















.")




 Медицина
МедицинаПохожие презентации:










Наследственные заболевания
1. НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ
2.
3. КЛАСЕФИКАЦИЯ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
Генные (моногенные - воснове патологии одна
пара аллельных генов)
Хромосомные
Болезни с наследственным
предрасположением
(мультифакториальные).
4. Фенилкетонурия
СТРАНАСИМПТОМЫ БОЛЕЗНИ
1.НАЛИЧИЕ В ОРГАНИЗМЕ
ФИНИЛПЕРОВЕНОГРАДНОЙ КИСЛОТЫ
КИТАЙ
2.СЛАБОУМИЕ
ОСНОВА БОЛЕЗНИ
ОТСУТСТВИЕ
ФЕРМЕНТА,КАТАЛИЗИРУЮЩАЯ
РЕАКЦИЮ ОКИСЛЕНИЯ
ФЕНИЛОЛИНА В ТЕРАЗИН
Встречаемост
ь заболевания
1 на 18000
Финляндия
менее 1 на
100000
Ирландия
1 на 4500
Япония
1 на 120000
Корея
1 на 41000
Норвегия
1 на 13000
Турция
1 на 2600
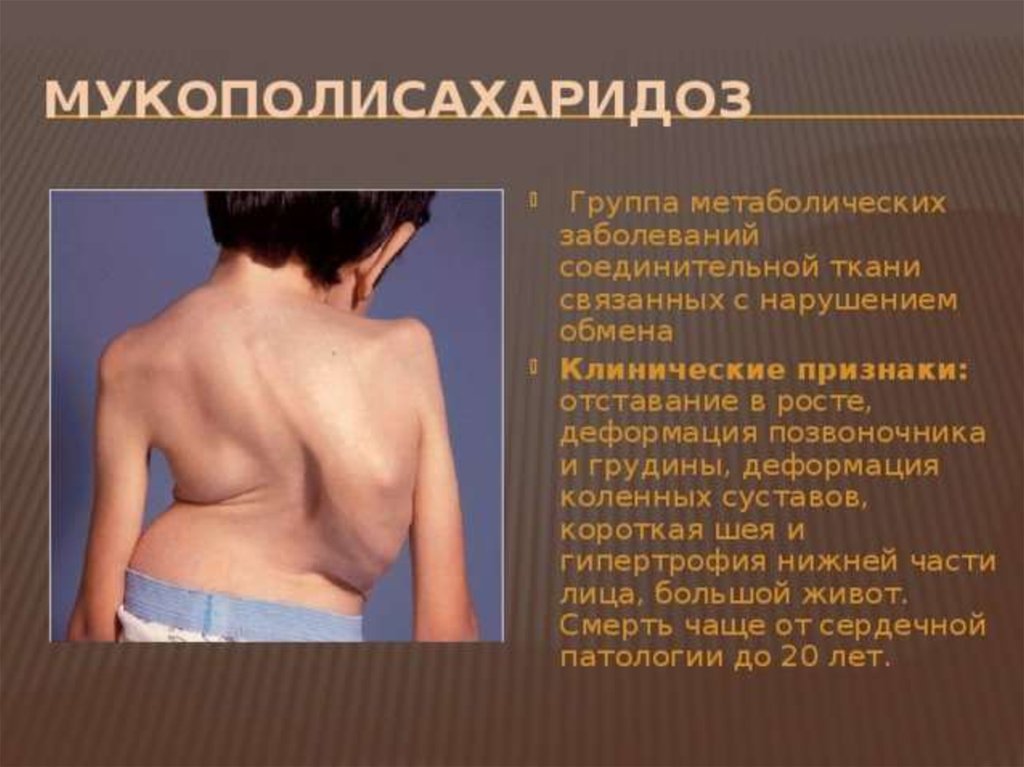
5. Алкаптонурия
6. Болезнь Ниманна — Пика
7. Болезнь Гоше
8. Синдром Лёша — Нихена
9.
10. Болезнь Вильсона — Коновалова
БОЛЕЗНЬ ВИЛЬСОНА —КОНОВАЛОВА
Болезнь Вильсона (Болезнь Вильсона — Коновалова,
гепатоцеребральная дистрофия, гепатолентикулярная
дегенерация, болезнь Вестфаля — Вильсона —
Коновалова) — врождённое нарушение метаболизма
меди, приводящее к тяжелейшим наследственным
болезням центральной нервной системы и внутренних
органов.
Диагностируется у 5-10 % больных циррозом печени
дошкольного и школьного возраста. Заболевание
передается по аутосомно-рецессивному типу. Ген
ATP7B, мутации которого вызывают заболевание,
расположен на 13-й хромосоме (участок 13q14-q21).
11. Первые симптомы односторонний птоз, или проблемы при открытии век, который постепенно прогрессирует и приводит к двустороннему птозу. Ког
Первые симптомы односторонний птоз, или проблемы приоткрытии век, который постепенно прогрессирует и приводит к
двустороннему птозу. Когда птоз усиливается, пострадавший
обычно запрокидывает шею, поднимая подбородок в попытке
предотвратить окклюзию зрительной оси опустившимися веками.
Наряду с коварным развитием птоза, движения глаз в конечном
итоге становятся ограниченными, в результате чего, лицо больше
полагается на поворот головы из стороны в сторону или вверх и
вниз для просмотра объектов в периферическом поле зрения.
12. 10 самых редких болезний
13. Фатальная семейная бессонница.
Редчайшая наследственная болезнь, при которой человек погибает отнеспособности заснуть. До сих пор она отмечалась лишь в 40 семьях по всему
миру. Фатальная бессонница обычно проявляется между 30 и 60 годами (чаще
всего — после 50 лет) и продолжается от 7 до 36 месяцев. По мере того, как
заболевание прогрессирует, пациент страдает от всё более тяжелых нарушений
сна, причем никакие снотворные ему не помогают. На первой стадии бессонница
сопровождается паническими атаками и фобиями, на второй к ним прибавляются
галлюцинации и повышенное потоотделение. На третьей стадии болезни человек
полностью теряет способность спать и начинает выглядеть намного старше своих
лет. Затем развивается деменция, и пациент погибает — как правило, от
истощения или пневмонии.
14. Нарколепсия-катаплексия.
Синдром нарколепсии-катаплексии, для которого характерны внезапныеприступы сна и расслабления мускулатуры тела, тоже имеет генетическую
природу и возникает из–за нарушений быстрой фазы сна. Он встречается
намного чаще фатальной семейной бессонницы: у 40 из каждых 100 тыс.
человек, в равной степени у мужчин и у женщин. Человек, страдающий
нарколепсией, способен внезапно заснуть на несколько минут посреди
дня. «Сонные атаки» напоминают фазу быстрого сна и могут случаться
очень часто: до 100 раз в день, с предшествующей им головной болью,
либо без неё. Они часто провоцируются бездеятельностью, но могут
возникать в совершенно неподходящее время: во время полового акта,
занятий спортом, вождения. Просыпается человек отдохнувшим.
15. Синдром Юнера Тана.
Синдром Юнера Тана (СЮТ) характерен прежде всего тем, что люди,страдающие им, ходят на четвереньках. Открыл его турецкий биолог Юнер Тан
после изучения пяти членов семьи Улас в сельской местности Турции. Чаще всего
люди с СЮТ пользуются примитивной речью и имеют врождённую мозговую
недостаточность. В 2006-м году о семье Улас был снят документальный фильм
под названием «Семья, ходящая на четвереньках». Тан описывает это так:
«Генетическая природа синдрома предполагает обратную ступень в эволюции
человека, вызванную, скорее всего, генетической мутацией, обратному процессу
перехода от квадропедализма (хождения на четырёх конечностях) к бипедализму
(хождению на двух). В этом случае синдром соответствует теории прерывистого
равновесия.
16. Наследственная сенсорная нейропатия первого типа.
Одно из самых редких в мире заболеваний: этот вид нейропатиидиагностируют у двух человек из миллиона. Аномалия возникает из-за
поражения периферической нервной системы, возникающего вследствие
переизбытка гена PMP22. Главным признаком развития наследственной
сенсорной нейропатии первого типа является потеря чувствительности рук
и ног. Человек перестаёт испытывать боль и ощущать изменение
температуры, что может привести к возникновению некроза тканей,
например, если вовремя не распознать перелом или другую травму. Боль —
одна из реакций организма, сигнализирующих о каких-либо «неполадках»,
поэтому потеря болевой чувствительности чревата слишком поздним
выявлением опасных заболеваний, будь то инфекции или язвы.
17. Гипертрихоз.
Гипертрихоз также называют «синдромом оборотня» или «синдромом Абрамса».Он проявляется только у одного человека из миллиарда, и только 50 случаев со
времён Средневековья были задокументированы. Люди, страдающие
гипертрихозом, отличаются чрезмерным количеством волос на лице, ушах и
плечах. Это происходит из-за нарушения связей между эпидермисом и дермой во
время формирования у трёхмесячного плода волосяных фолликул. Как правило,
сигналы от образующейся дермы «сообщают» фолликулам их форму. Фолликулы
тоже, в свою очередь, сигнализируют кожным слоям, что в этой области одна
фолликула уже есть, и это приводит к тому, что на теле волоски растут на
приблизительно одинаковом расстоянии друг от друга. В случае с гипертрихозом
эти связи нарушены, что приводит к образованию слишком плотного волосяного
покрова на тех участках тела, где его быть не должно.
18. Конгенитальная миотония.
Если вы когда-нибудь слышали о козьем обмороке, то примерно знаете, каквыглядит конгенитальная миотония — из-за мышечных спазмов человек на
некоторое время будто замирает. Причиной возникновения конгенитальной
(врождённой) миотонии является генетическое отклонение: вследствие
мутации нарушается работа хлорных каналов скелетных мышц. Мышечная
ткань оказывается «сбитой с толку», возникают произвольные сокращения и
расслабления, причём патология может затрагивать мускулатуру ног, рук,
челюстей и диафрагмы.
19. Фибродисплазия оссифицирующая прогрессирующая (ФОП).
Редкое генетическое заболевание, при котором организм начинаетформировать новые кости — оссификаты — в неположенных местах:
внутри мышц, связок, сухожилий и других соединительных тканей. К
их образованию может привести любая травма: ушиб, порез, перелом,
внутримышечная инъекция или операция. Из–за этого удалять
оссификаты нельзя: после хирургического вмешательства кость может
только сильнее разрастись. Физиологически оссификаты не
отличаются от обыкновенных костей и могут выдерживать
значительные нагрузки, вот только находятся не там, где надо.
ФОП возникает из–за мутации в гене ACVR1/ALK2, кодирующем
рецептор костного морфогенетического белка. Она передается
человеку по наследству от одного из родителей, если он тоже болен.
Быть носителем этого заболевания нельзя: пациент либо болен, либо
нет. Пока ФОП относится к числу неизлечимых болезней, однако
сейчас проводится вторая серия испытаний препарата под названием
паловаротен, который позволяет заблокировать ген, ответственный за
патологию.
20. Пигментная ксеродерма
Это наследственное заболевание кожи проявляется в повышеннойчувствительности человека к ультрафиолетовым лучам. Возникает оно из-за
мутации белков, ответственных за исправление повреждений ДНК,
появляющихся при воздействии ультрафиолетового излучения. Первые
симптомы обычно проявляются в раннем детстве (до 3-х лет): когда ребёнок
находится на солнце, у него возникают серьёзные ожоги уже после
нескольких минут воздействия солнечных лучей. Также заболевание
характеризуется появлением веснушек, сухостью кожи и неравномерным
изменением цвета кожного покрова. Согласно статистике, люди с
пигментной ксеродермой более других подвержены риску развития
онкологических заболеваний: при отсутствии надлежащих
профилактических мер, примерно у половины детей, страдающих
ксеродермой, к десяти годам развиваются те или иные раковые
заболевания. Существует восемь видов этого недуга различной тяжести и
симптомов. По данным европейских и американских медиков, болезнь
встречается примерно у четырёх человек из миллиона.
21. Географический язык.
Любопытное название для болезни, не так ли? Впрочем,существует и научный термин для обозначения этой «болячки» —
десквамативный глоссит. Географический язык проявляется
примерно у 2,58% людей, причём чаще всего заболевание имеет
хронические свойства и обостряется после еды, во время стресса
или гормональных стрессов. Симптомы проявляются в
возникновении на языке обесцвеченных гладких пятен,
напоминающих острова, потому заболевание и получило столь
необыкновенное прозвище, причём со временем некоторые
«острова» меняют свою форму и расположение, в зависимости от
того, какие из вкусовых сосочков, расположенных на языке,
заживают, а какие, наоборот, раздражаются.
Географический язык практически безвреден, если не брать в
расчёт повышенную чувствительность к острой пище или
некоторый дискомфорт, который он может причинять. Медицине
неизвестны причины возникновения этой болезни, но есть данные
о генетической предрасположенности к её развитию.
22. Синдром «Серебряного человека»
Научное название этого заболевания — аргироз. Симптомы такой редкойпатологии возникают из-за переизбытка в организме серебра и проявляются
синим цветом кожных покровов. Так, житель Казани, Валерий Вершинин
лечил обычный насморк каплями, в состав которых входило серебро. Дерма,
волосяные луковицы, потовые железы и капилляры кожи накопили в себе
гранулы серебра, в результате чего его кожа стала серебристо-синего цвета,
а волосы побелели. Также самые редкие заболевания кожи, связанные с
переизбытком в организме серебра, могут проявляться у людей,
работающих в сфере добычи или обработки серебра.