаторе, так и комплексными гидридами металлов (LiAlH4,
NaBH4).
4. Восстановлению до спиртов поддаются также производные карбоновых
кислот – хлорангидриды и сложные эфиры.
14.
Свойства спиртов. Кислотность• Величины рКа алканолов, измеренные в водных растворах растут от
метанола с увеличением длины углеродной цепи и со степенью
замещенности α-углеродного атома.
Протон может быть удален из гидроксильной группы действием достаточно
сильного основания. Спирты взаимодействуют, например, с металлическим
натрием, гидридом натрия, реактивами Гриньяра, образуя соответствующие
алкоголяты или алкоксиды
15.
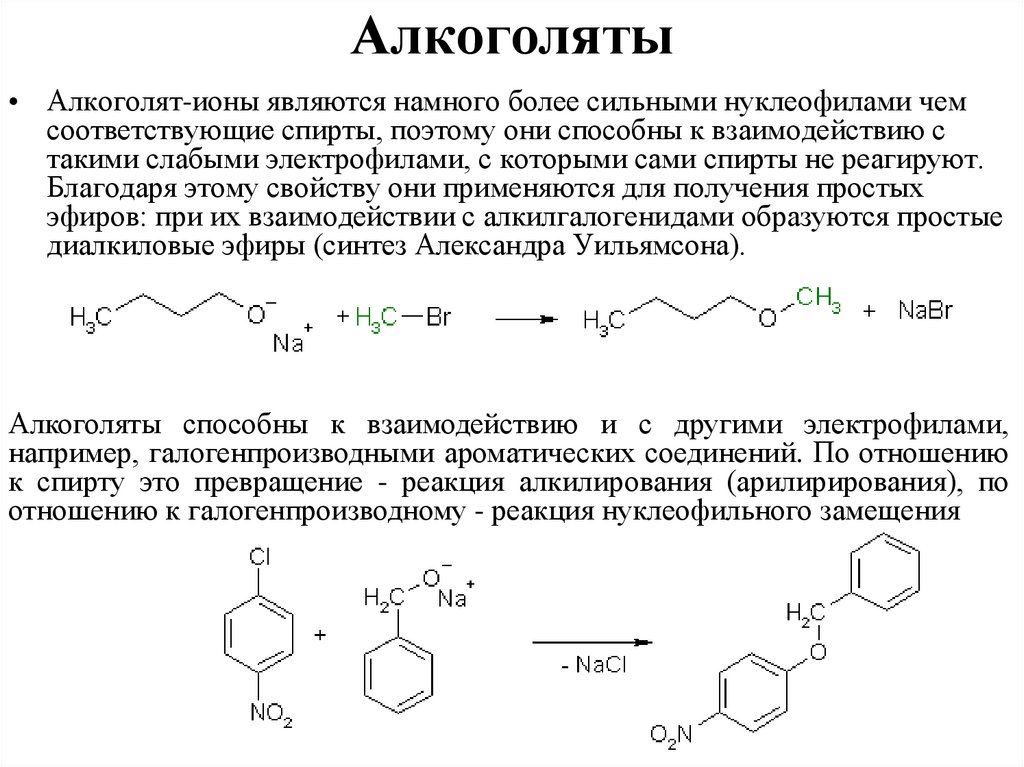
Алкоголяты• Алкоголят-ионы являются намного более сильными нуклеофилами чем
соответствующие спирты, поэтому они способны к взаимодействию с
такими слабыми электрофилами, с которыми сами спирты не реагируют.
Благодаря этому свойству они применяются для получения простых
эфиров: при их взаимодействии с алкилгалогенидами образуются простые
диалкиловые эфиры (синтез Александра Уильямсона).
Алкоголяты способны к взаимодействию и с другими электрофилами,
например, галогенпроизводными ароматических соединений. По отношению
к спирту это превращение - реакция алкилирования (арилирирования), по
отношению к галогенпроизводному - реакция нуклеофильного замещения
16.
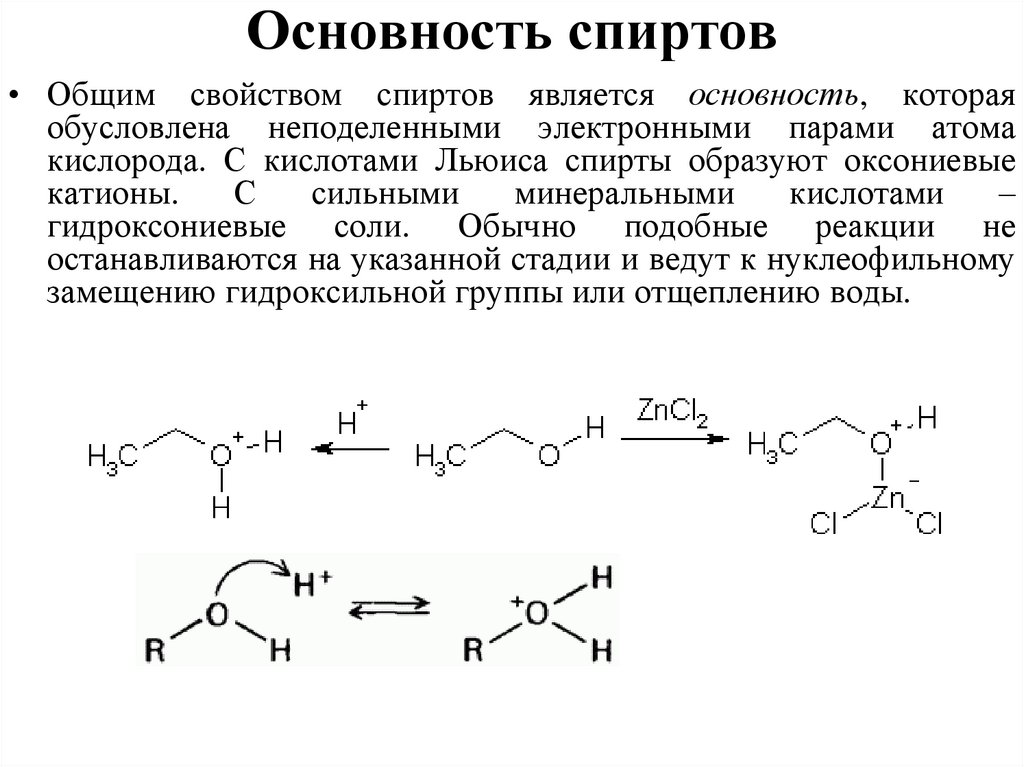
Основность спиртов• Общим свойством спиртов является основность, которая
обусловлена неподеленными электронными парами атома
кислорода. С кислотами Льюиса спирты образуют оксониевые
катионы.
С
сильными
минеральными
кислотами
–
гидроксониевые соли. Обычно подобные реакции не
останавливаются на указанной стадии и ведут к нуклеофильному
замещению гидроксильной группы или отщеплению воды.
17.
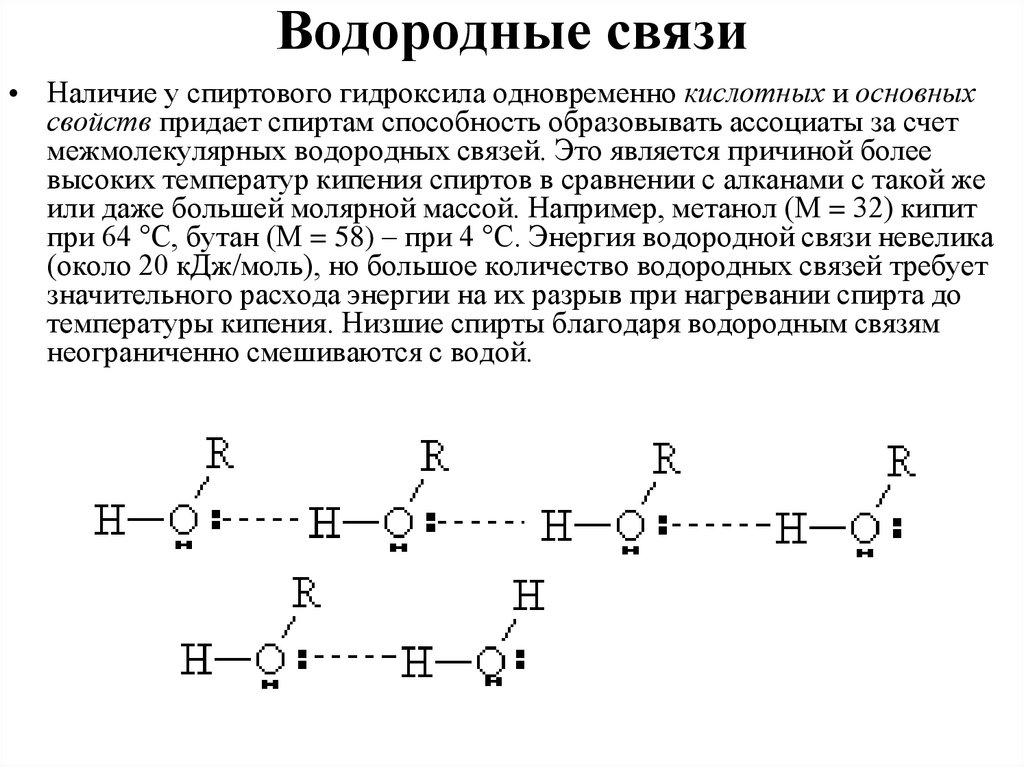
Водородные связи• Наличие у спиртового гидроксила одновременно кислотных и основных
свойств придает спиртам способность образовывать ассоциаты за счет
межмолекулярных водородных связей. Это является причиной более
высоких температур кипения спиртов в сравнении с алканами с такой же
или даже большей молярной массой. Например, метанол (М = 32) кипит
при 64 °С, бутан (М = 58) – при 4 °С. Энергия водородной связи невелика
(около 20 кДж/моль), но большое количество водородных связей требует
значительного расхода энергии на их разрыв при нагревании спирта до
температуры кипения. Низшие спирты благодаря водородным связям
неограниченно смешиваются с водой.
18.
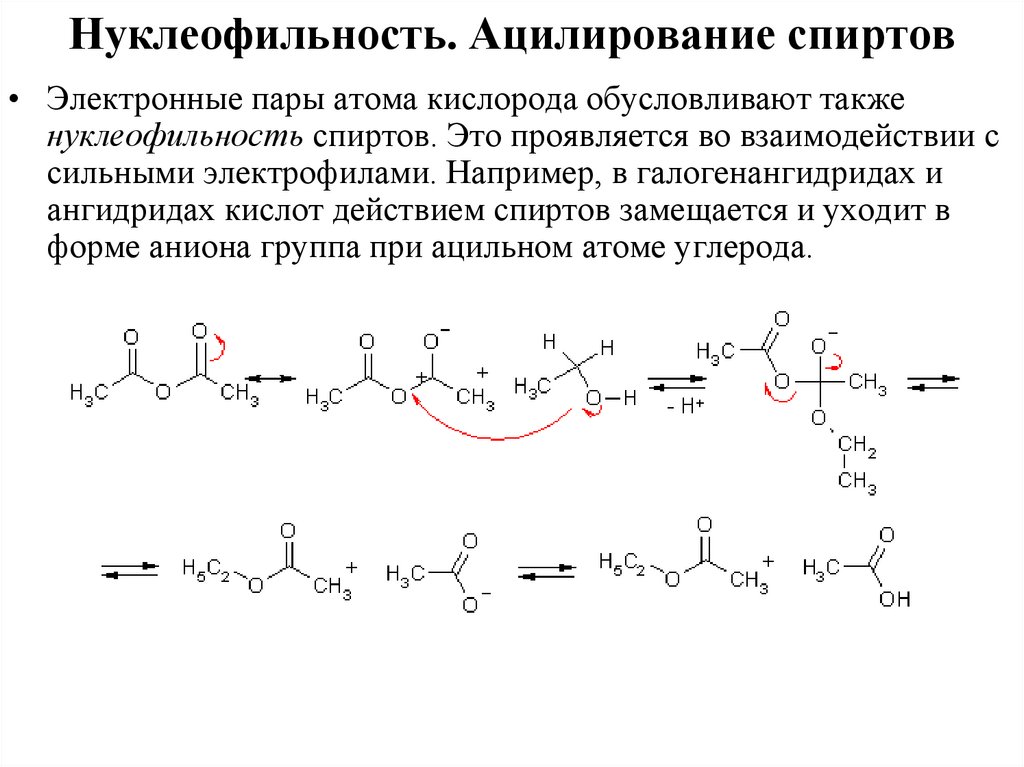
Нуклеофильность. Ацилирование спиртов• Электронные пары атома кислорода обусловливают также
нуклеофильность спиртов. Это проявляется во взаимодействии с
сильными электрофилами. Например, в галогенангидридах и
ангидридах кислот действием спиртов замещается и уходит в
форме аниона группа при ацильном атоме углерода.
19.
Нуклеофильность. Ацилирование спиртов• Образование сложных эфиров при действии ангидридов,
галогенангидридов происходит легче, когда реакцию проводят в
присутствии основания, роль которого состоит в переводе
гидроксипроизводного в форму более нуклеофильного
алкоголята. Это явление известно как основной катализ
ацилирования.
+
C
H
O
N
a
+
C
H
C
(
O
)
C
l
2
5
3
C
H
O
C
(
O
)
C
H
+
N
a
C
l
2
5
3
+
C
H
O
N
a
+
H
O
C
H
O
H
+
N
a
O
H
2
5
2
2
5
20.
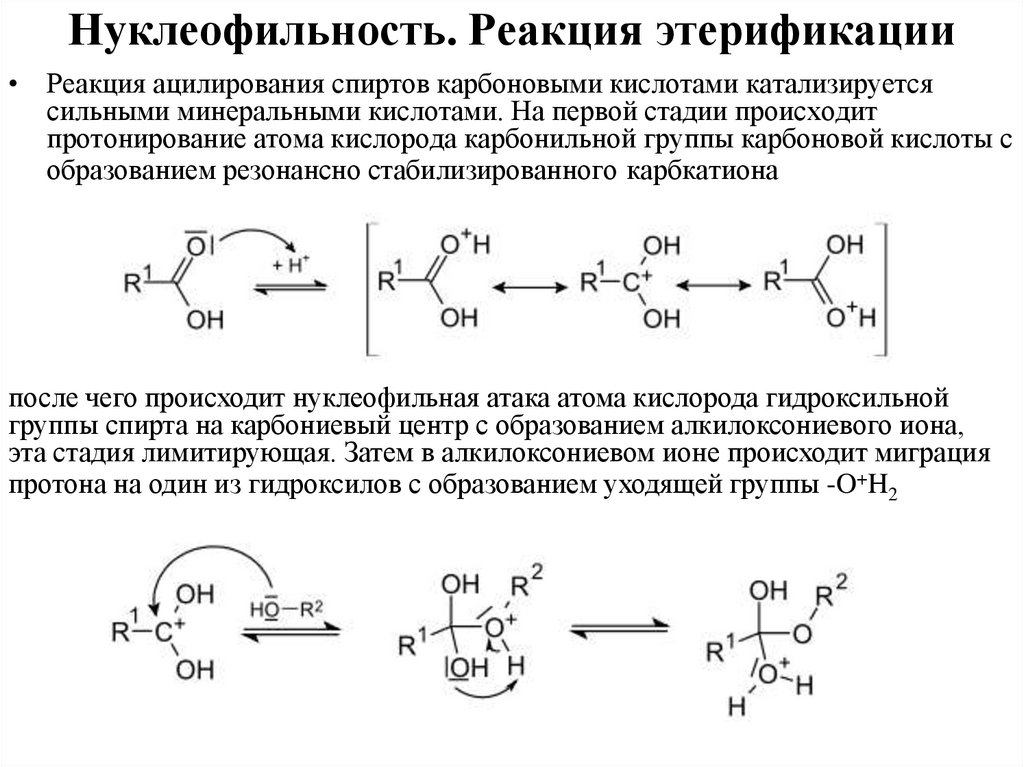
Нуклеофильность. Реакция этерификации• Реакция ацилирования спиртов карбоновыми кислотами катализируется
сильными минеральными кислотами. На первой стадии происходит
протонирование атома кислорода карбонильной группы карбоновой кислоты с
образованием резонансно стабилизированного карбкатиона
после чего происходит нуклеофильная атака атома кислорода гидроксильной
группы спирта на карбониевый центр с образованием алкилоксониевого иона,
эта стадия лимитирующая. Затем в алкилоксониевом ионе происходит миграция
протона на один из гидроксилов с образованием уходящей группы -O+H2
21.
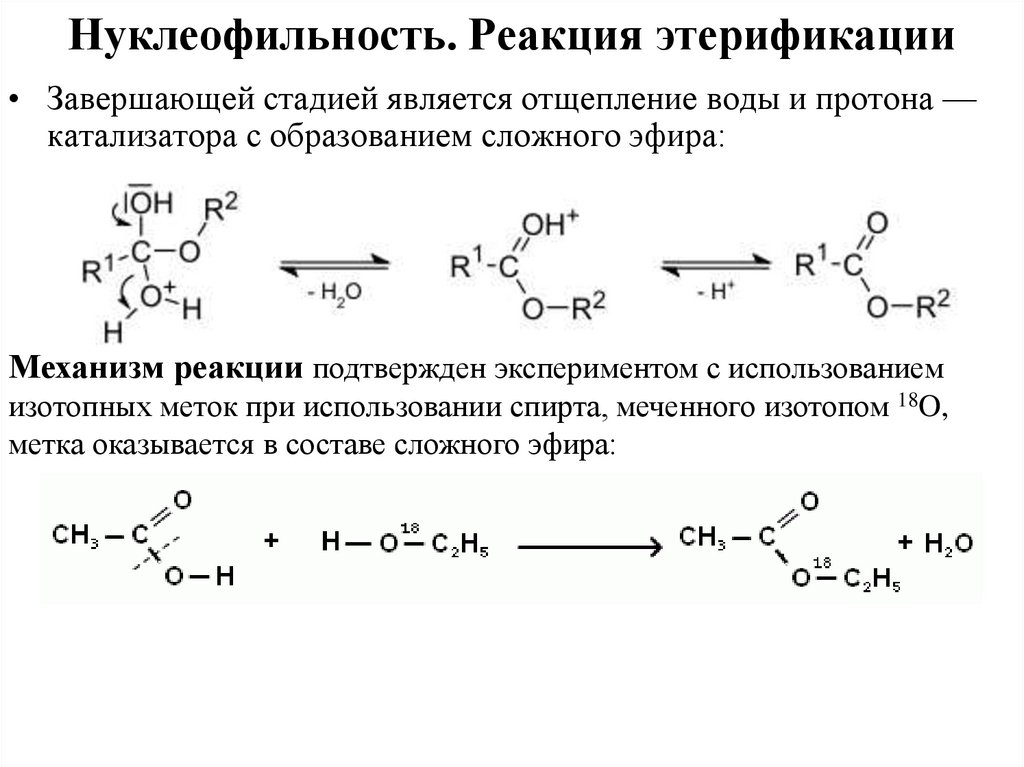
Нуклеофильность. Реакция этерификации• Завершающей стадией является отщепление воды и протона —
катализатора с образованием сложного эфира:
Механизм реакции подтвержден экспериментом с использованием
изотопных меток при использовании спирта, меченного изотопом 18O,
метка оказывается в составе сложного эфира:
22.
Переэтерификация жиров• Переэтерификация — химическая реакция обмена структурных элементов
сложных эфиров и гидроксильных групп спиртов.
• Виды реакции:
• 1. Обмен радикалами жирных кислот между молекулами двух разных
глицеридов (межмолекулярная переэтерификация)
2. При внутримолекулярной переэтерификации изменяется взаимное положение
ацильных групп внутри одной молекулы триглицерида
23.
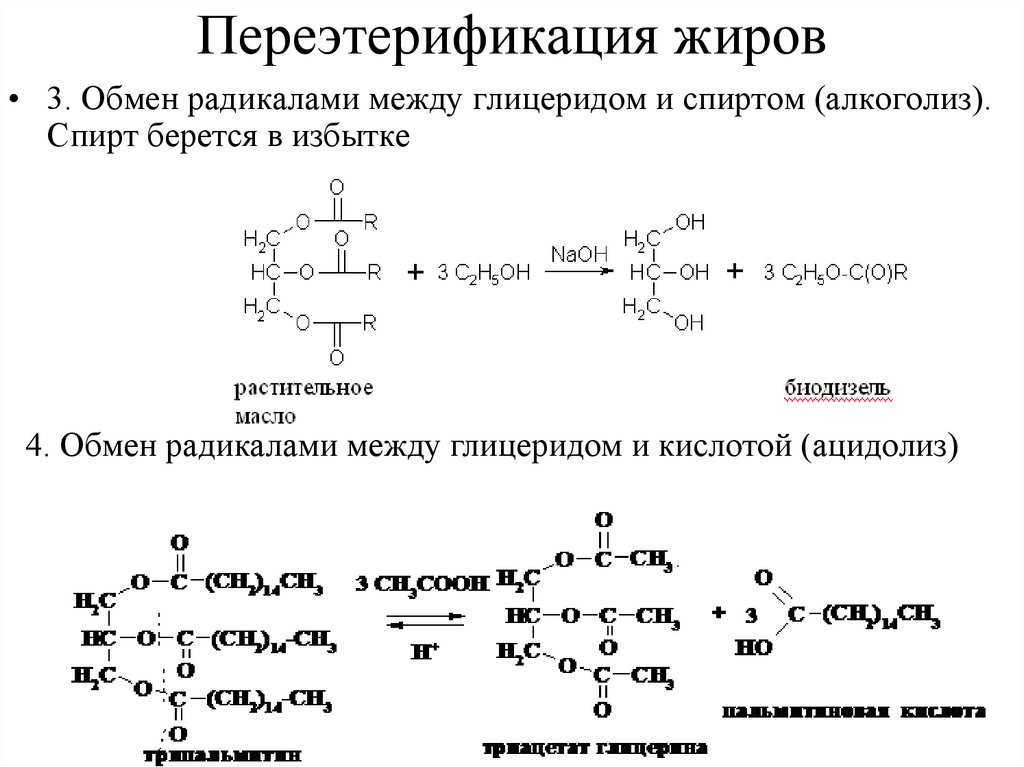
Переэтерификация жиров• 3. Обмен радикалами между глицеридом и спиртом (алкоголиз).
Спирт берется в избытке
4. Обмен радикалами между глицеридом и кислотой (ацидолиз)
24.
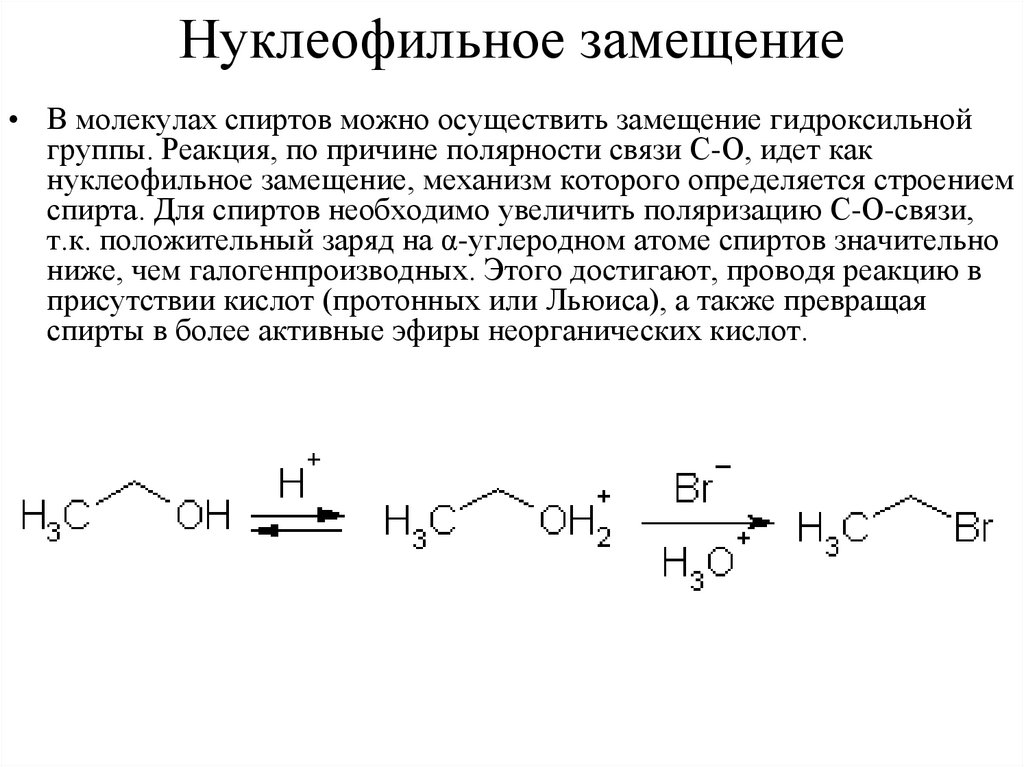
Нуклеофильное замещение• В молекулах спиртов можно осуществить замещение гидроксильной
группы. Реакция, по причине полярности связи С-О, идет как
нуклеофильное замещение, механизм которого определяется строением
спирта. Для спиртов необходимо увеличить поляризацию С-О-связи,
т.к. положительный заряд на α-углеродном атоме спиртов значительно
ниже, чем галогенпроизводных. Этого достигают, проводя реакцию в
присутствии кислот (протонных или Льюиса), а также превращая
спирты в более активные эфиры неорганических кислот.
25.
Нуклеофильное замещениеВ промышленности амины получают аммонолизом спиртов в
присутствии катализаторов дегидратации (Al2O3, SiO2, ThO2 и т. п.)
при 300–500 °С. При этом также образуется смесь первичных,
вторичных и третичных аминов:
Этот метод, называемый так же аминолизом, применяют для
производства N-алкил- и N,N-диалкиланилинов. Обычно в таких
случаях алкилирование спиртами ведут в присутствии кислот:
26.
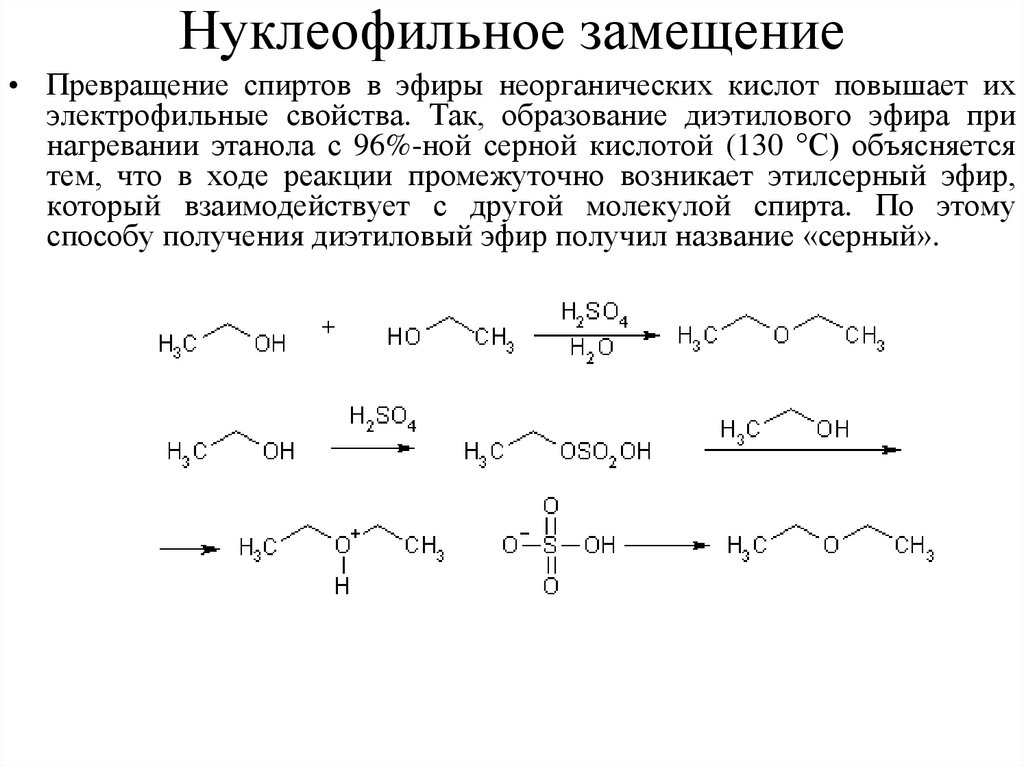
Нуклеофильное замещение• Превращение спиртов в эфиры неорганических кислот повышает их
электрофильные свойства. Так, образование диэтилового эфира при
нагревании этанола с 96%-ной серной кислотой (130 °С) объясняется
тем, что в ходе реакции промежуточно возникает этилсерный эфир,
который взаимодействует с другой молекулой спирта. По этому
способу получения диэтиловый эфир получил название «серный».
27.
Реакции спиртов с йодом и его производными• Иодоводородная кислота обладает достаточной кислотностью и иодидион является сильным нуклеофилом, но его сильные
восстановительные свойства не всегда позволяют с высокими
выходами превращать спирты в алкилйодиды. Поэтому замещение
гидроксила проводят действием трииодида фосфора, который
генерируют из красного фосфора и йода непосредственно в
реакционном растворе.
На первой стадии реакции трииодид фосфора образует со спиртом эфир
(сродство фосфора к кислороду очень велико), который в качестве
электрофила участвует в реакции нуклеофильного замещения.
28.
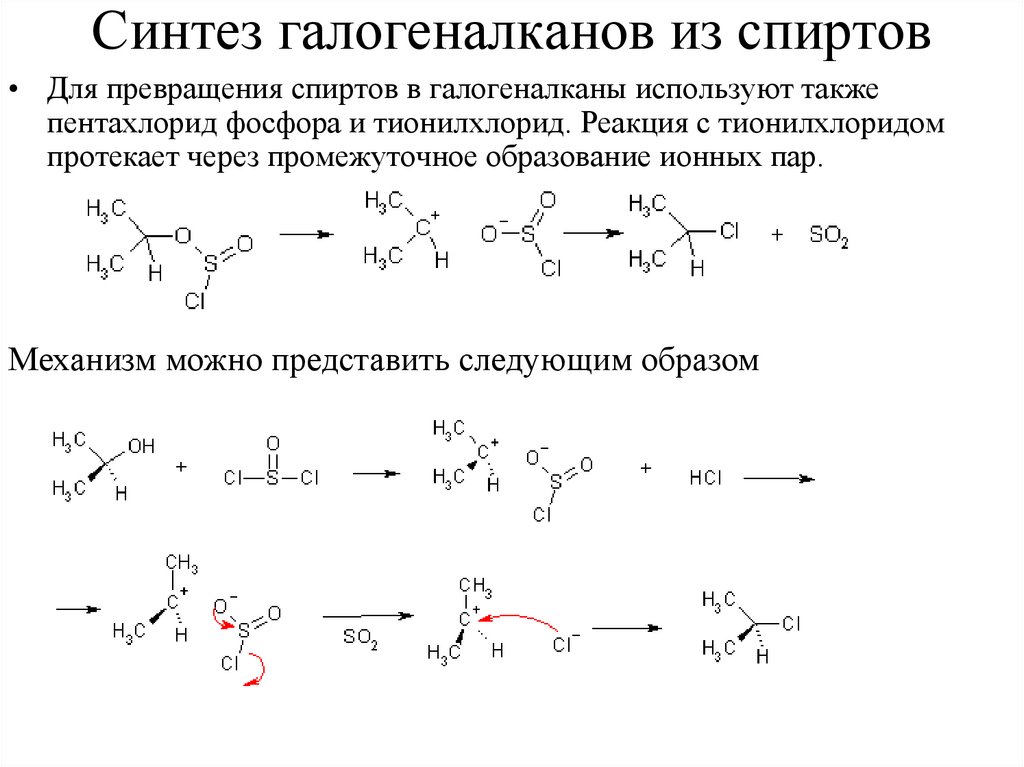
Синтез галогеналканов из спиртов• Для превращения спиртов в галогеналканы используют также
пентахлорид фосфора и тионилхлорид. Реакция с тионилхлоридом
протекает через промежуточное образование ионных пар.
Механизм можно представить следующим образом
29.
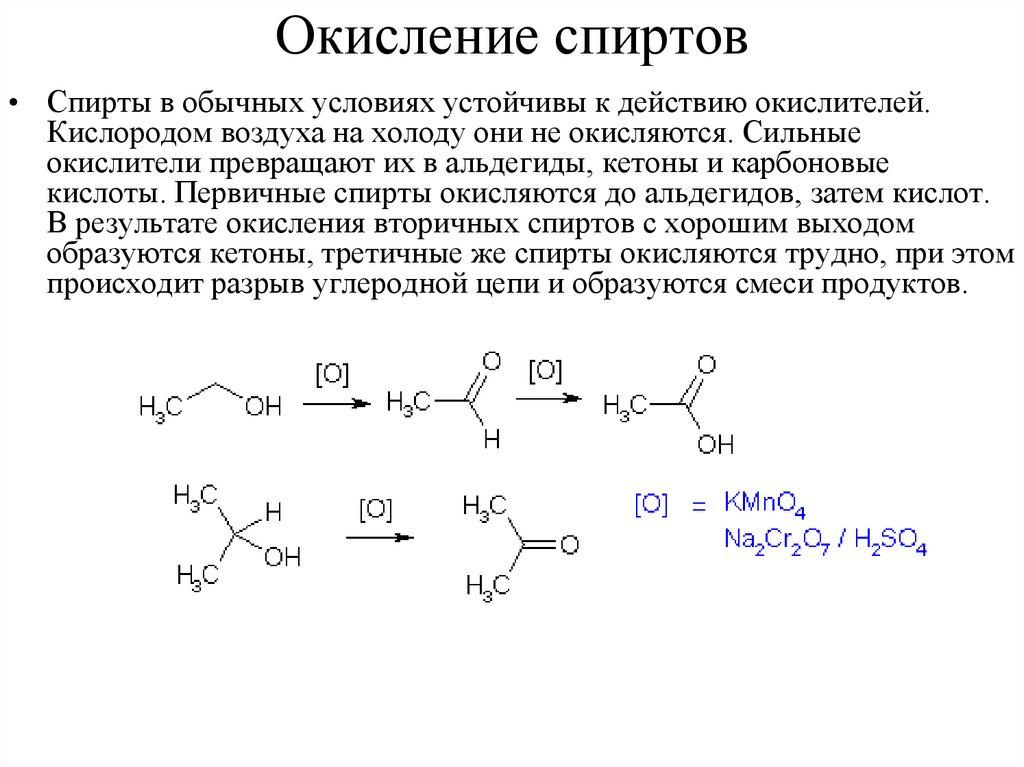
Окисление спиртов• Спирты в обычных условиях устойчивы к действию окислителей.
Кислородом воздуха на холоду они не окисляются. Сильные
окислители превращают их в альдегиды, кетоны и карбоновые
кислоты. Первичные спирты окисляются до альдегидов, затем кислот.
В результате окисления вторичных спиртов с хорошим выходом
образуются кетоны, третичные же спирты окисляются трудно, при этом
происходит разрыв углеродной цепи и образуются смеси продуктов.
30.
Многоатомные спирты• Первый член ряда многоатомных спиртов – этандиол-1,2 обычно
называют по тривиальной номенклатуре – этиленгликоль, поэтому все
1,2-двухатомные спирты объединяют под названием гликоли.
Трехатомные спирты называются глицеринами по названию самого
простого из них – пропантриола-1,2,3.
• По своим свойствам этиленгликоль, глицерин и другие полиатомные
спирты большей частью похожи на алканолы.
• При действии на гликоли сильных оснований образуется два ряда
алкоголятов: по одному и по обоим гидроксилам в зависимости от
количества основания.
31.
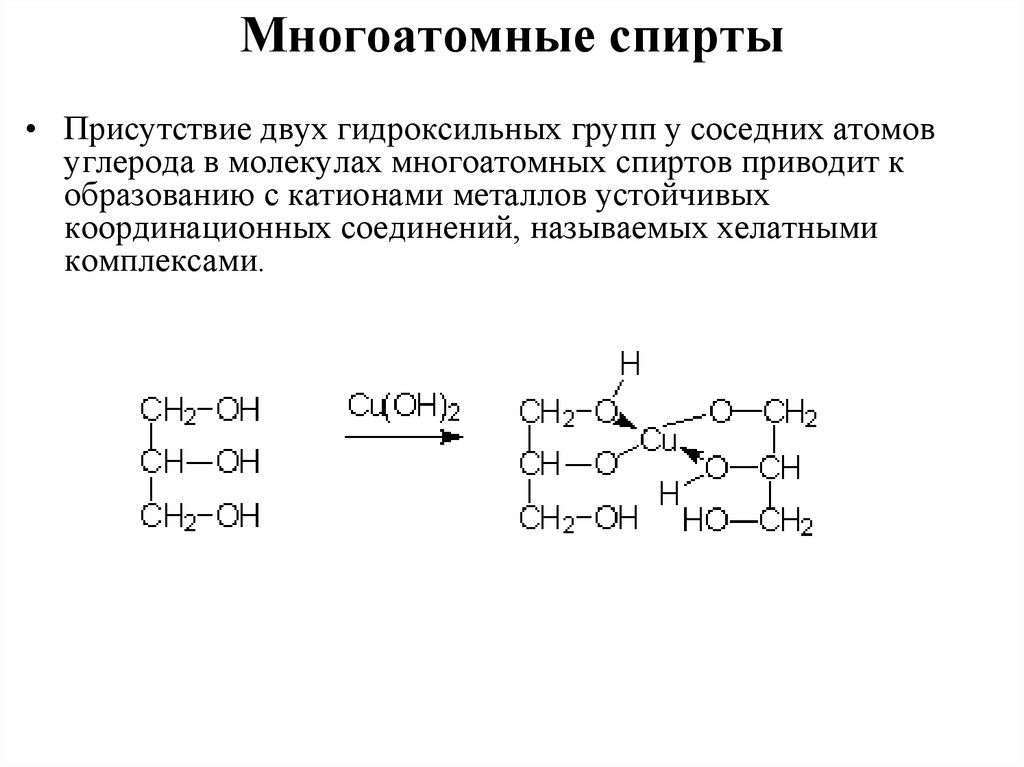
Многоатомные спирты• Присутствие двух гидроксильных групп у соседних атомов
углерода в молекулах многоатомных спиртов приводит к
образованию с катионами металлов устойчивых
координационных соединений, называемых хелатными
комплексами.
32.
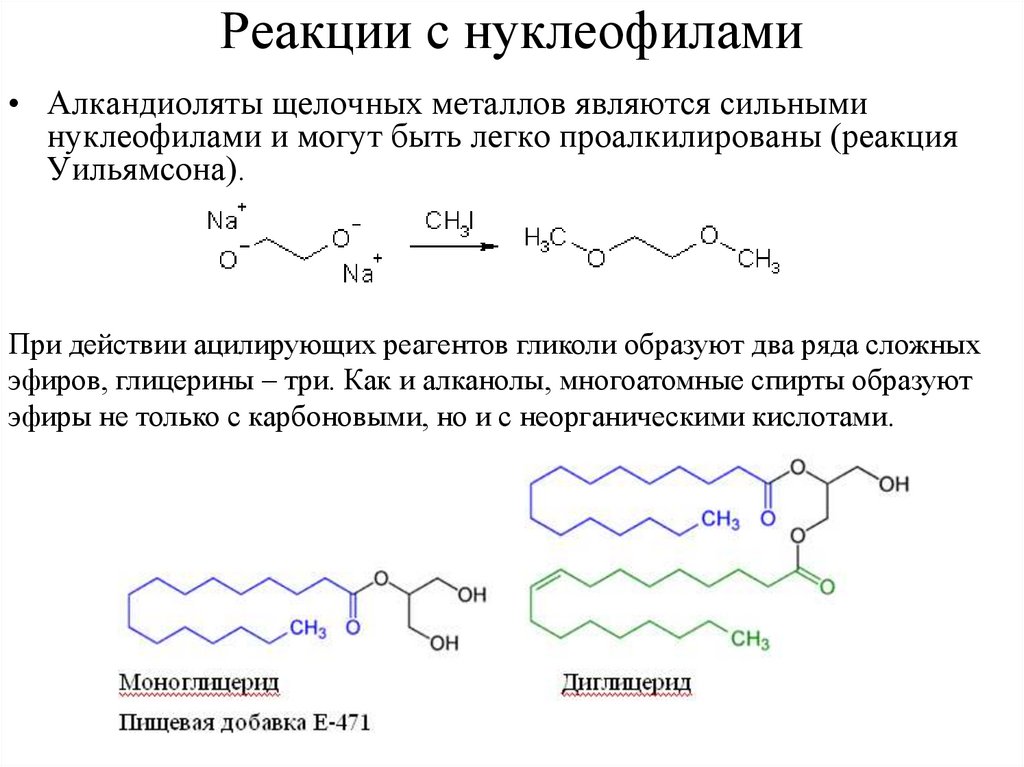
Реакции с нуклеофилами• Алкандиоляты щелочных металлов являются сильными
нуклеофилами и могут быть легко проалкилированы (реакция
Уильямсона).
При действии ацилирующих реагентов гликоли образуют два ряда сложных
эфиров, глицерины – три. Как и алканолы, многоатомные спирты образуют
эфиры не только с карбоновыми, но и с неорганическими кислотами.
33.
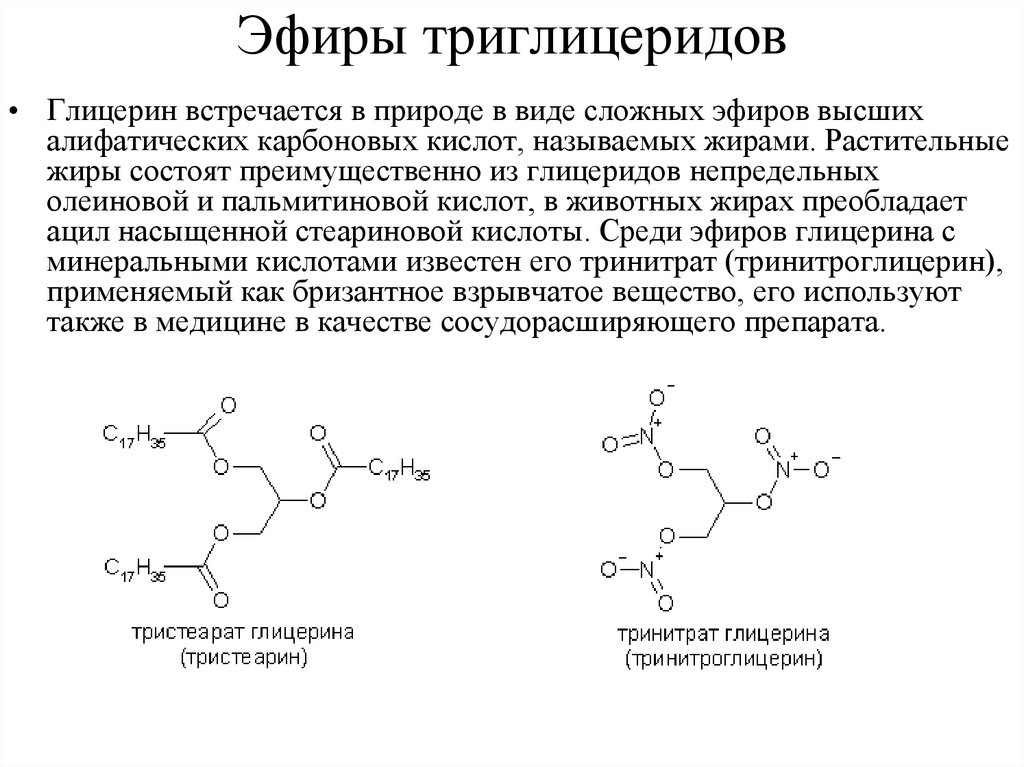
Эфиры триглицеридов• Глицерин встречается в природе в виде сложных эфиров высших
алифатических карбоновых кислот, называемых жирами. Растительные
жиры состоят преимущественно из глицеридов непредельных
олеиновой и пальмитиновой кислот, в животных жирах преобладает
ацил насыщенной стеариновой кислоты. Среди эфиров глицерина с
минеральными кислотами известен его тринитрат (тринитроглицерин),
применяемый как бризантное взрывчатое вещество, его используют
также в медицине в качестве сосудорасширяющего препарата.
34.
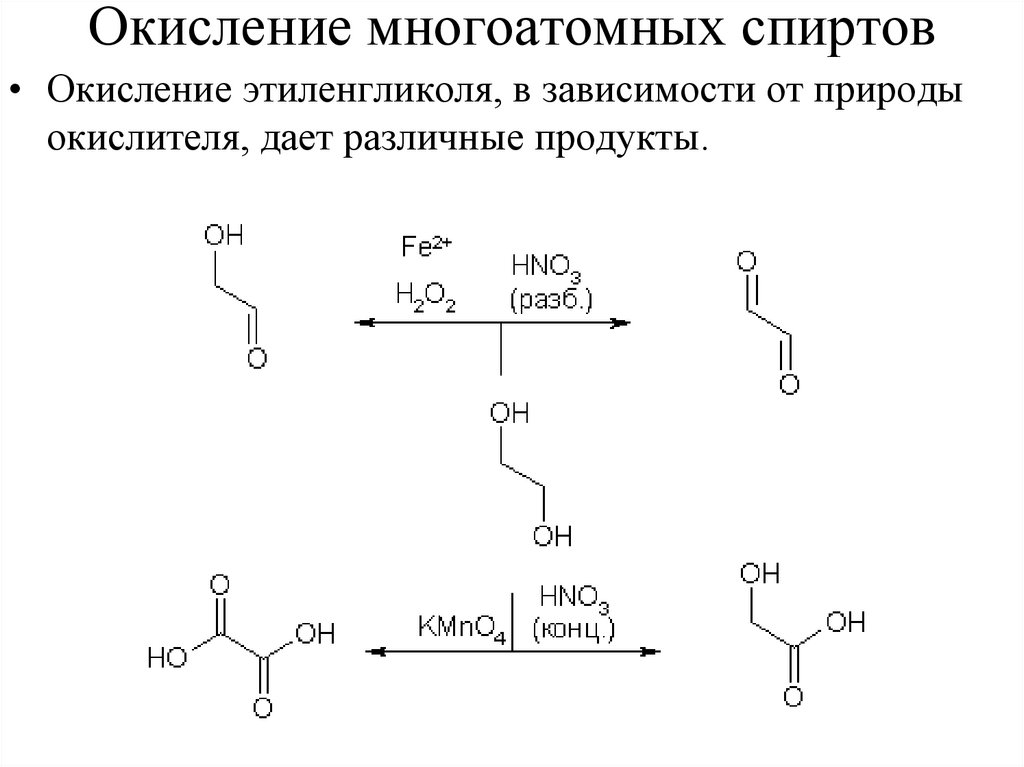
Окисление многоатомных спиртов• Окисление этиленгликоля, в зависимости от природы
окислителя, дает различные продукты.
35.
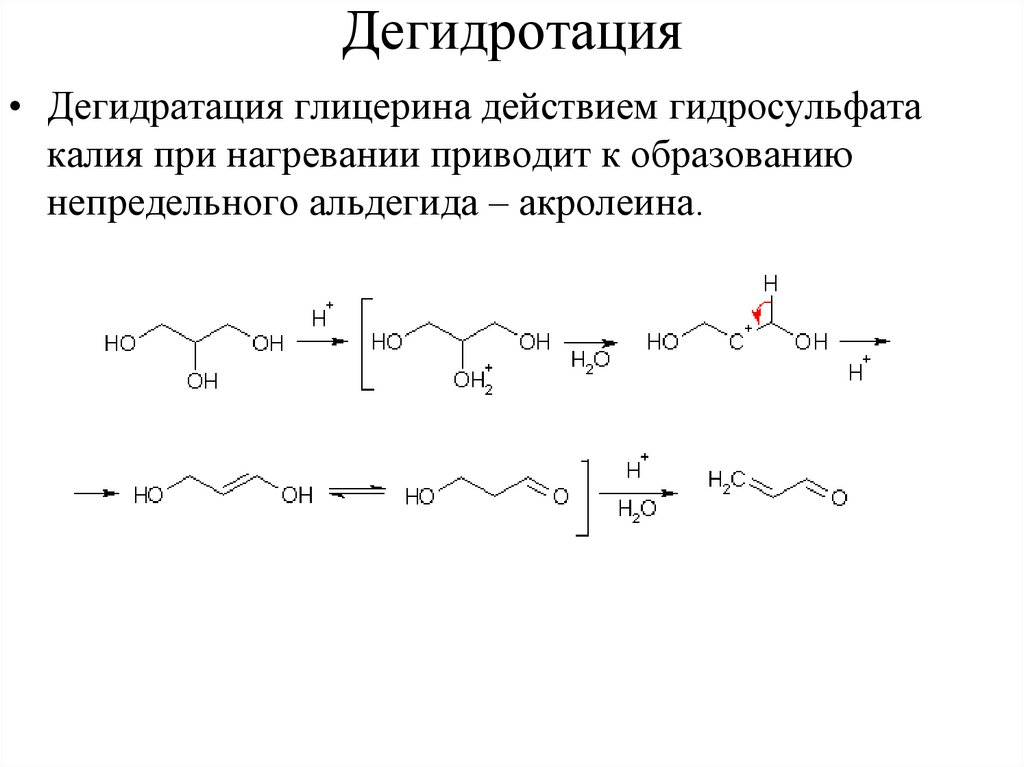
Дегидротация• Дегидратация глицерина действием гидросульфата
калия при нагревании приводит к образованию
непредельного альдегида – акролеина.
36.
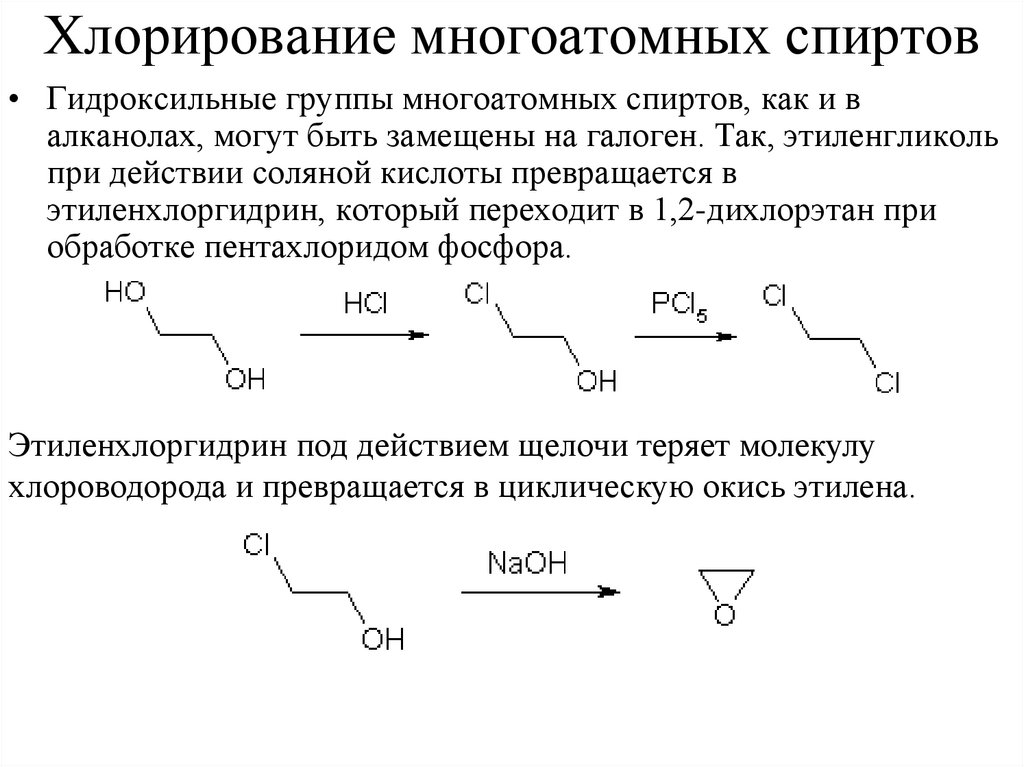
Хлорирование многоатомных спиртов• Гидроксильные группы многоатомных спиртов, как и в
алканолах, могут быть замещены на галоген. Так, этиленгликоль
при действии соляной кислоты превращается в
этиленхлоргидрин, который переходит в 1,2-дихлорэтан при
обработке пентахлоридом фосфора.
Этиленхлоргидрин под действием щелочи теряет молекулу
хлороводорода и превращается в циклическую окись этилена.
37.
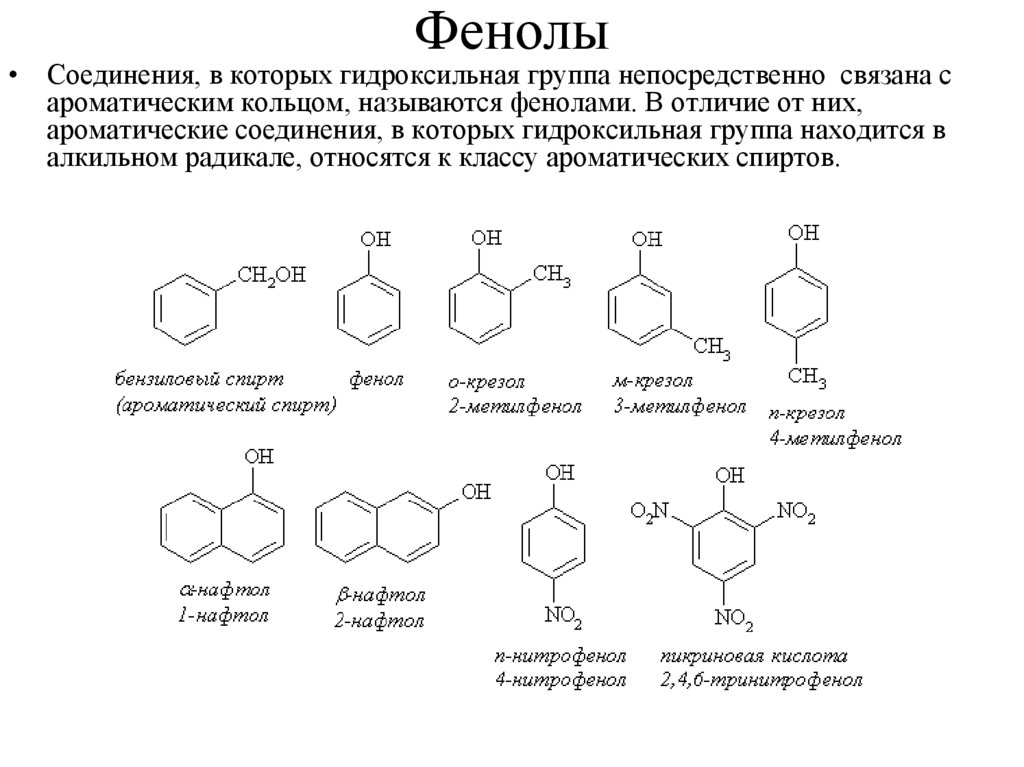
Фенолы• Соединения, в которых гидроксильная группа непосредственно связана с
ароматическим кольцом, называются фенолами. В отличие от них,
ароматические соединения, в которых гидроксильная группа находится в
алкильном радикале, относятся к классу ароматических спиртов.
38.
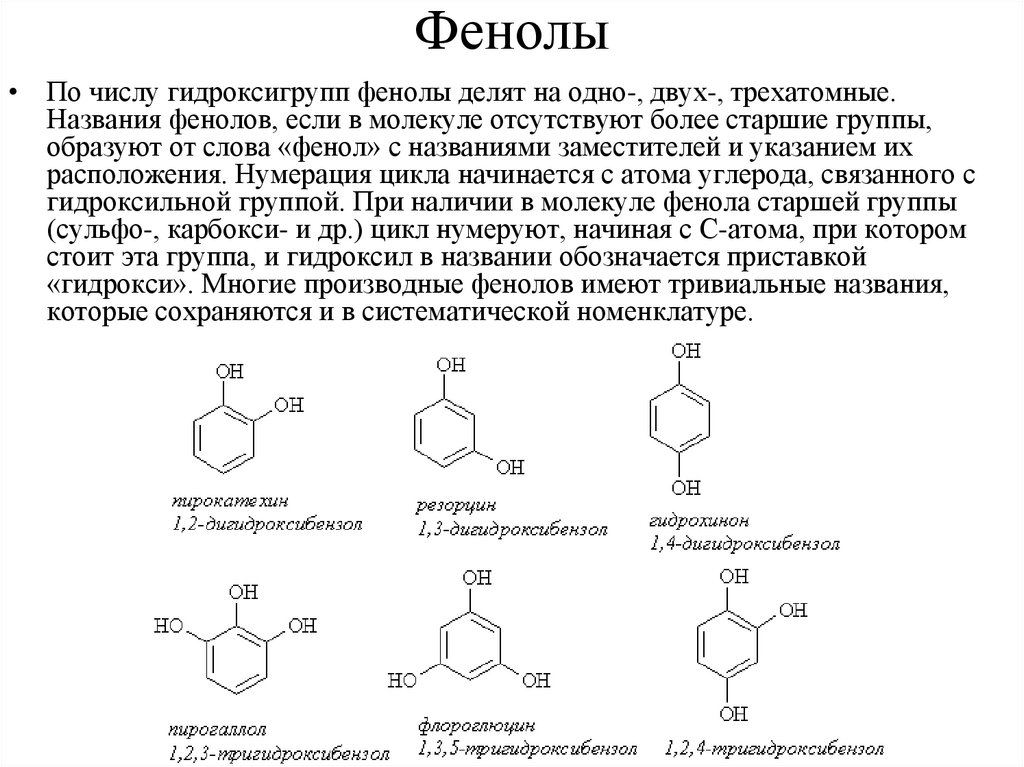
Фенолы• По числу гидроксигрупп фенолы делят на одно-, двух-, трехатомные.
Названия фенолов, если в молекуле отсутствуют более старшие группы,
образуют от слова «фенол» с названиями заместителей и указанием их
расположения. Нумерация цикла начинается с атома углерода, связанного с
гидроксильной группой. При наличии в молекуле фенола старшей группы
(сульфо-, карбокси- и др.) цикл нумеруют, начиная с С-атома, при котором
стоит эта группа, и гидроксил в названии обозначается приставкой
«гидрокси». Многие производные фенолов имеют тривиальные названия,
которые сохраняются и в систематической номенклатуре.
39.
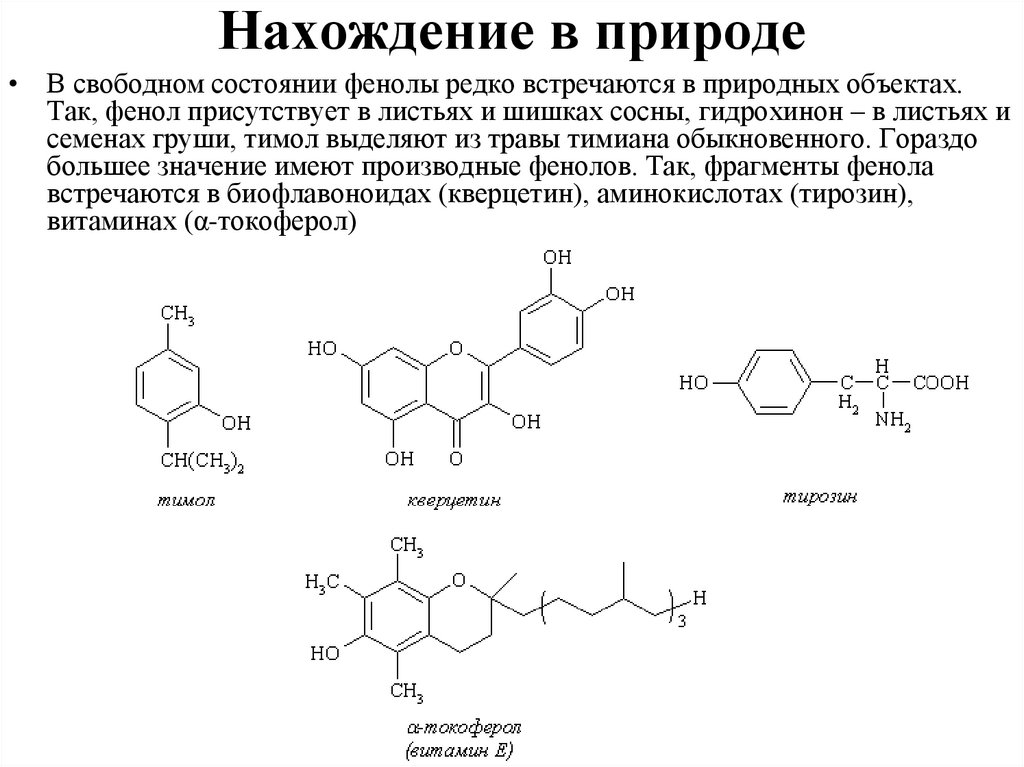
Нахождение в природе• В свободном состоянии фенолы редко встречаются в природных объектах.
Так, фенол присутствует в листьях и шишках сосны, гидрохинон – в листьях и
семенах груши, тимол выделяют из травы тимиана обыкновенного. Гораздо
большее значение имеют производные фенолов. Так, фрагменты фенола
встречаются в биофлавоноидах (кверцетин), аминокислотах (тирозин),
витаминах (α-токоферол)
40.
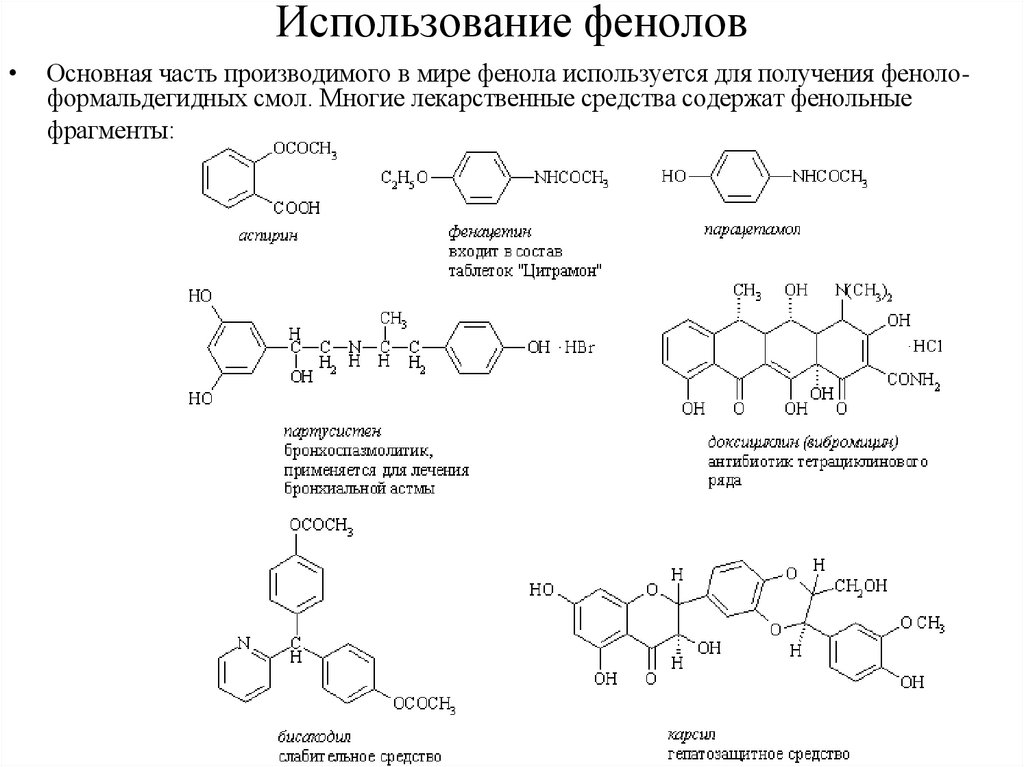
Использование феноловОсновная часть производимого в мире фенола используется для получения фенолоформальдегидных смол. Многие лекарственные средства содержат фенольные
фрагменты:
41.
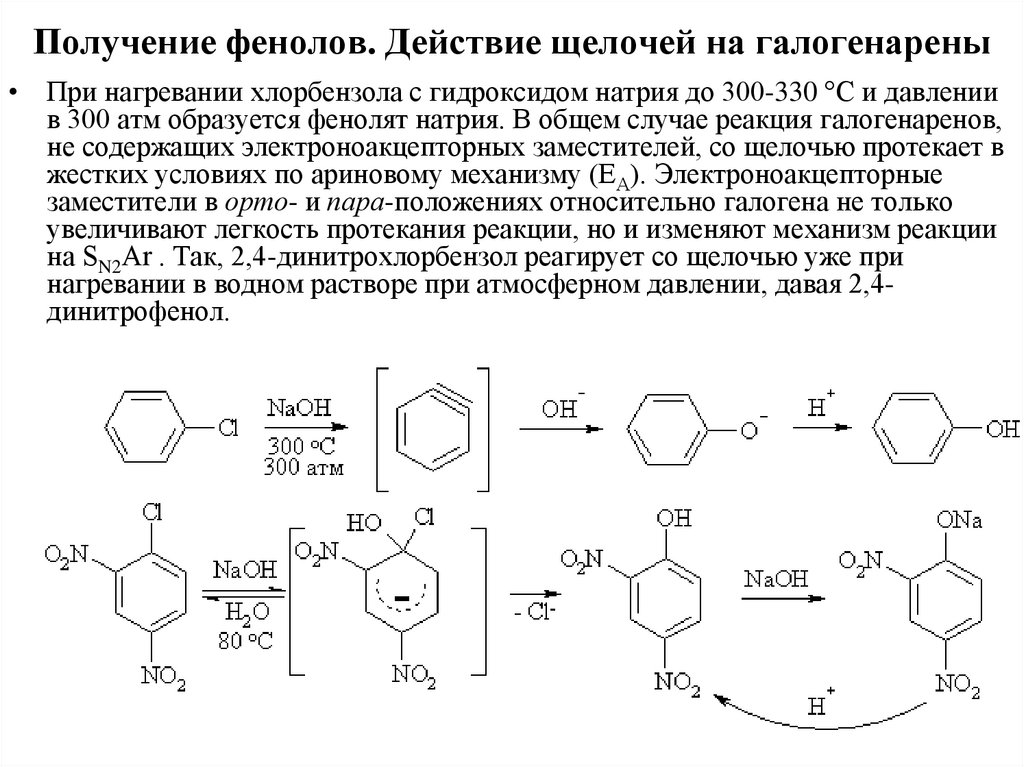
Получение фенолов. Действие щелочей на галогенарены• При нагревании хлорбензола с гидроксидом натрия до 300-330 °С и давлении
в 300 атм образуется фенолят натрия. В общем случае реакция галогенаренов,
не содержащих электроноакцепторных заместителей, со щелочью протекает в
жестких условиях по ариновому механизму (ЕА). Электроноакцепторные
заместители в орто- и пара-положениях относительно галогена не только
увеличивают легкость протекания реакции, но и изменяют механизм реакции
на SN2Ar . Так, 2,4-динитрохлорбензол реагирует со щелочью уже при
нагревании в водном растворе при атмосферном давлении, давая 2,4динитрофенол.
42.
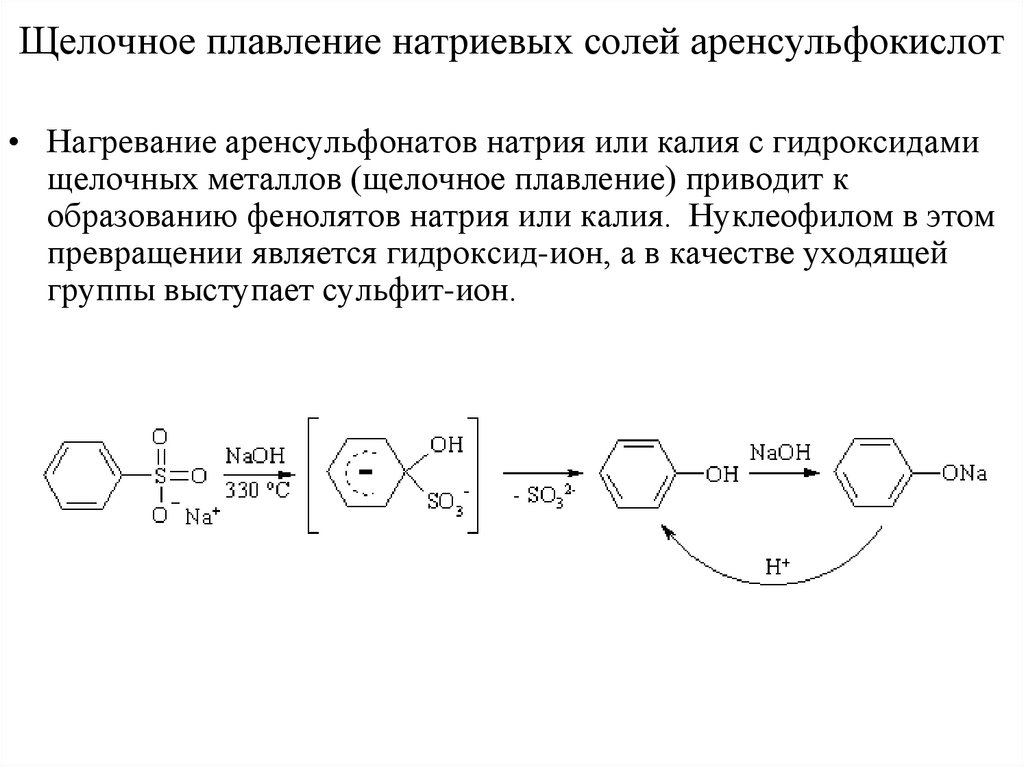
Щелочное плавление натриевых солей аренсульфокислот• Нагревание аренсульфонатов натрия или калия с гидроксидами
щелочных металлов (щелочное плавление) приводит к
образованию фенолятов натрия или калия. Нуклеофилом в этом
превращении является гидроксид-ион, а в качестве уходящей
группы выступает сульфит-ион.
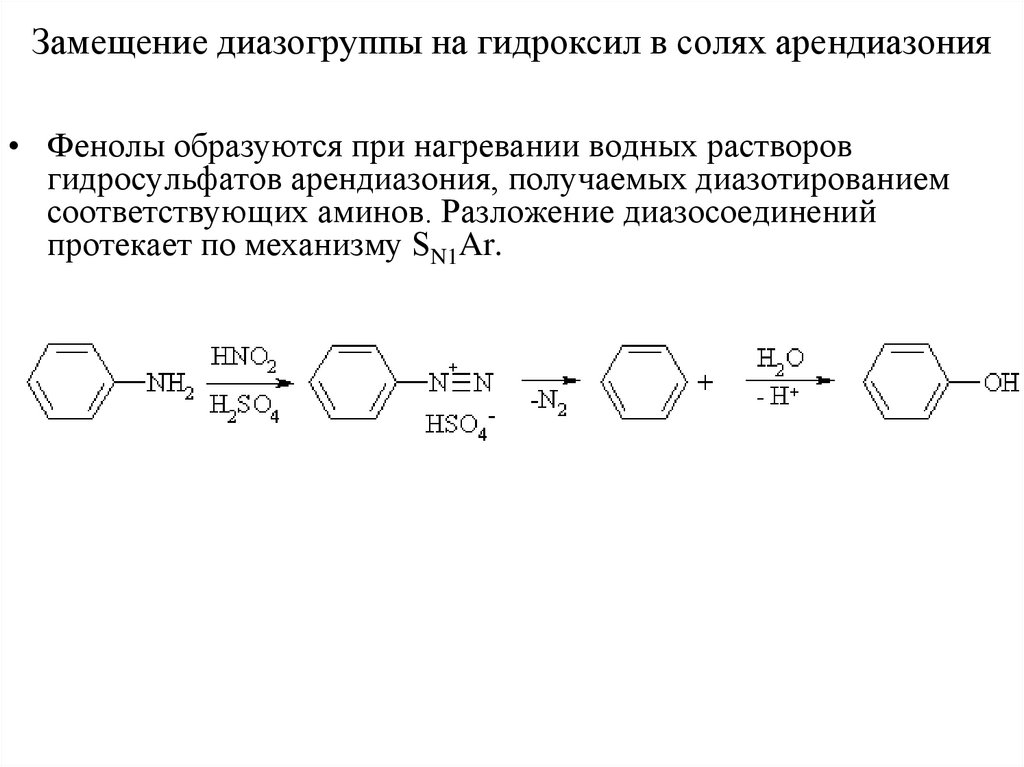
43.
Замещение диазогруппы на гидроксил в солях арендиазония• Фенолы образуются при нагревании водных растворов
гидросульфатов арендиазония, получаемых диазотированием
соответствующих аминов. Разложение диазосоединений
протекает по механизму SN1Ar.
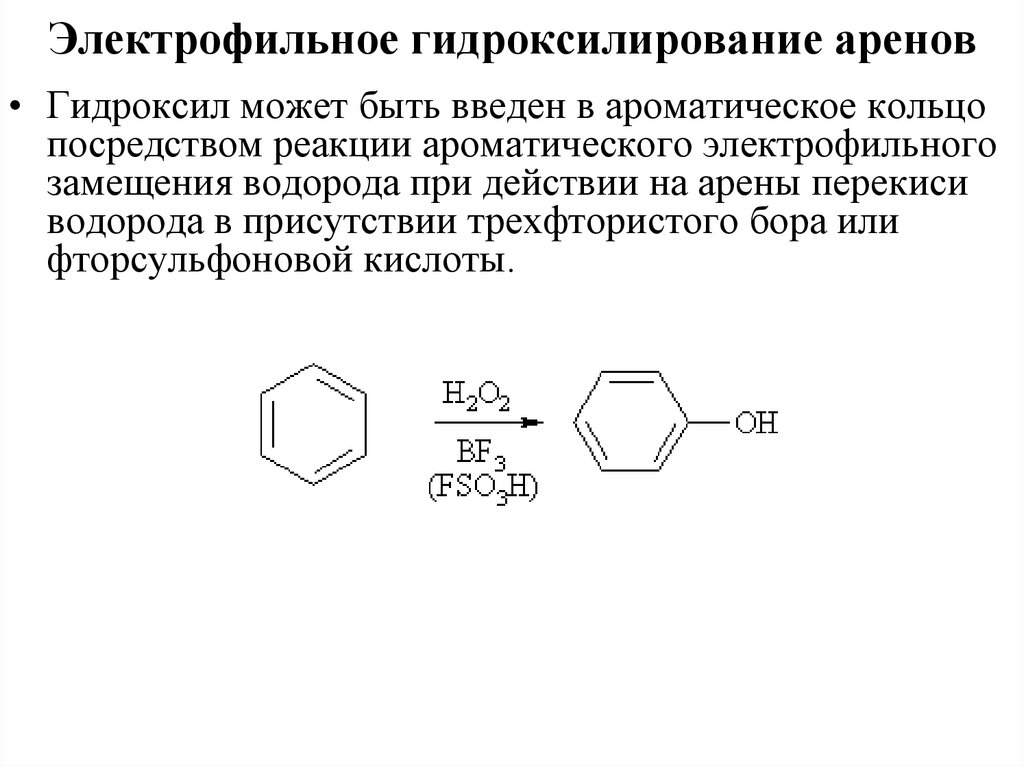
44.
Электрофильное гидроксилирование аренов• Гидроксил может быть введен в ароматическое кольцо
посредством реакции ароматического электрофильного
замещения водорода при действии на арены перекиси
водорода в присутствии трехфтористого бора или
фторсульфоновой кислоты.
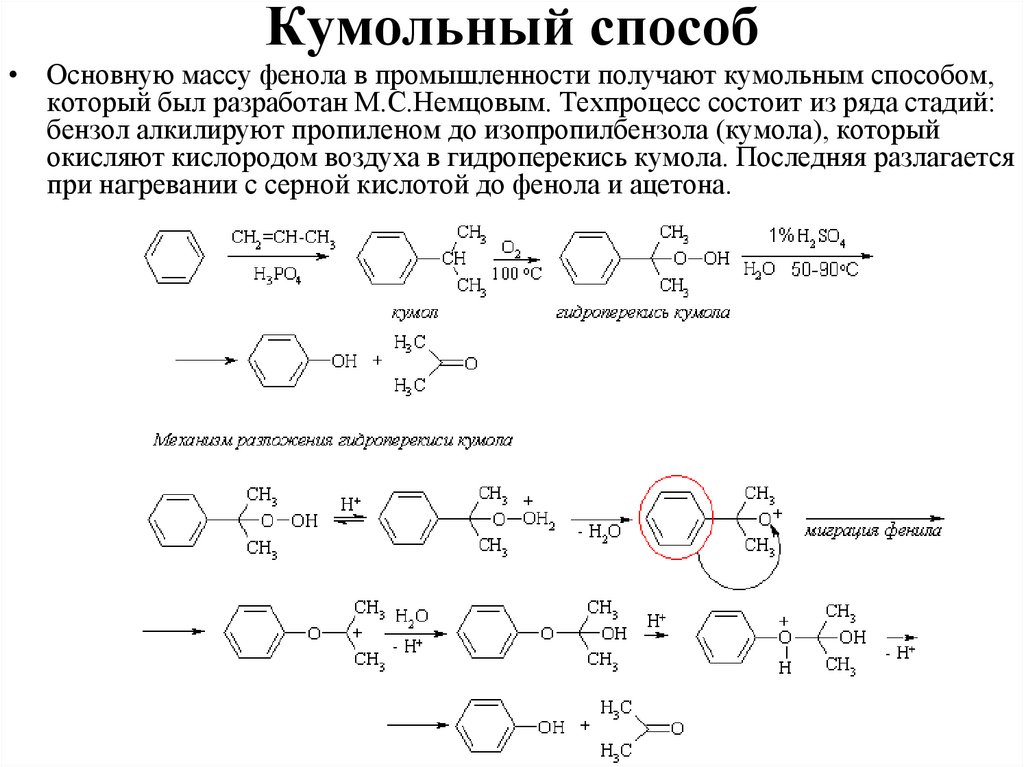
45.
Кумольный способ• Основную массу фенола в промышленности получают кумольным способом,
который был разработан М.С.Немцовым. Техпроцесс состоит из ряда стадий:
бензол алкилируют пропиленом до изопропилбензола (кумола), который
окисляют кислородом воздуха в гидроперекись кумола. Последняя разлагается
при нагревании с серной кислотой до фенола и ацетона.
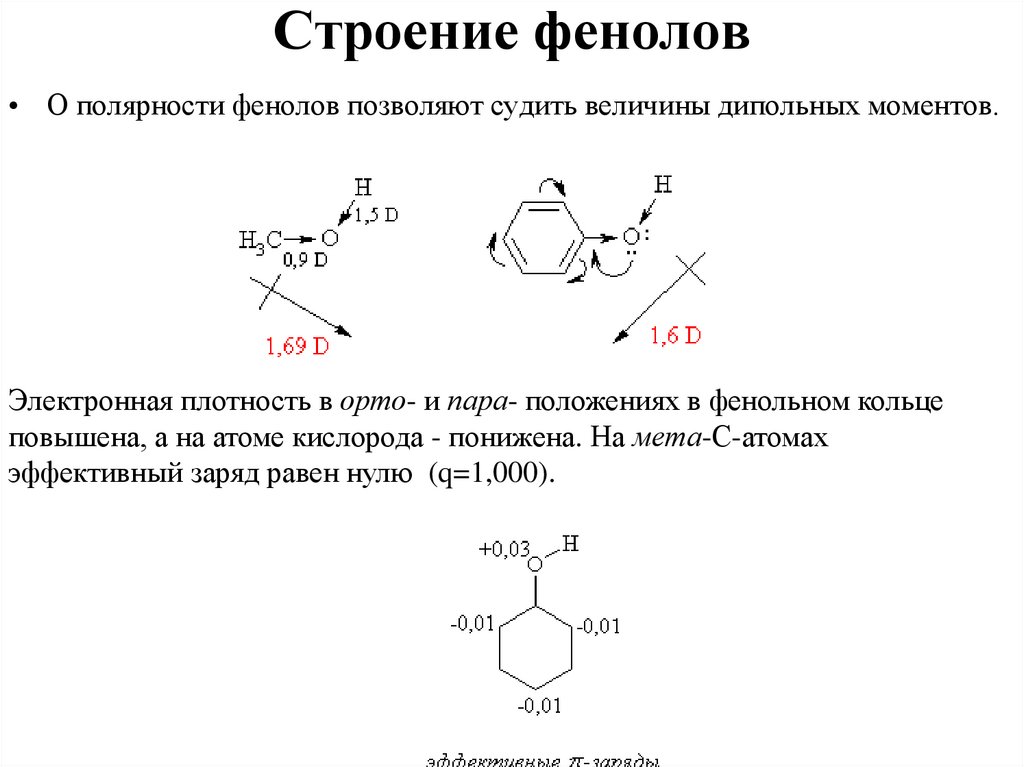
46.
Строение фенолов• О полярности фенолов позволяют судить величины дипольных моментов.
Электронная плотность в орто- и пара- положениях в фенольном кольце
повышена, а на атоме кислорода - понижена. На мета-С-атомах
эффективный заряд равен нулю (q=1,000).
47.
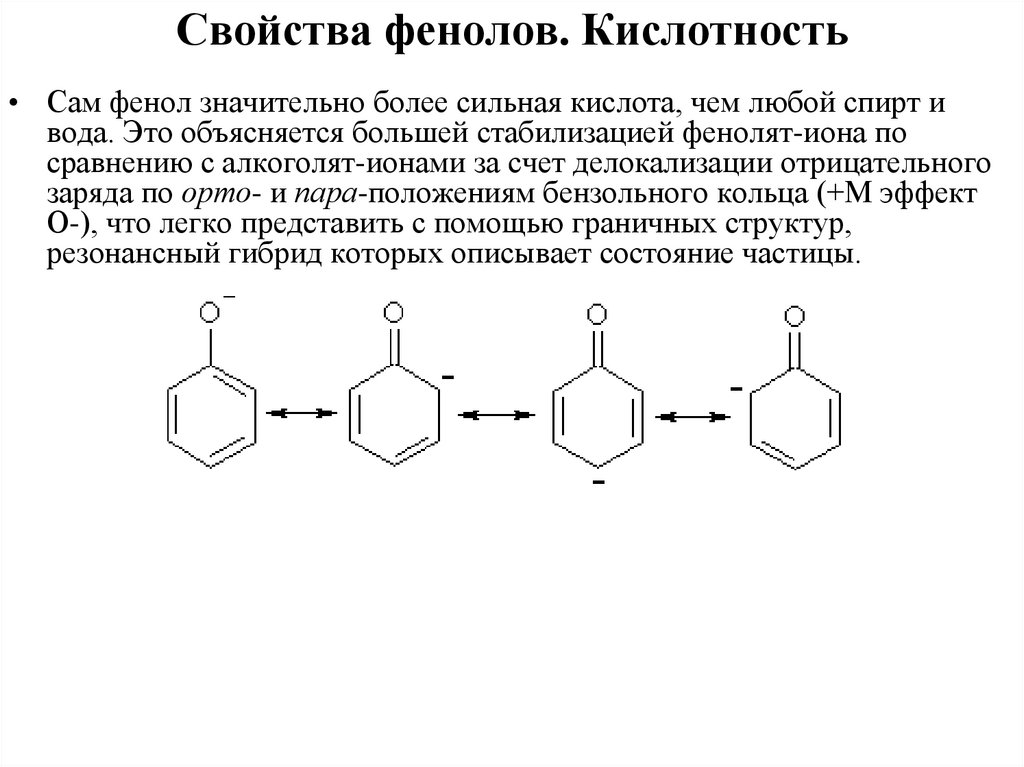
Свойства фенолов. Кислотность• Сам фенол значительно более сильная кислота, чем любой спирт и
вода. Это объясняется большей стабилизацией фенолят-иона по
сравнению с алкоголят-ионами за счет делокализации отрицательного
заряда по орто- и пара-положениям бензольного кольца (+М эффект
О-), что легко представить с помощью граничных структур,
резонансный гибрид которых описывает состояние частицы.
48.
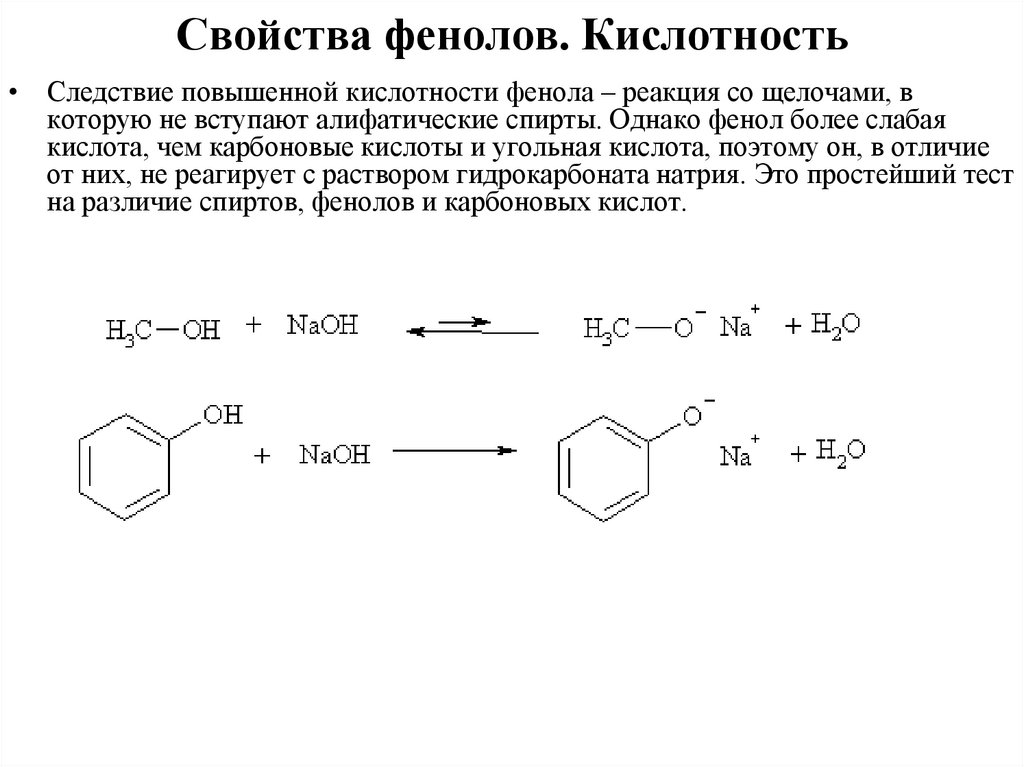
Свойства фенолов. Кислотность• Следствие повышенной кислотности фенола – реакция со щелочами, в
которую не вступают алифатические спирты. Однако фенол более слабая
кислота, чем карбоновые кислоты и угольная кислота, поэтому он, в отличие
от них, не реагирует с раствором гидрокарбоната натрия. Это простейший тест
на различие спиртов, фенолов и карбоновых кислот.
49.
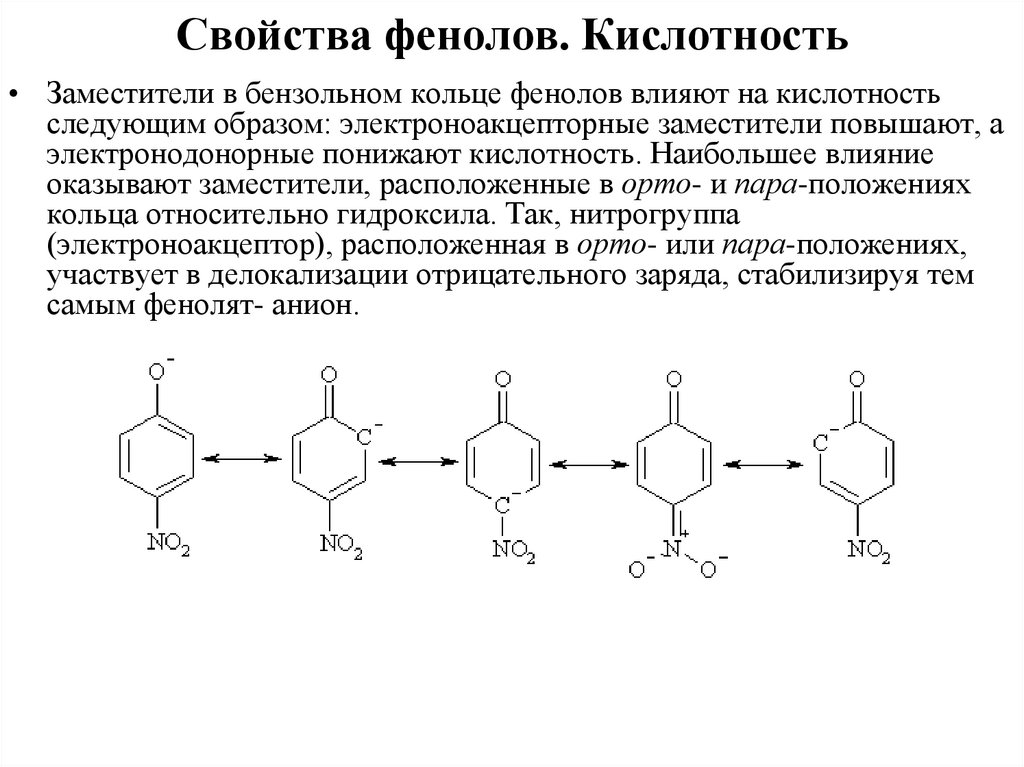
Свойства фенолов. Кислотность• Заместители в бензольном кольце фенолов влияют на кислотность
следующим образом: электроноакцепторные заместители повышают, а
электронодонорные понижают кислотность. Наибольшее влияние
оказывают заместители, расположенные в орто- и пара-положениях
кольца относительно гидроксила. Так, нитрогруппа
(электроноакцептор), расположенная в орто- или пара-положениях,
участвует в делокализации отрицательного заряда, стабилизируя тем
самым фенолят- анион.
50.
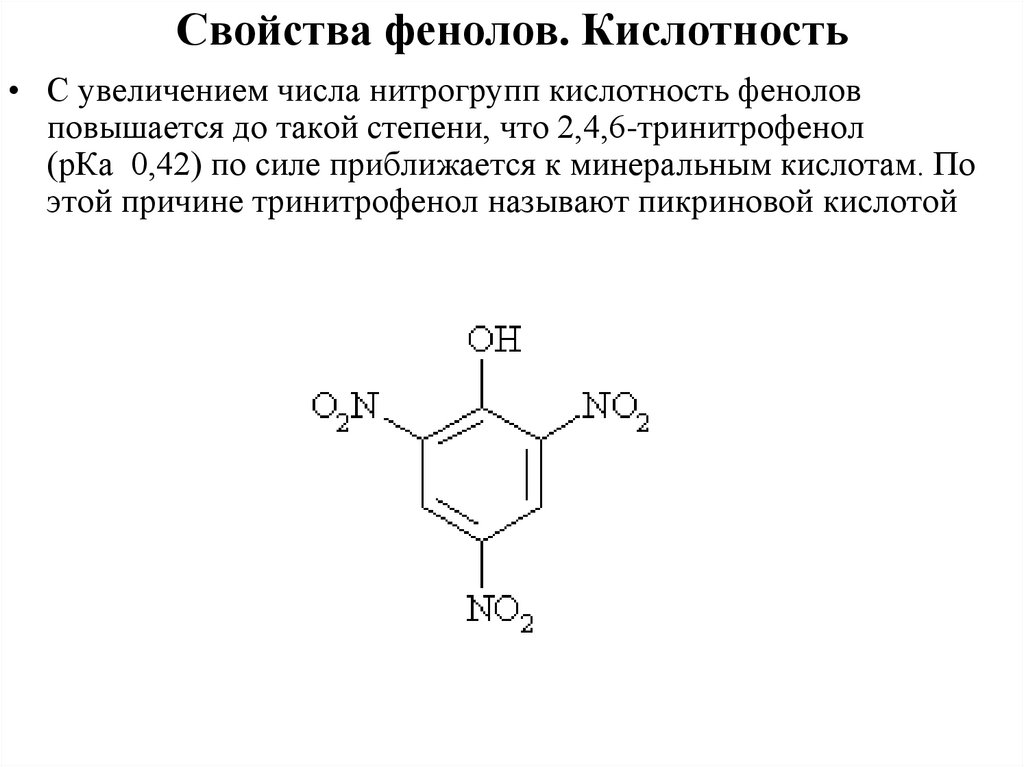
Свойства фенолов. Кислотность• С увеличением числа нитрогрупп кислотность фенолов
повышается до такой степени, что 2,4,6-тринитрофенол
(рКа 0,42) по силе приближается к минеральным кислотам. По
этой причине тринитрофенол называют пикриновой кислотой
51.
Значения рКа некоторых спиртов и фенолов52.
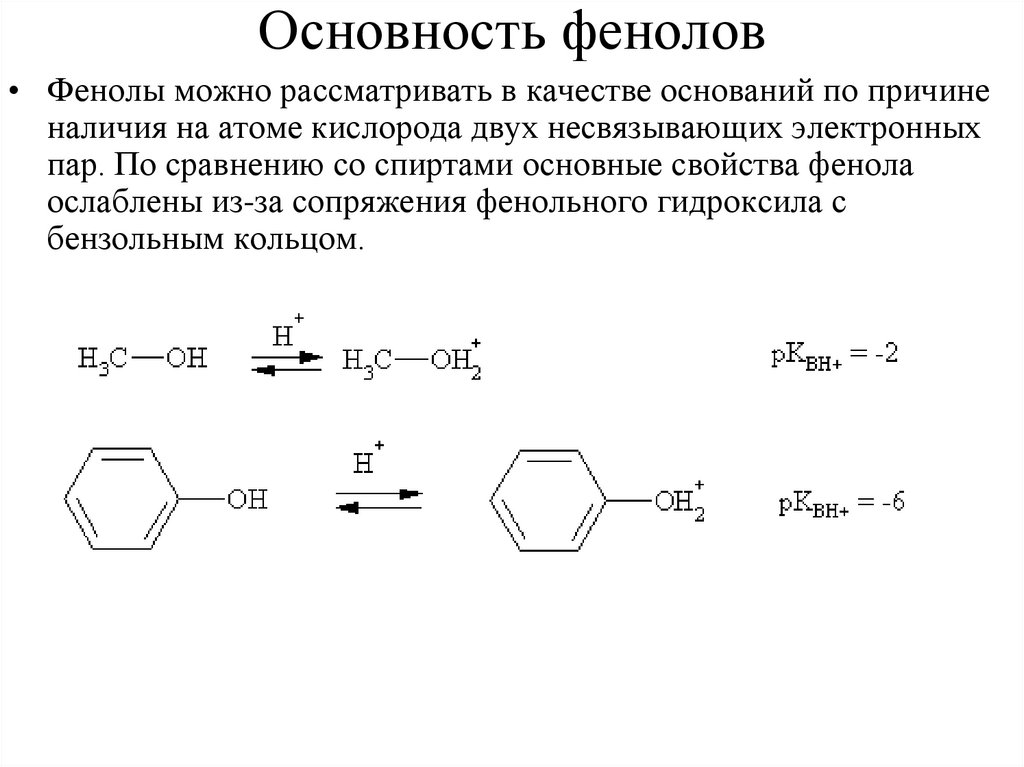
Основность фенолов• Фенолы можно рассматривать в качестве оснований по причине
наличия на атоме кислорода двух несвязывающих электронных
пар. По сравнению со спиртами основные свойства фенола
ослаблены из-за сопряжения фенольного гидроксила с
бензольным кольцом.
53.
Нуклеофильность фенолов и фенолят-ионов• С основностью тесно связана нуклеофильность. Фенольный
гидроксил проявляет слабый нуклеофильный характер, тогда как
анионы фенолов являются сильными нуклеофилами и могут
участвовать в этом качестве в реакциях
• алкилирования,
• арилирования
• и ацилирования по атому кислорода
54.
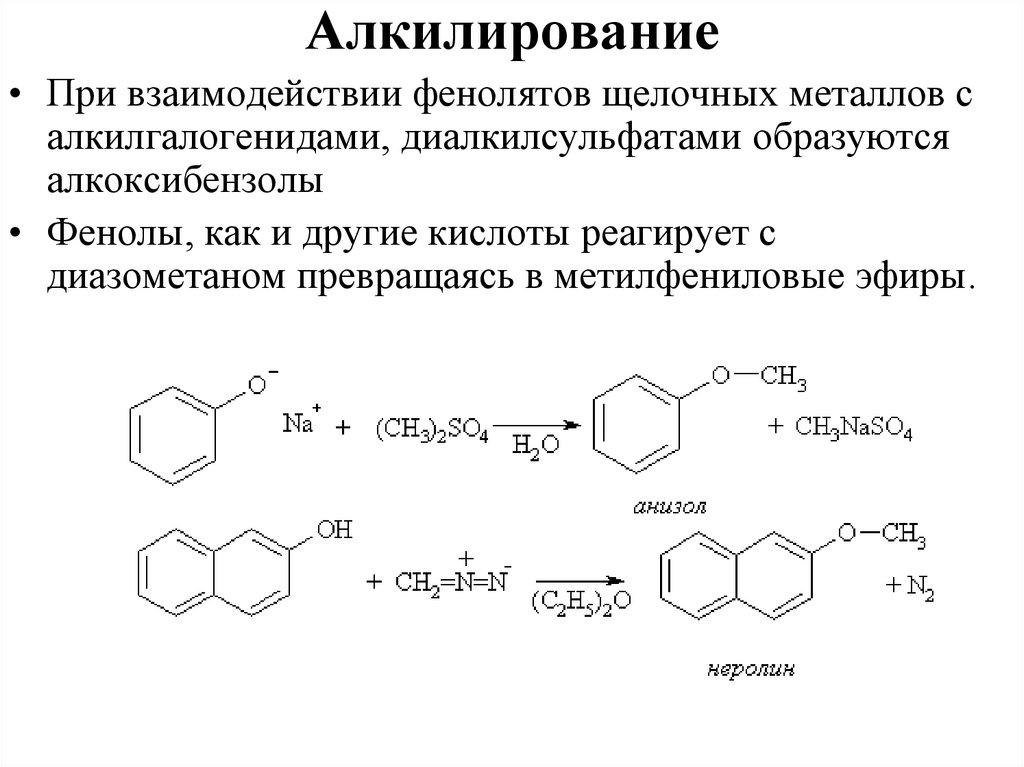
Алкилирование• При взаимодействии фенолятов щелочных металлов с
алкилгалогенидами, диалкилсульфатами образуются
алкоксибензолы
• Фенолы, как и другие кислоты реагирует с
диазометаном превращаясь в метилфениловые эфиры.
55.
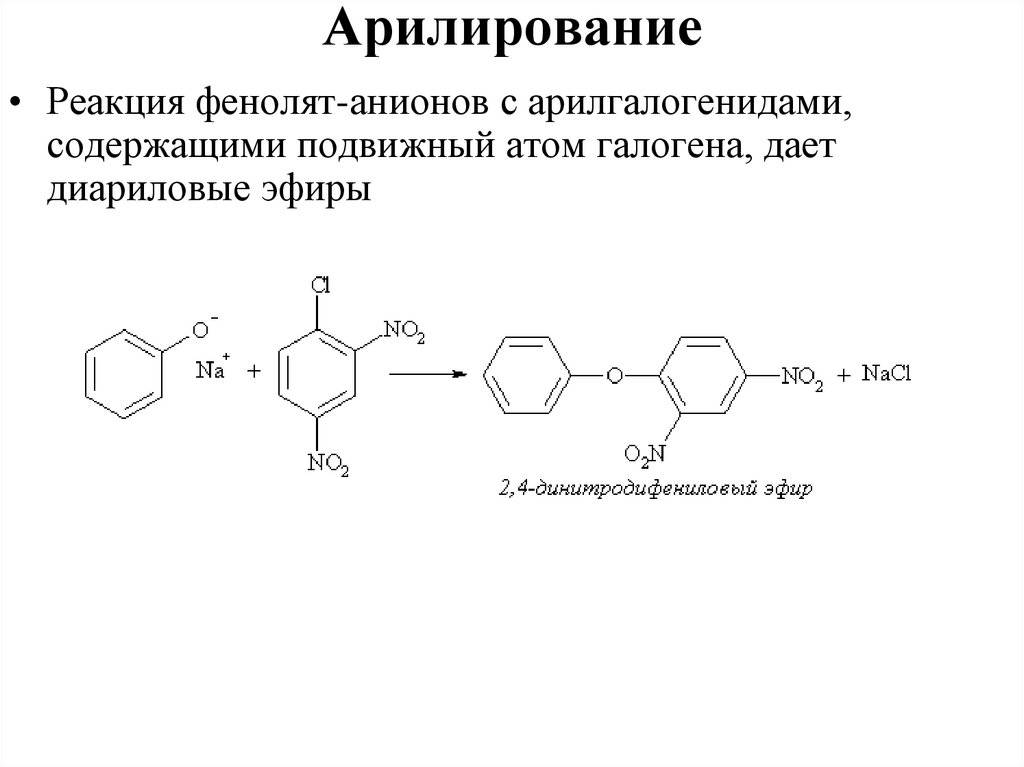
Арилирование• Реакция фенолят-анионов с арилгалогенидами,
содержащими подвижный атом галогена, дает
диариловые эфиры
56.
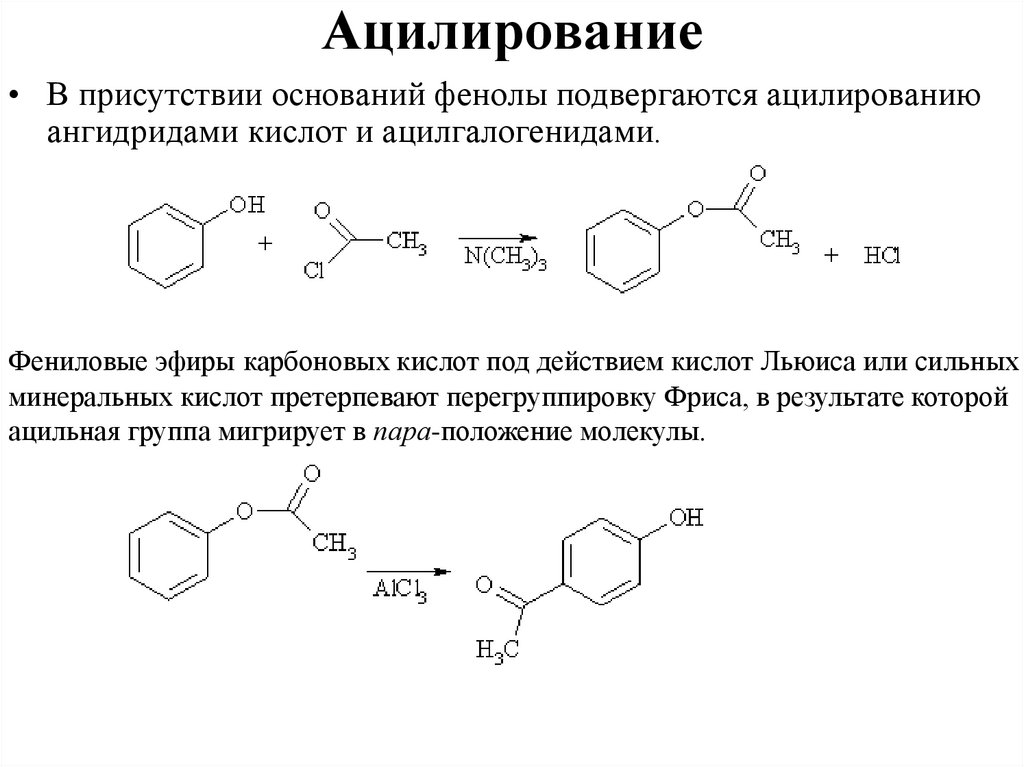
Ацилирование• В присутствии оснований фенолы подвергаются ацилированию
ангидридами кислот и ацилгалогенидами.
Фениловые эфиры карбоновых кислот под действием кислот Льюиса или сильных
минеральных кислот претерпевают перегруппировку Фриса, в результате которой
ацильная группа мигрирует в пара-положение молекулы.
57.
Замещение гидроксила галогенами• В отличие от спиртов, гидроксильная группа которых легко
замещается нуклеофильными реагентами (Br-, Cl-), замещение
фенольного гидроксила галогеном протекает лишь в жестких
условиях при действии галогенангидридов неорганических
кислот. Например, реакция пентахлорида фосфора с фенолом
дает хлорбензол, а взаимодействие пикриновой кислоты с
хлорокисью фосфора – пикрилхлорид.
58.
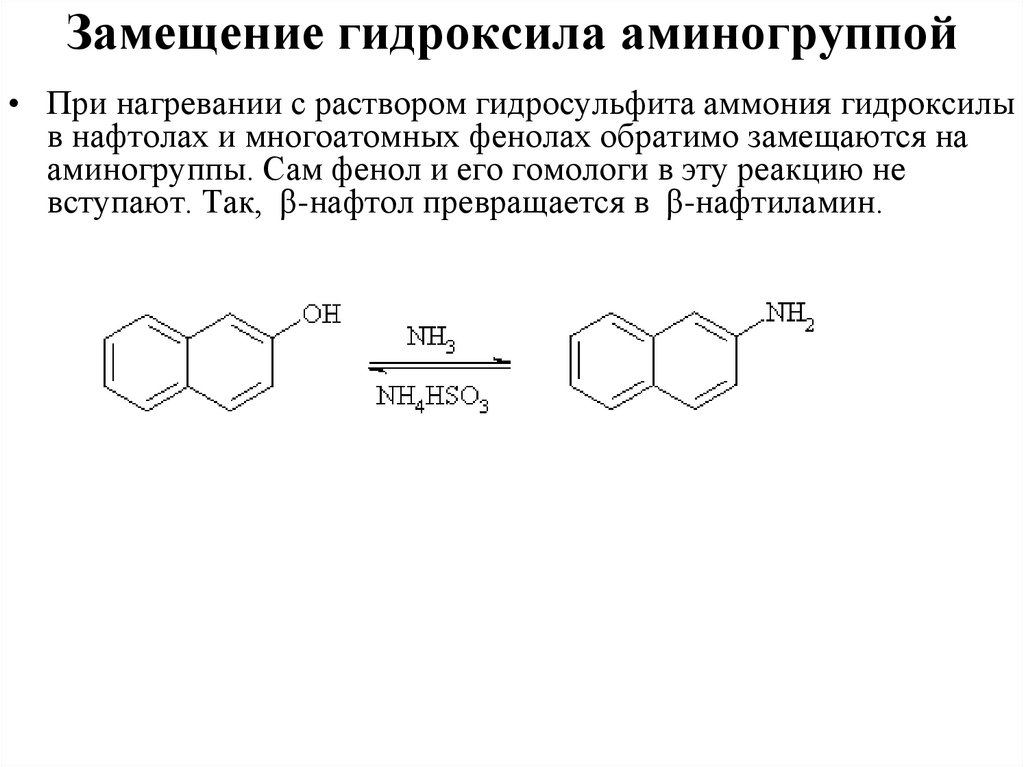
Замещение гидроксила аминогруппой• При нагревании с раствором гидросульфита аммония гидроксилы
в нафтолах и многоатомных фенолах обратимо замещаются на
аминогруппы. Сам фенол и его гомологи в эту реакцию не
вступают. Так, β-нафтол превращается в β-нафтиламин.
59.
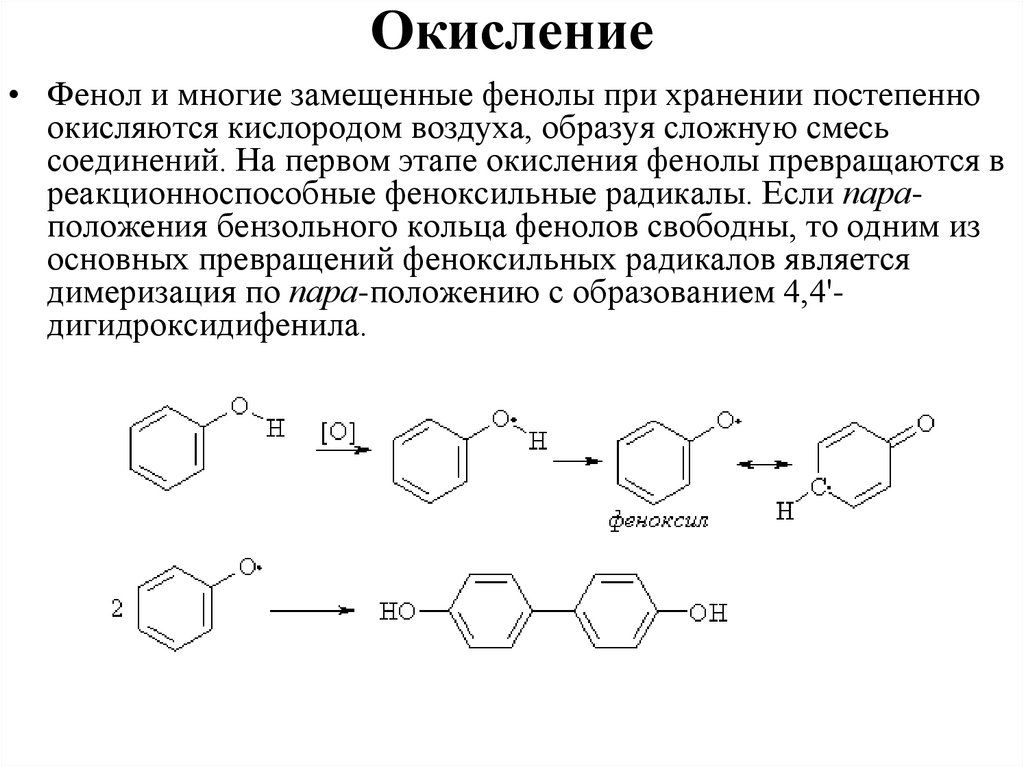
Окисление• Фенол и многие замещенные фенолы при хранении постепенно
окисляются кислородом воздуха, образуя сложную смесь
соединений. На первом этапе окисления фенолы превращаются в
реакционноспособные феноксильные радикалы. Если параположения бензольного кольца фенолов свободны, то одним из
основных превращений феноксильных радикалов является
димеризация по пара-положению с образованием 4,4'дигидроксидифенила.
60.
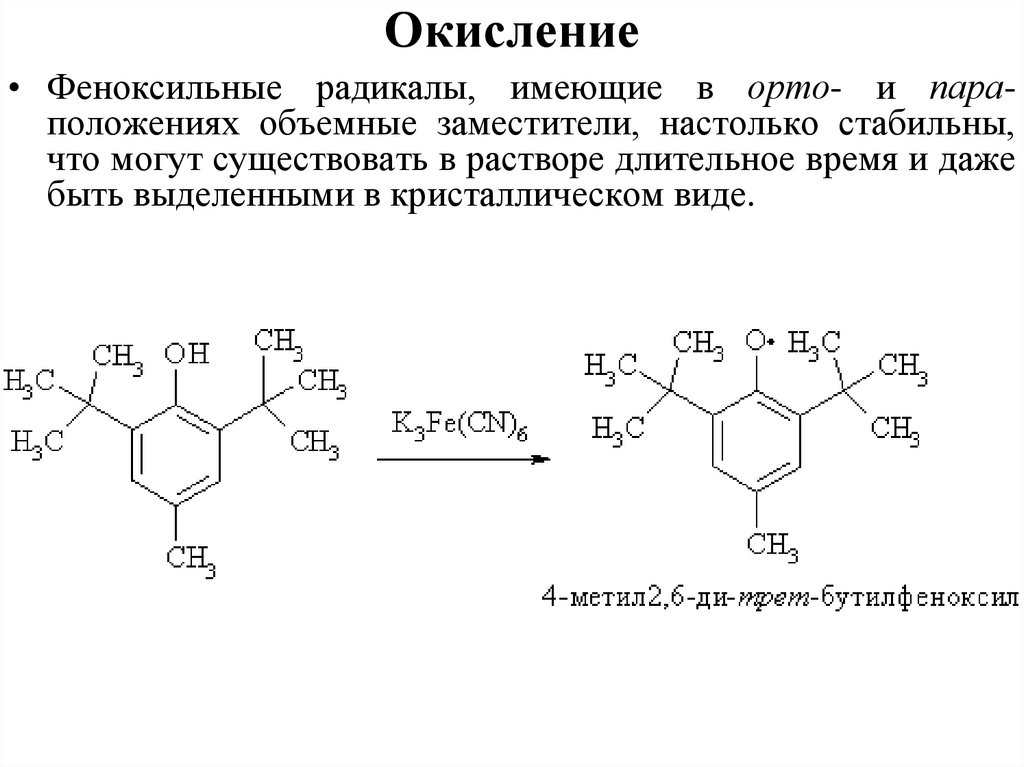
Окисление• Феноксильные радикалы, имеющие в орто- и параположениях объемные заместители, настолько стабильны,
что могут существовать в растворе длительное время и даже
быть выделенными в кристаллическом виде.
61.
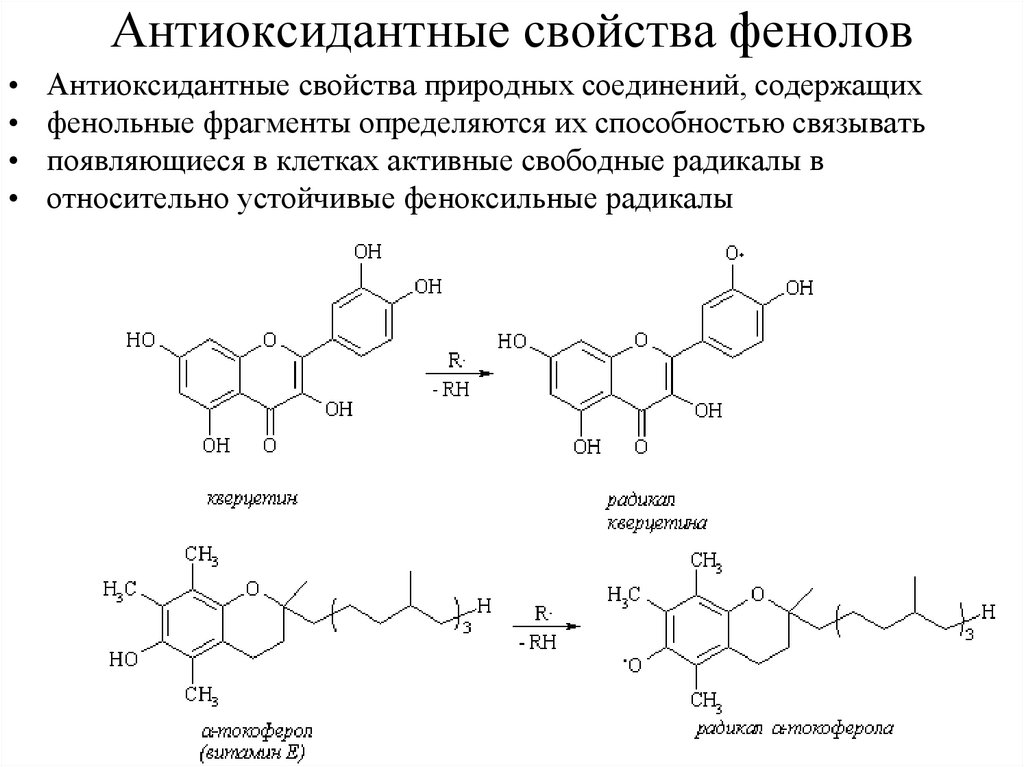
Антиоксидантные свойства феноловАнтиоксидантные свойства природных соединений, содержащих
фенольные фрагменты определяются их способностью связывать
появляющиеся в клетках активные свободные радикалы в
относительно устойчивые феноксильные радикалы
62.
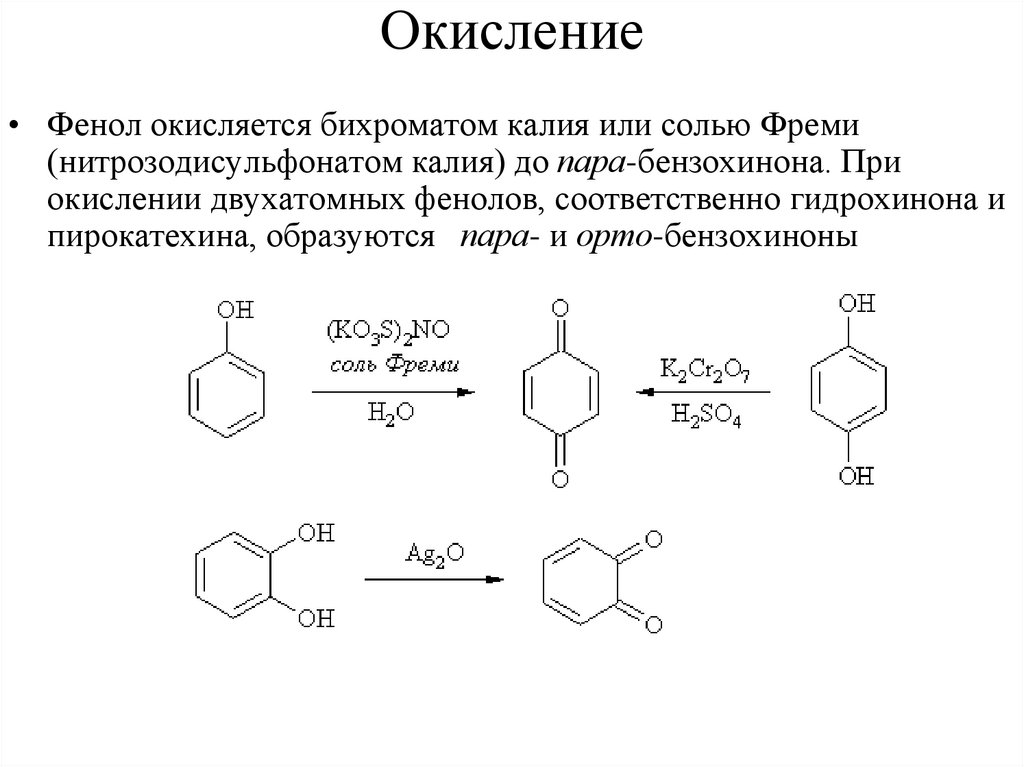
Окисление• Фенол окисляется бихроматом калия или солью Фреми
(нитрозодисульфонатом калия) до пара-бензохинона. При
окислении двухатомных фенолов, соответственно гидрохинона и
пирокатехина, образуются пара- и орто-бензохиноны
63.
Электрофильное замещение в ароматическом кольце фенолов• Реакции ароматического кольца фенолов протекают по
механизму
электрофильного
ароматического
замещения.
Гидроксильная группа ориентирует вступление электрофильного
реагента в орто- и пара-положения бензольного кольца. Из-за
донорного влияния гидроксигруппы (+М эффект) реакции
электрофильного замещения в фенолах протекают значительно
легче, чем в бензоле и его гомологах. Фенолы и фенолят-анионы
вступают во взаимодействие даже с очень слабыми
электрофилами
64.
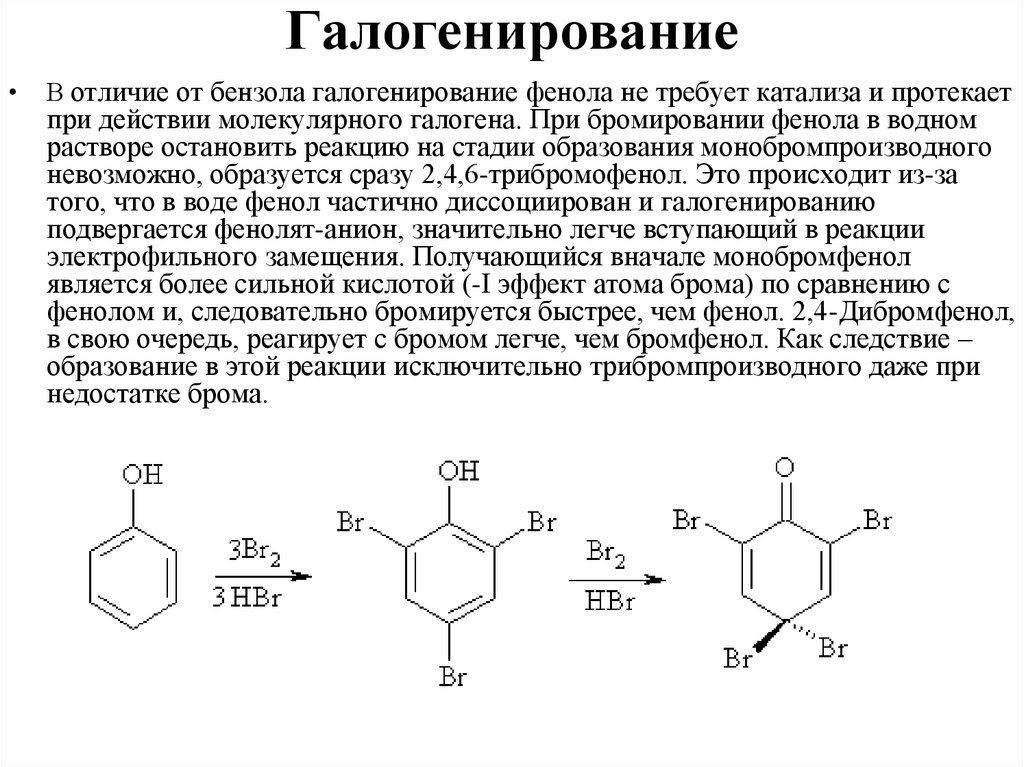
ГалогенированиеВ отличие от бензола галогенирование фенола не требует катализа и протекает
при действии молекулярного галогена. При бромировании фенола в водном
растворе остановить реакцию на стадии образования монобромпроизводного
невозможно, образуется сразу 2,4,6-трибромофенол. Это происходит из-за
того, что в воде фенол частично диссоциирован и галогенированию
подвергается фенолят-анион, значительно легче вступающий в реакции
электрофильного замещения. Получающийся вначале монобромфенол
является более сильной кислотой (-I эффект атома брома) по сравнению с
фенолом и, следовательно бромируется быстрее, чем фенол. 2,4-Дибромфенол,
в свою очередь, реагирует с бромом легче, чем бромфенол. Как следствие –
образование в этой реакции исключительно трибромпроизводного даже при
недостатке брома.
65.
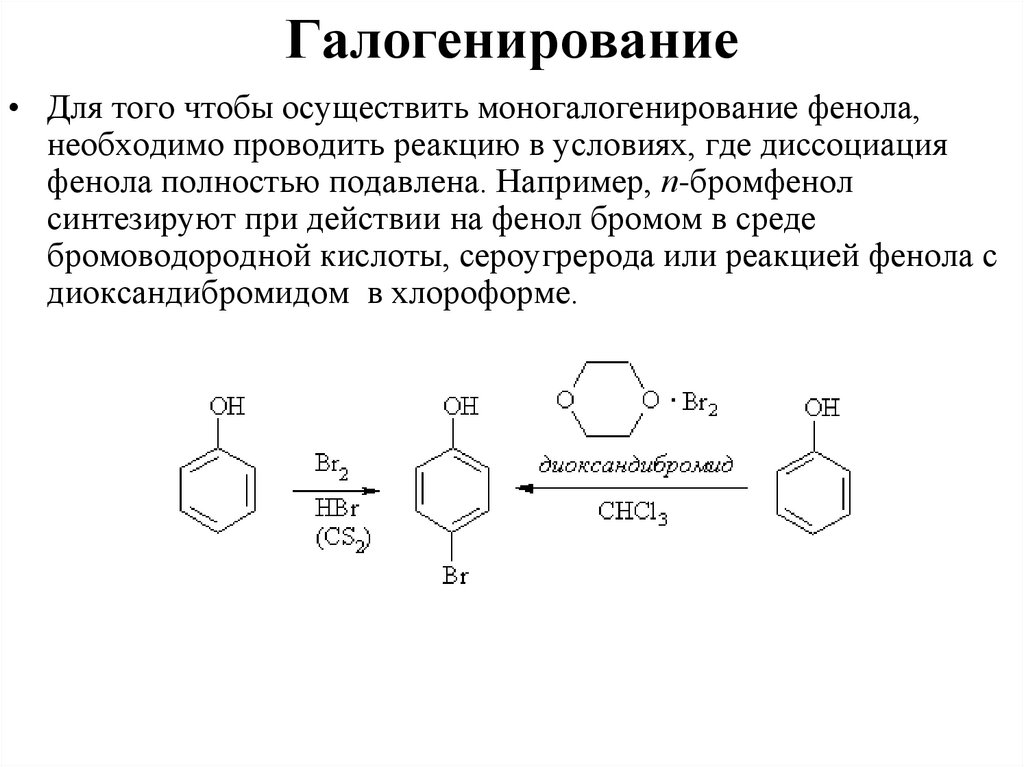
Галогенирование• Для того чтобы осуществить моногалогенирование фенола,
необходимо проводить реакцию в условиях, где диссоциация
фенола полностью подавлена. Например, п-бромфенол
синтезируют при действии на фенол бромом в среде
бромоводородной кислоты, сероугрерода или реакцией фенола с
диоксандибромидом в хлороформе.
66.
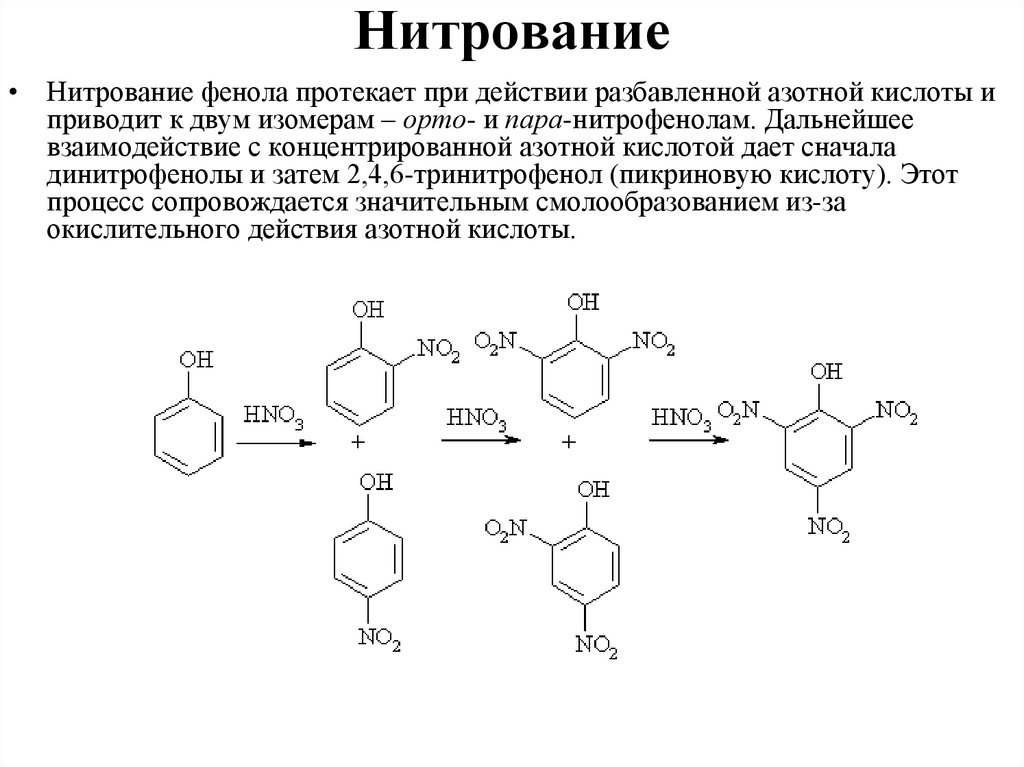
Нитрование• Нитрование фенола протекает при действии разбавленной азотной кислоты и
приводит к двум изомерам – орто- и пара-нитрофенолам. Дальнейшее
взаимодействие с концентрированной азотной кислотой дает сначала
динитрофенолы и затем 2,4,6-тринитрофенол (пикриновую кислоту). Этот
процесс сопровождается значительным смолообразованием из-за
окислительного действия азотной кислоты.
67.
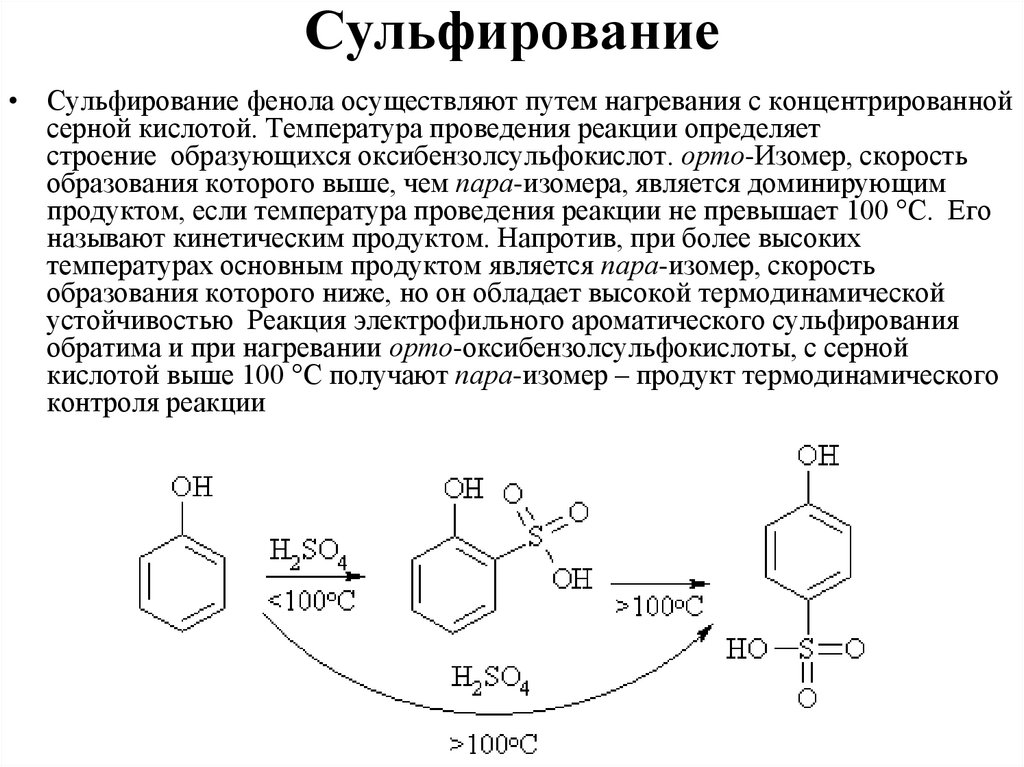
Сульфирование• Сульфирование фенола осуществляют путем нагревания с концентрированной
серной кислотой. Температура проведения реакции определяет
строение образующихся оксибензолсульфокислот. орто-Изомер, скорость
образования которого выше, чем пара-изомера, является доминирующим
продуктом, если температура проведения реакции не превышает 100 °С. Его
называют кинетическим продуктом. Напротив, при более высоких
температурах основным продуктом является пара-изомер, скорость
образования которого ниже, но он обладает высокой термодинамической
устойчивостью Реакция электрофильного ароматического сульфирования
обратима и при нагревании орто-оксибензолсульфокислоты, с серной
кислотой выше 100 °С получают пара-изомер – продукт термодинамического
контроля реакции
68.
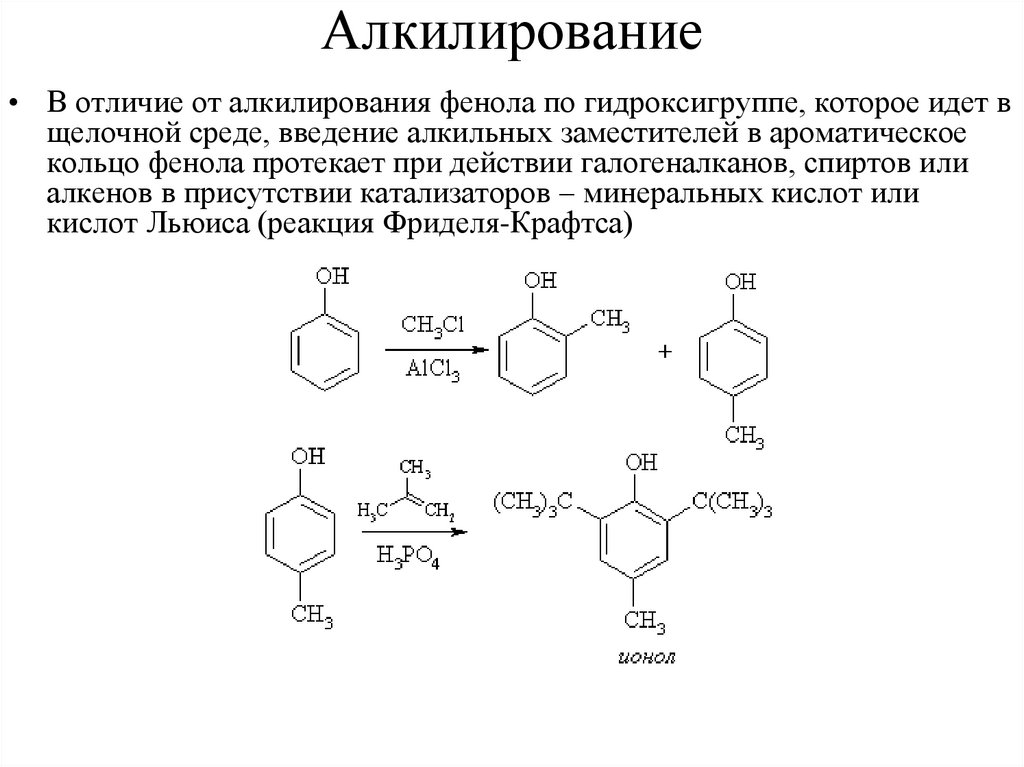
Алкилирование• В отличие от алкилирования фенола по гидроксигруппе, которое идет в
щелочной среде, введение алкильных заместителей в ароматическое
кольцо фенола протекает при действии галогеналканов, спиртов или
алкенов в присутствии катализаторов – минеральных кислот или
кислот Льюиса (реакция Фриделя-Крафтса)
69.
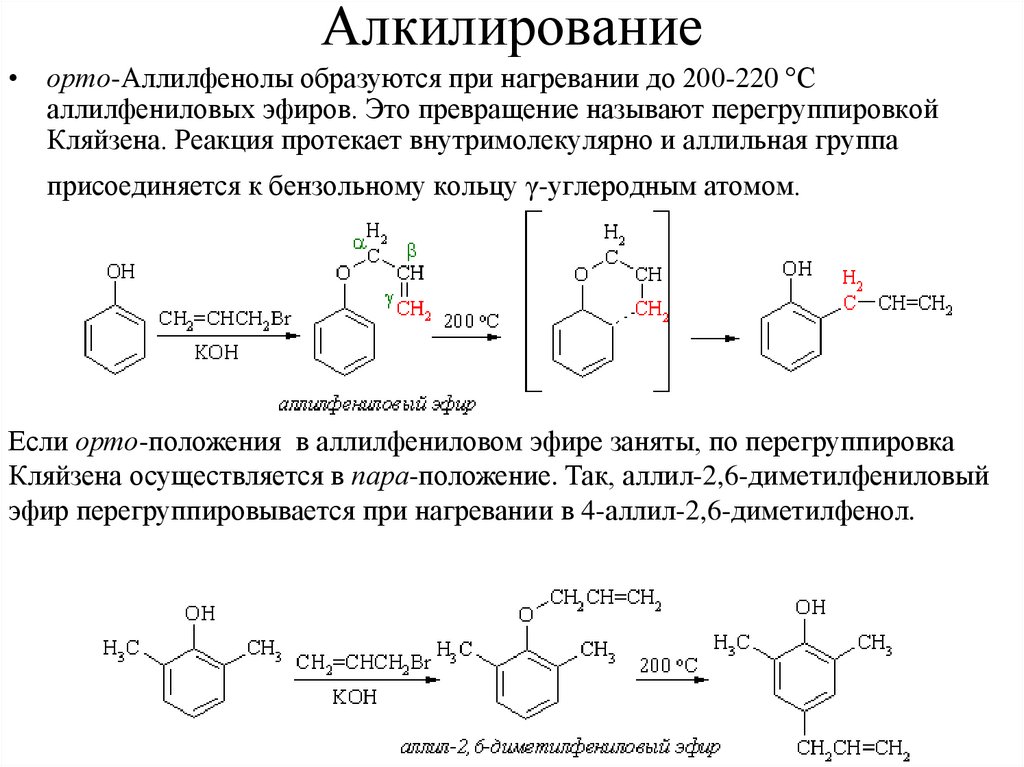
Алкилирование• орто-Аллилфенолы образуются при нагревании до 200-220 °С
аллилфениловых эфиров. Это превращение называют перегруппировкой
Кляйзена. Реакция протекает внутримолекулярно и аллильная группа
присоединяется к бензольному кольцу γ-углеродным атомом.
Если орто-положения в аллилфениловом эфире заняты, по перегруппировка
Кляйзена осуществляется в пара-положение. Так, аллил-2,6-диметилфениловый
эфир перегруппировывается при нагревании в 4-аллил-2,6-диметилфенол.
70.
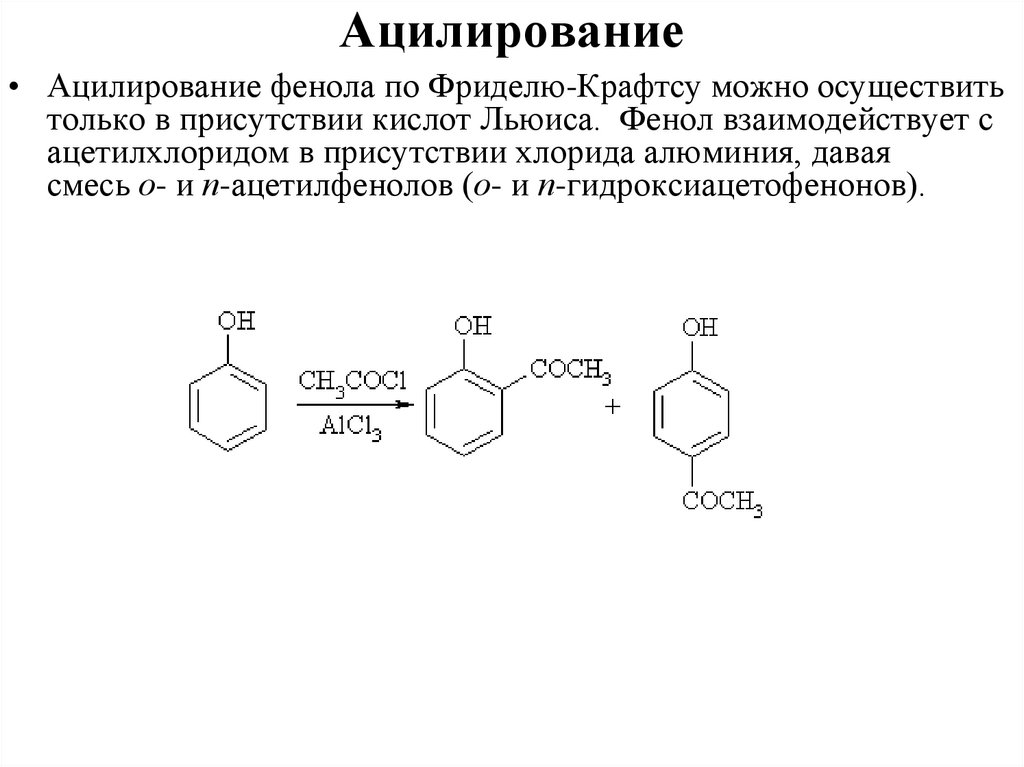
Ацилирование• Ацилирование фенола по Фриделю-Крафтсу можно осуществить
только в присутствии кислот Льюиса. Фенол взаимодействует с
ацетилхлоридом в присутствии хлорида алюминия, давая
смесь о- и п-ацетилфенолов (о- и п-гидроксиацетофенонов).
71.
Ацилирование• Другой способ получения С-ацилпроизводных фенолов основан на
упомянутой выше перегруппировке Фриса. Ариловые эфиры карбоновых
кислот при нагревании в присутствии AlCl3 или AlBr3 превращаются в ортои пара-ацилфенолы. При этом, варьируя температуру и растворитель, можно
изменить соотношение изомеров в пользу одного из них. Механизм реакции
включает две стадии: 1) межмолекулярное ацилирование бензольного кольца
одной молекулы сложного эфира комплексом второй молекулы с кислотой
Льюиса; 2) межмолекулярный перенос О-ацильной группы к молекуле
фенола.
72.
Ацилирование• Ацилирование фенолов фталевым ангидридом в присутствии
серной кислоты или хлорида цинка приводит к образованию
фталеинов, многие из которых являются индикаторами
(фенолфталеин, тимолфталеин) или красителями (флуоресцеин).
73.
Формилирование• Введение формильной группы в ароматическое кольцо фенолов
осуществляется различными методами – для этого применимы
реакции Реймера-Тимана, Вильсмайера-Хаака, Гаттермана.
• Взаимодействие фенола с хлороформом в щелочной среде
(реакция Реймера-Тимана) дает орто-гидроксибензальдегид
(салициловый альдегид).
74.
Реакция Реймера-Тимана• Электрофильной частицей в реакции выступает дихлоркарбен,
который генерируется из хлороформа под действием щелочи.
Промежуточно образующийся орто-(дихлорметил)фенол
гидролизуется в ходе реакции до альдегида. По Реймеру-Тиману
формилируется большинство С-замещенных фенолов, нафтолы и
многоатомные фенолы, при этом формильная группа вступает
всегда в орто-положение относительно гидроксила
75.
Реакция Вильсмайера-Хаака• Для формилирования фенолов по Вильсмайеру-Хааку используют
замещенные амиды муравьиной кислоты – N,N-диметилформамид и Nметилформанилид, которые в ходе реакции превращают в электрофильные
иминиевые соли взаимодействием с хлорокисью фосфора, тионилхлоридом
или фосгеном. Последние, хотя и являются слабыми электрофилами, атакуют
ароматическое ядро, что приводит к соответствующим иммониевым солям,
которые далее гидролизуются до альдегидов. Формильная группа вводится
исключительно в пара-положение относительно гидроксильной группы
76.
Формилирование по Гаттерману-Коху• Классическое формилирование по Гаттерману-Коху (СО и HCl),
протекающее для бензола, нафталина и их гомологов, для фенолов
неосуществимо. Однако сам Л. Гаттерман предложил модификацию
метода, приемлемую для фенолов, в том числе и многоатомных, и
нафтолов. При действии на фенол б/в HCN и газообразного
хлороводорода (реакция Гаттермана) образуется парагидроксибензальдегид. В некоторых случаях вместо токсичного
цианистого водорода используют цианид цинка.
77.
Карбоксилирование (реакция Кольбе)• Нагревание фенолята натрия с диоксидом углерода при давлении в 5 атм
приводит к образованию салицилата натрия. Интересно, что при
использовании в качестве исходного соединения фенолята калия
карбоксильная группа вступает в пара-положение бензольного кольца и
образуется пара-гидроксибензоат калия. Электрофилом в реакции Кольбе
выступает диоксид углерода, который атакует электроноизбыточные ортоили пара-положения фенолят-аниона. Предполагают, что если в реакционной
смеси присутствуют ионы натрия, то за счет комплексообразования
происходит стабилизация переходного состояния, ведущего к образованию
салициловой кислоты.
78.
Карбоксилирование (реакция Кольбе)• Многоатомные фенолы реагируют с диоксидом углерода в более
мягких условиях. Например, резорцин карбоксилируется уже при
кипячении с раствором карбоната натрия