





















")

























 и стрипы (В)")















 Семиотика - раздел медицинской генетики, изучающий симптомы наследственных болезней,")



")











 Биология
БиологияПохожие презентации:








")

Введение в медицинскую генетику. Методы медицинской генетики. Семиотика наследственных болезней. Лекция 1
1. ЛЕКЦИЯ1. Введение в медицинскую генетику. Методы медицинской генетики. Семиотика наследственных болезней. Наследственные
2. ВВЕДЕНИЕ В МЕДИЦИНСКУЮ ГЕНЕТИКУ
Генетика - раздел биологии, наука о генах,наследственности
и
изменчивости. Это «теоретический фундамент
современной медицины» (Бочков Николай
Павлович).
Медицинская генетика (МГ) – область
медицины, наука о роли наследственности и
изменчивости
в
популяциях
людей,
закономерностях развития нормальных и
патологических признаков.
Клиническая генетика - прикладная МГ,
направленная на
применение достижений
генетики и МГ для решения клинических
проблем пациентов и их семей.
1931-2011
3. Этапы развития медицинской генетики
1. Доменделевский периодГиппократ – роль наследственности в
происхождении болезней.
Пьер Луи де Мопертюи (французский
естествоиспытатель и математик), в 1745
году издал книгу «Научная Венера, или
Рассуждения о начале людей и животных,
где описал, что новый организм наследует
признаки одного из родителей, в качестве
примера проанализировал генеалогию
семьи с полидактилией. Впервые
использовал термин «доминирование»,
например, в отношении темного цвета
кожи и альбинизма.
460 – 370 гг. до н.э.
1698 – 1759 гг.
4.
1810 –1882
гг.
Теодор Шванн – 1839 г. – клеточная
теория структуры живого.
Чарльз Дарвин в 1859г. –
«Происхождение видов».
Василий Макарович Флоринский и,
1809 –
1882
гг.
1834 –
1899гг.
независимо, Френсис Га́льтон,
в 1865 г. – концепции о
патологической наследственности и
вырождении человеческого рода.
1822 –
1911гг.
5.
а. Первый закона Менделя – законаединообразия гибридов первого
поколения. Тот признак, который
проявлялся у гибридов первого
поколения, был назван Менделем
доминантным, а не проявляющийся
признак – рецессивным.
Доминирование не всегда является
абсолютным.
б. Второй закон Менделя – закон
расщепления признаков. Во втором
поколении гибридов появлялись
растения, как с доминантным, так и
с рецессивным признаком, в
среднем, в соотношении 3:1.
в. Закон независимого
комбинирования признаков.
6. Переоктрытие законов Менделя 1900 г.
Гуго де Фриз(Голландия)
Карл Эрих Корренс
(Германия)
Эрих фон Чермак
(Австрия)
1848 –
1935гг.
1848 –
1935гг.
1871 –
1962гг.
7.
Выделенный «нуклеин» обладал кислотными свойствами и неподвергался разложению протеолитическими ферментами.
8.
Альбрехт Коссель (немецкий биохимик) показал,что вещество, названное «нуклеином», состоит из
белкового и небелкового компонента. Выделил и
описал небелковый компонент, который был назван
«нуклеиновая кислота».
С 1885 по 1901 годы Коссель выедил и описал 5
органических соединений в составе нуклеиновых
кислот:
Аденин,
Цитозин,
Гуанин,
Тимин,
Урацил.
В 1910 году был удостоен Нобелевской премией по
физиологии и медицине за исследования клеточной
биологии, химического состава клеточного ядра и
выделение и описание нуклеиновых кислот.
А. Коссель
(1853-1927)
9.
Фридих Энгельс – в работе «АнтиДюринг» (1878 г):Жизнь – есть способ существования
белковых тел, существенным
моментом которого является
постоянный обмен веществ с
окружающей их внешней природой,
причём с прекращением этого обмена
веществ прекращается и жизнь, что
приводит к разложению белка.
Несколько десятилетий длилось
всеобщее представление, что носитель
генетической информации – белки…
Ф. Энгельс (1820 – 1895)
10.
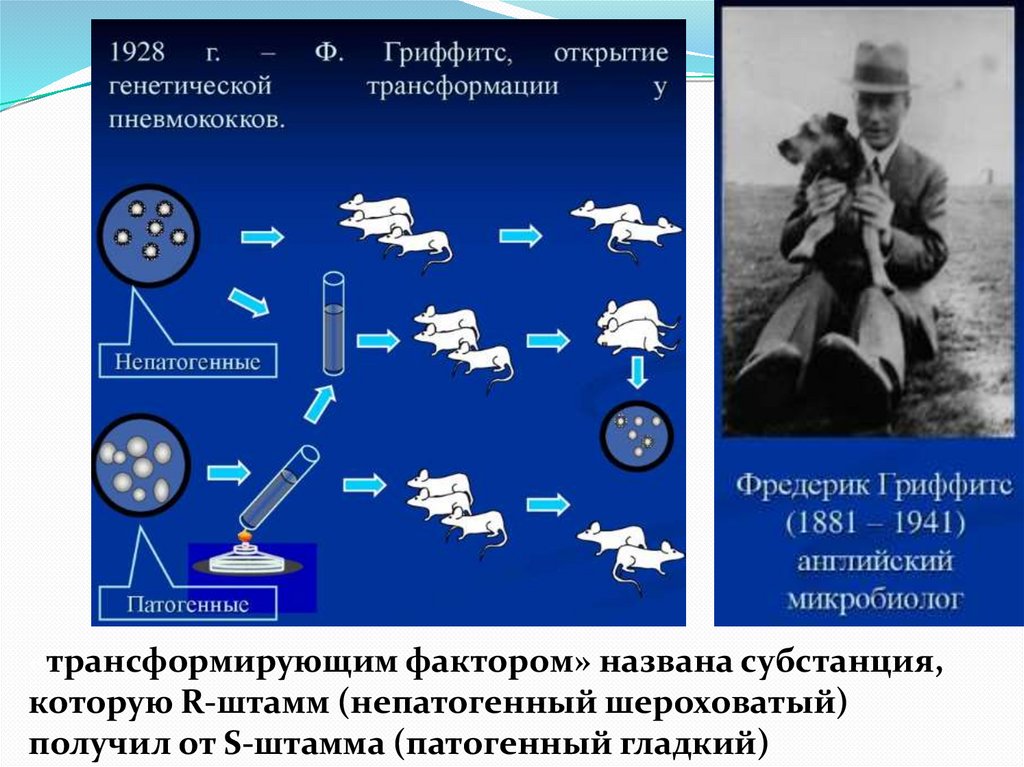
«трансформирующим фактором» названа субстанция,которую R-штамм (непатогенный шероховатый)
получил от S-штамма (патогенный гладкий)
11.
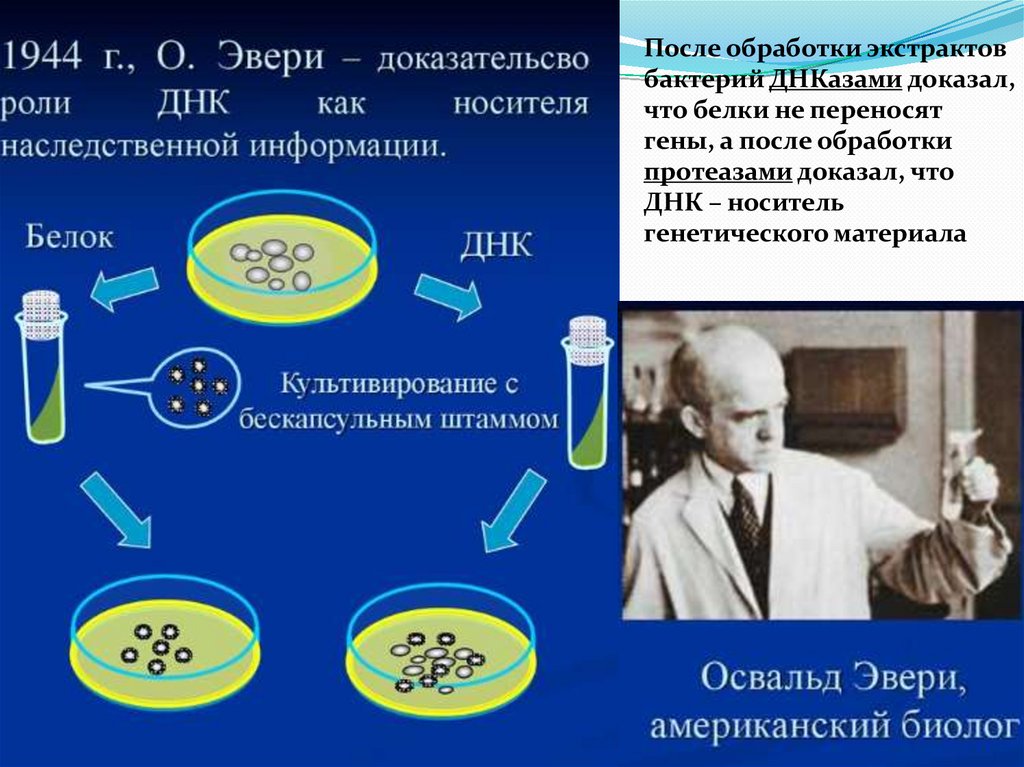
После обработки экстрактовбактерий ДНКазами доказал,
что белки не переносят
гены, а после обработки
протеазами доказал, что
ДНК – носитель
генетического материала
12.
13. К основным задачам медицинской генетики относятся:
анализ причин возникновения наследственных болезней(НБ) , характера их наследования в разных семьях,
распространенность НП в человеческих популяциях,
изучение специфических молекулярных механизмов,
запускающих патологический процесс.
поиск возможных подходов к профилактике и лечению
наследственных болезней.
14.
Ген – участок хромосомы или ДНК, несущий в себе информацию оструктуре одного белка или функциональной РНК.
Генотип - совокупность генов.
Фенотип - совокупность признаков организма.
Варианты наследственных факторов или альтернативные
состояния генов (доминантный, рецессивный) носят названия
аллелей.
Генотип может быть гомозиготным при наличии двух одинаковых
аллелей (АА или аа) или гетерозиготным, если аллели разные
(Аа). Иногда отношения доминантности и рецессивности
отсутствуют и оба аллеля проявляются в фенотипе. Этот тип
взаимоотношения аллелей называется кодоминированием.
Геном - полный состав ДНК клетки, то есть совокупность всех
генов и межгенных участков. Можно считать, что геном - полный
набор инструкций для формирования и функционирования
индивида.
15. Методы генетики человека
Популяционно-статистическийБлизнецовый метод
Клинико-генеалогический
Цитогенетический
Молекулярно-цитогенетический
Молекулярно-генетический
Биохимический
16. Популяционно-статистический метод
Методы генетики популяций широко применяют висследованиях человека для решения следующих
задач:
изучения частоты генов, аллелей и генотипов в
популяции (в т.ч. – генов, вызывающих
наследственные болезни);
анализа закономерности мутационного процесса в
популяциях;
сопоставления роли генотипа и средовых
факторов в формировании фенотипического
полиморфизма человека по многим признакам, а
также в возникновении болезней с наследственной
предрасположенностью.
17. Популяционно-статистический метод
Для идеальной популяции бесконечно большого размера, вкоторой не действует естественный отбор, нет обмена особями с
другими популяциями и мутационного процесса, все
скрещивания случайны и нет дрейфа генов, действует закон
Харди-Вайнберга:
p+q=1
p – частота (доля) доминантного аллеля,
q – частота (доля) рецессивного аллеля.
p2+2pq+q2=1
р2 – доля гомозигот по доминантному аллелю,
2pq – доля гетерозигот,
q2 –доля гомозигот по рецессивному аллелю.
Благодаря закону Харди-Вайнберга можно оценить
популяционный риск НБ, т.к. каждая популяция обладает
определенными частотами неблагоприятных аллелей. Позволяет
предсказать риск рождения больного ребенка и рассчитать
структуру аллелефонда по ЧВ болезни.
18. Популяционно-статистический метод
В основе метода лежит закон Харди — Вайнберга.Этот закон оказывается вполне пригодным для
анализа генетических процессов в крупных
популяциях (большими считаются популяции,
насчитывающие свыше 4,5 тыс. людей) где идет
относительно свободное скрещивание.
Статистический анализ распространения
отдельных наследственных признаков (генов) в
популяциях людей в разных странах позволяет
определить адаптивную ценность конкретных
генотипов.
19. Близнецовый метод введен в медицинскую практику Ф. Гальтоном в 1875г.
основан на явлениимногоплодной
беременности у человека
и позволяет определить
соотносительную роль
генотипа и среды в
проявлении признаков.
Различают монозиготных
и дизиготных близнецов.
Частота появления
близнецов у людей
составляет около 1% (1/3
монозиготных, 2/3
дизиготных).
20.
Монозиготные (однояйцовые) близнецы развиваются изодной оплодотворенной яйцеклетки. Монозиготные
близнецы имеют совершенно одинаковый ядерный
генотип, но могут отличаться по фенотипу, что
обусловлено воздействием факторов внешней среды.
Монозиготные близнецы имеют большую степень сходства
по признакам, которые определяются в основном
генотипом. Например, они всегда однополы, у них
одинаковые группы крови по разным системам (ABO, Rh,
MN и др.), одинаковый цвет глаз, однотипные
дерматоглифические узоры на пальцах и ладонях и др. Эти
фенотипические признаки используются в качестве
критериев диагностики зиготности близнецов.
Дизиготные (двуяйцовые, двойни) близнецы развиваются
после оплодотворения сперматозоидами нескольких
одновременно созревших яйцеклеток (полиовуляция).
Такие близнецы имеют разный генотип, и их
фенотипические отличия обусловлены как генотипом, так
и факторами внешней среды.
21.
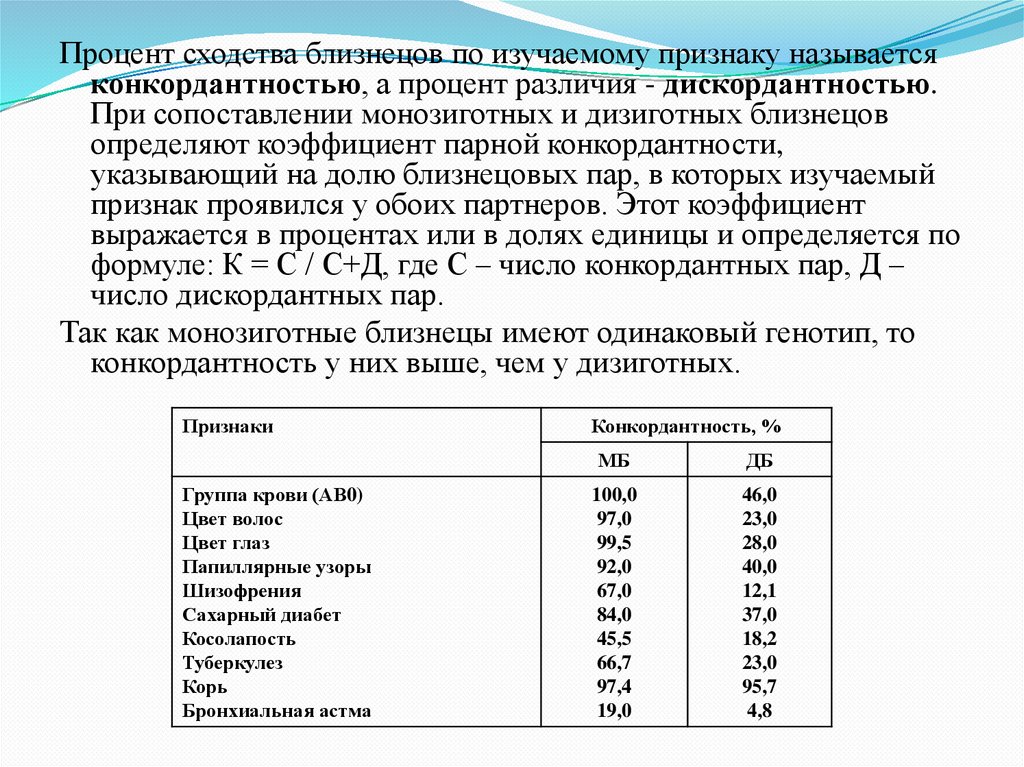
Процент сходства близнецов по изучаемому признаку называетсяконкордантностью, а процент различия - дискордантностью.
При сопоставлении монозиготных и дизиготных близнецов
определяют коэффициент парной конкордантности,
указывающий на долю близнецовых пар, в которых изучаемый
признак проявился у обоих партнеров. Этот коэффициент
выражается в процентах или в долях единицы и определяется по
формуле: К = С / С+Д, где С – число конкордантных пар, Д –
число дискордантных пар.
Так как монозиготные близнецы имеют одинаковый генотип, то
конкордантность у них выше, чем у дизиготных.
Признаки
Группа крови (АВ0)
Цвет волос
Цвет глаз
Папиллярные узоры
Шизофрения
Сахарный диабет
Косолапость
Туберкулез
Корь
Бронхиальная астма
Конкордантность, %
МБ
ДБ
100,0
97,0
99,5
92,0
67,0
84,0
45,5
66,7
97,4
19,0
46,0
23,0
28,0
40,0
12,1
37,0
18,2
23,0
95,7
4,8
22.
Для количественной оценки ролинаследственности и среды в развитии того или
иного признака обычно используется коэффициент
наследуемости, вычисляемый по формуле
Хольцингера:
Н = КМБ-КДБ /(100%-КДБ)
где Н – коэффициент наследуемости, КМБ -
конкордантность монозиготных близнецов, КДБ конкордантность дизиготных близнецов.
Если результат расчетов по формуле Хольцингера
приближается к единице, то основная роль в
развитии признака принадлежит
наследственности, и наоборот, чем ближе результат
к нулю, тем больше роль средовых факторов.
23. БЛИЗНЕЦОВЫЙ МЕТОД
Процентное соотношение общих генов для кровныхродственников:
Монозиготные близнецы (МБ) имеют 100% общих
генов.
I степень родства (дизиготные близнецы (ДБ), сибсы
за исключением МБ, родители-дети) – 50%.
II степень родства (дядя/тетя-племянники,
бабушка/дедушка-внуки) – 25%.
III степень родства (двоюродные братья/сестры) –
12,5%.
IV степень родства (троюродные братья/сестры) –
3,125%.
24. Клинико-генеалогический метод
был предложен в 1883г. Ф. Гальтоном.Основан на построении родословных и прослеживании в
ряду поколений передачи определенного признака. Этот
метод относится к наиболее универсальным методам
генетики человека. Он широко применяется для решения
теоретических и прикладных проблем.
Метод позволяет установить:
1) является ли данный признак наследственным (по
проявлению его у родственников);
2) тип и характер наследования (доминантный или
рецессивный, аутосомный или гоносомный);
3) зиготность лиц родословной (гомо- или гетерозиготы);
4) пенетрантность гена (частота его проявления);
5) вероятность рождения ребенка с наследственной
патологией (генетический риск).
25.
Родословная собирается по одному или несколькимпризнакам.
В зависимости от цели исследования родословная
может быть полной или ограниченной.
Пробанд должен хорошо знать родственников по линии
матери и отца не менее трех поколений и состояние их
здоровья, что бывает крайне редко.
Этапы генеалогического анализа складываются из
1) сбора данных обо всех кровных родственниках
обследуемого (пробанда) с максимально широким охватом
сведений по восходящей и нисходящей линии, а также по
боковым направлениям;
2) графического построения родословной, сопровождаемого
поясняющими описаниями (легенда);
3) анализа родословной и формулированием выводов.
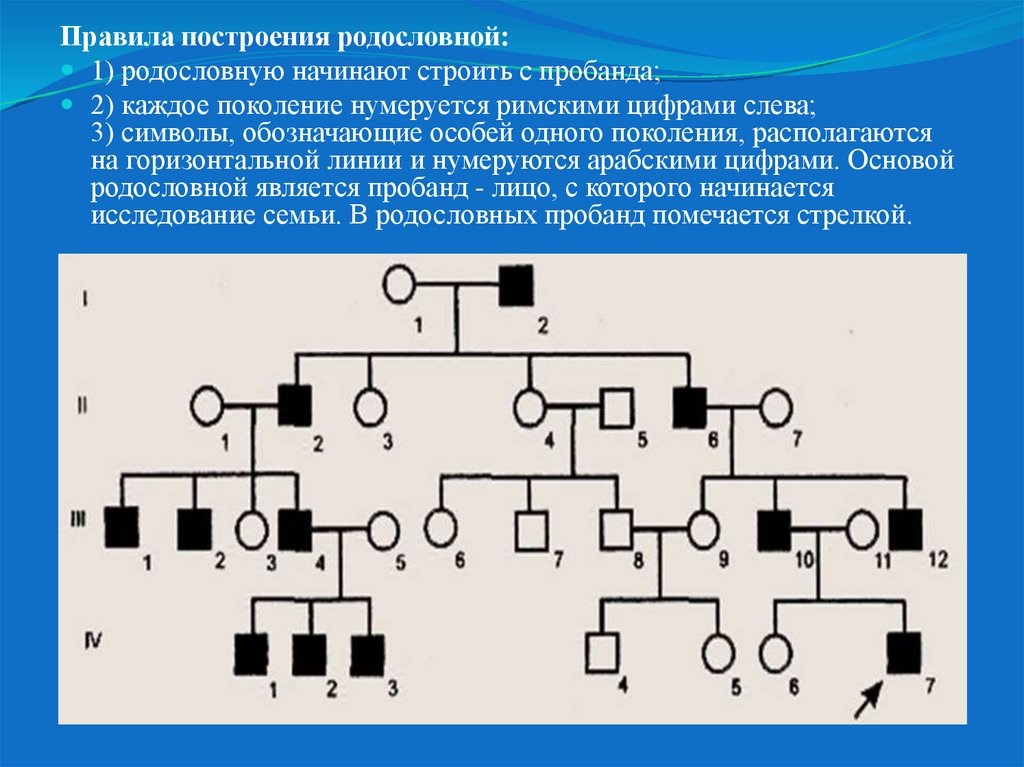
26.
Правила построения родословной:1) родословную начинают строить с пробанда;
2) каждое поколение нумеруется римскими цифрами слева;
3) символы, обозначающие особей одного поколения, располагаются
на горизонтальной линии и нумеруются арабскими цифрами. Основой
родословной является пробанд - лицо, с которого начинается
исследование семьи. В родословных пробанд помечается стрелкой.
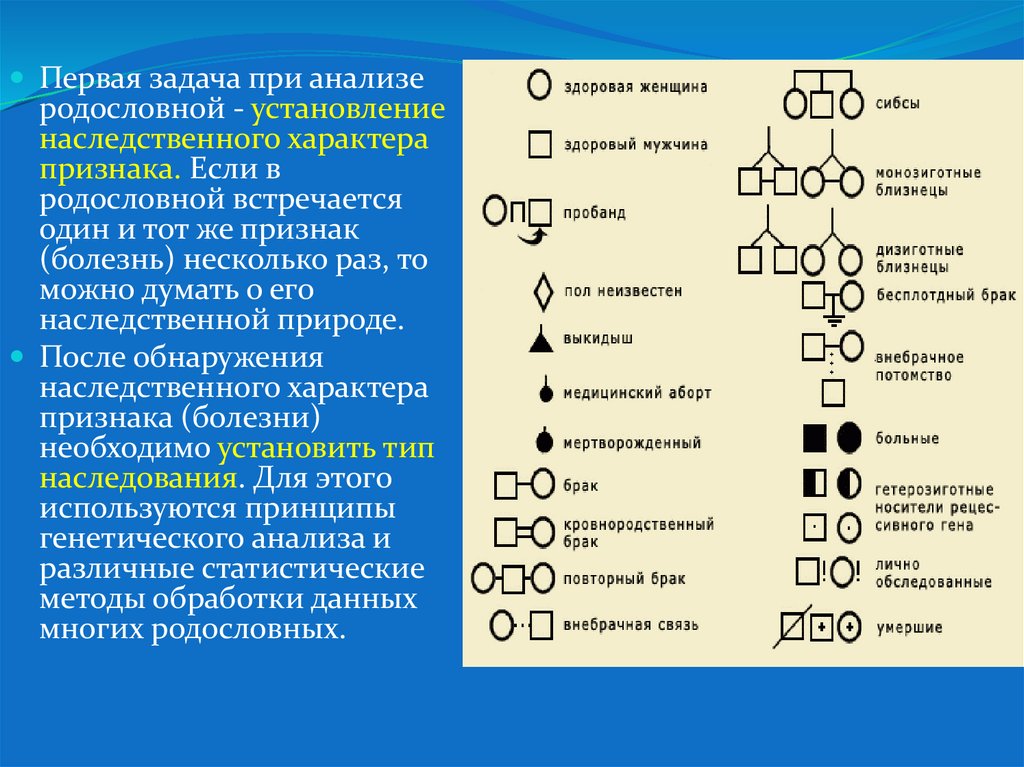
27.
Первая задача при анализеродословной - установление
наследственного характера
признака. Если в
родословной встречается
один и тот же признак
(болезнь) несколько раз, то
можно думать о его
наследственной природе.
После обнаружения
наследственного характера
признака (болезни)
необходимо установить тип
наследования. Для этого
используются принципы
генетического анализа и
различные статистические
методы обработки данных
многих родословных.
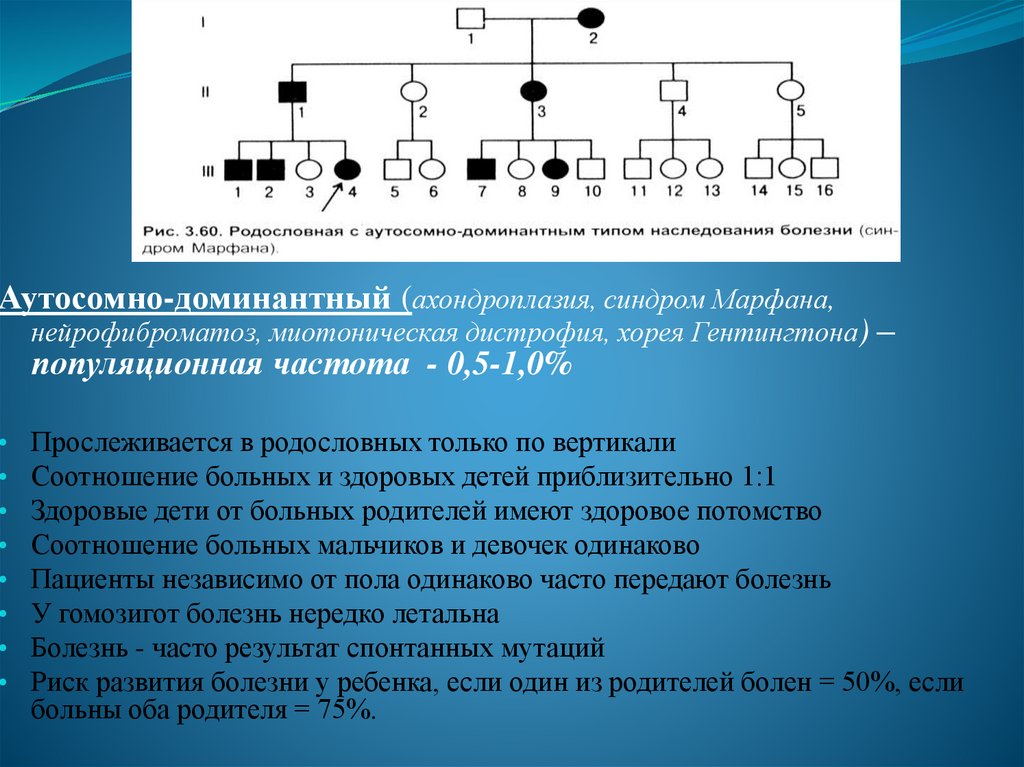
28.
Аутосомно-доминантный (ахондроплазия, синдром Марфана,нейрофиброматоз, миотоническая дистрофия, хорея Гентингтона) –
популяционная частота - 0,5-1,0%
Прослеживается в родословных только по вертикали
Соотношение больных и здоровых детей приблизительно 1:1
Здоровые дети от больных родителей имеют здоровое потомство
Соотношение больных мальчиков и девочек одинаково
Пациенты независимо от пола одинаково часто передают болезнь
У гомозигот болезнь нередко летальна
Болезнь - часто результат спонтанных мутаций
Риск развития болезни у ребенка, если один из родителей болен = 50%, если
больны оба родителя = 75%.
29. Аутосомно-рецессивный тип наследования
Характерен для большинствамоногенных болезней обмена
веществ, популяционная частота
около 0,25%
Прослеживается в родословных по горизонтали (не во всех поколениях)
От здоровых родителей могут родиться больные дети.
У здоровых детей от больных родителей могут родиться больные дети
Соотношение больных мальчиков и девочек одинаково
Пациенты независимо от пола одинаково часто передают болезнь
Болезнь чаще встречается в кровно-родственных браках
Если больны оба родителя, то все дети будут больными
Риск развития болезни у ребенка, если один из родителей болен, а второй – здоров и не является
носителем = 0%, но при этом все дети будут носителями
Если оба супруга носители, то риск развития болезни у ребенка = 25%
Если один из супругов болен, а второй – носитель, то риск развития болезни у ребенка=50%
Если один из супругов носитель, а второй – здоровый гомозиготный, то риск развития болезни у
ребенка = 0%, но вероятность 50%, что он будет носителем.
30. Х-сцепленный рецессивный тип наследования (миодистрофия Дюшенна, гемофилии А и В, синдромы Леша-Нихена, Хантера, болезнь Фабри)
Болеют в основном лица мужского полаБольные мальчики могут родиться от фенотипически здоровых
родителей, если мать является носителем мутантного аллеля
В браке больного мужчины и здоровой матери, не являющейся
носительницей, все дети будут здоровыми, но все дочери становятся
носителями
В браке больного мужчины и здоровой матери-носительницы риск
развития болезни у сыновей = 50%, у дочерей = 50%.
Болезнь передается по горизонтали (не в каждом поколении)
Около 1/3 случаев болезни являются спорадическими (вызваны
неомутациями)
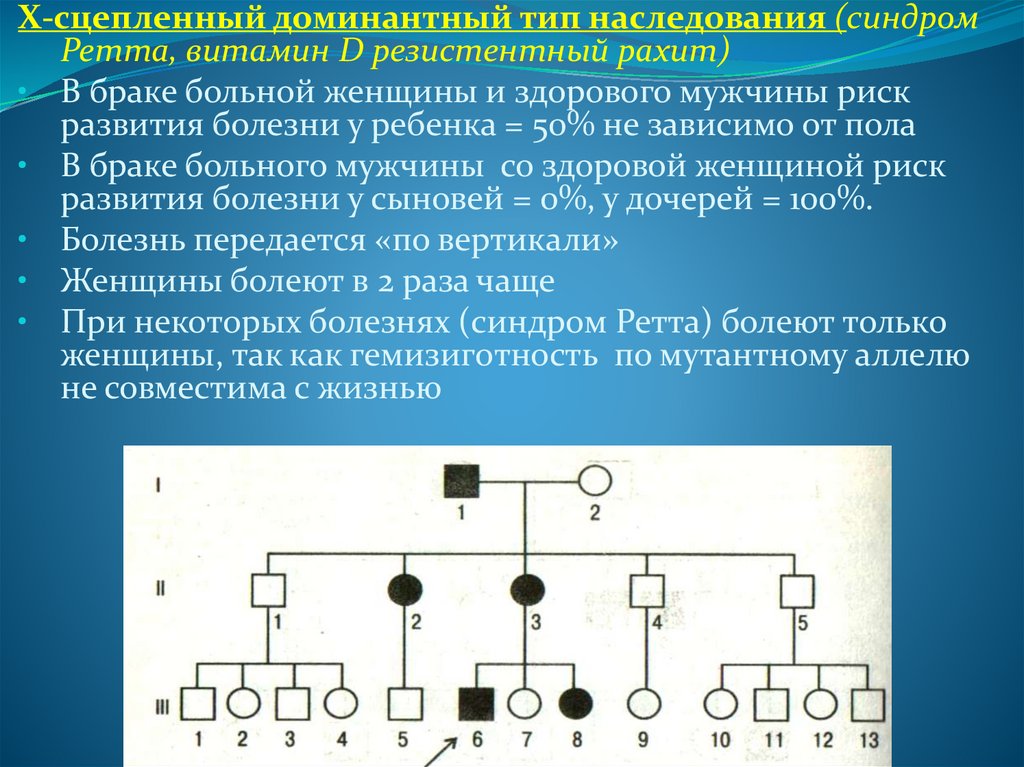
31.
Х-сцепленный доминантный тип наследования (синдромРетта, витамин D резистентный рахит)
• В браке больной женщины и здорового мужчины риск
развития болезни у ребенка = 50% не зависимо от пола
• В браке больного мужчины со здоровой женщиной риск
развития болезни у сыновей = 0%, у дочерей = 100%.
• Болезнь передается «по вертикали»
• Женщины болеют в 2 раза чаще
• При некоторых болезнях (синдром Ретта) болеют только
женщины, так как гемизиготность по мутантному аллелю
не совместима с жизнью
32. Другие типы наследования
Y-сцепленные (нарушения сперматогенеза, росттела, конечностей, зубов, гипертрихоз ушных
раковин)
Митохондриальные болезни (атрофия зрительного
нерва Лебера, синдром Кернса-Сейра, синдромы
MELAS и MERRF)
• Болезнь передается только по материнской линии
• Болеют мальчики и девочки
• Больные мужчины не передают болезнь потомству
• Характерна гетероплазмия (не все митохондрии несут
мутации)
33. Цитогенетический метод
Используют для изучения нормального кариотипачеловека,
а также при диагностике наследственных
заболеваний, связанных с геномными и
хромосомными мутациями.
Кроме того, этот метод применяют при исследовании
мутагенного действия различных химических
веществ, пестицидов, инсектицидов,
лекарственных препаратов и др.
34. Этапы цитогенетического метода
35. Подробное описание цитогенетического метода
1) Для цитогенетического исследования достаточно 0,5 млвенозной крови.
2) Для культивирования во флакон с культуральной средой (с
глутамином) и сывороткой крови (крупного рогатого скота)
добавляют 0,5 мл исследуемой крови + антибиотик (гентамицин)
+ фитогемагглютинин (стимулирует митоз). Перемешивают,
помещают в термостат на 72 часа (3 суток) при 37оС.
3) За 2 часа до фиксации добавляют колхицин. Непосредственно
перед фиксацией клетки обрабатывают 0,56% раствором KCl
при 37оС. Фиксацию производят смесью уксусной кислоты и
метанола (1:3) в течение 60 минут. Далее на предметное стекло
наносят 2 капли осадка, высушивают и окрашивают.
При дифференциальной G-окраске (по Гимзе) хромосомы
предварительно обрабатывают протеазой.
Состав красителя Гимза: Метанол – 750 мл, Глицерин – 256 мл,
Азур-1 – 3,772 г, Эозин – 2,165 г, Метиленовый синий – 1,563 г.
Готовый жидкий краситель перед окрашиванием разводят из
расчета 2 капли на 1 мл дистиллированной воды.
36. Методы дифференциального окрашивания хромосом человека
В70-е
годы
ХХ
века
были
разработаны
методы
дифференциального окрашивания хромосом человека, которые
показали, что каждая пара хромосом имеет свой специфический
характер чередования неокрашенных, светло- и темноокрашенных
дисков
(Парижская
классификация
хромосом
человека).
Разработка специальных методов окраски значительно упростила
распознавание всех хромосом человека, а в совокупности с
генеалогическим методом и методами клеточной и генной
инженерии дала возможность соотносить гены с конкретными
участками хромосом. Комплексное применение этих методов
лежит в основе составления карт хромосом человека.
37. Методы дифференциального окрашивания хромосом человека
Схематическоеизображение
дифференциально
окрашенных
хромосом человека
согласно
международной
классификации
38. Молекулярно-цитогенетический метод
Основан на технологии флюоресцентной гибридизации in situ(FISH).
FISH-метод в зависимости от фазы клеток:
1) На интерфазных клетках.
2) На метафазных клетках - можно определять локализацию
генов в хромосомах и все хромосомные аберрации (этапы
соответствуют цитогенетическому методу + обработка зондом и
гибридизация).
3) На клетках в стадию поздней профазы (вместо колхицина
используют метотрексат).
FISH-метод в зависимости от типа мечения зонда:
А – прямое мечение (непосредственно к зонду присоединяют
флюоресцентный краситель).
В – непрямое мечение (используется чаще, более чувствительная
методика).
39.
НЕПРЯМОЙ МЕТОД:Для исследуемой хромосомы или ее участка готовят однонитевой
участок ДНК (ЗОНД), к которому присоединяют биотин и
дигоксигенин.
На микроскопическом препарате in situ денатурируют
хромосомную ДНК щелочной обработкой, то есть разрывают
связи между двумя цепочками ДНК.
Препарат обрабатывают зондом. Так как последовательность
нуклеотидов зонда и соответствующего участка исследуемой
хромосомы комплементарны, то зонд присоединяется к хромосоме.
В этом участке происходит ренатурация ДНК.
Далее препарат обрабатывают стрептовидином (присоединяется к
биотину) или антидигоксигениновым антителом (к дигоксигенину).
К этим веществам присоединяют флюоресцентные красители
(родамин - красный цвет или флюоресцеин - зеленый цвет).
40. Молекулярно-цитогенетическая диагностика
Fluorescent in situ hybridisation (FISH)Подготовка цитологического препарата
Выбор и подготовка ДНК-пробы
Обработка препарата перед гибридизацией
Проведение гибридизации in situ
Постгибридизационная обработка препарата
Анализ результатов
Заключение по результатам исследования
41. Молекулярно-цитогенетические методы
24-цветная FISH хромосом человека:a - метафазная пластинка;
b - pаскладка хромосом.
(из Рубцов Н. Б., Карамышева Т. В. Вестн. ВОГиС, 2000).
42. FISH с хромосом-специфическими ДНК-пробами на “прямых” препаратах из хориона/плаценты
WCP-7FISH
с хромосом-специфическими
ДНК-пробами
на “прямых” препаратах из
хориона/плаценты
WCP-4
D18Z1
DXZ1
45,X
47,ХY,+mar
47,XX,+i(18p)
46,XY,t(4;15)
43. ЭТАПЫ FISH-МЕТОДА
1. Конструирование зондов (размер не более 1000п.н.).
2. Приготовление препарата метафазных хромосом
(глава 1.4) или интерфазных ядер, фиксация на
предметном стекле.
3. Денатурация ДНК (воздействие формамда при
70оС).
4. Добавление ДНК-зонда и гибридазация в
течение 12 часов.
5. Отмывка, удаление негибридизировавших
зондов.
6. Визуализация люминесцентным микроскопом.
44.
Молекулярно-генетические методы направлены на выявлениеизменений или особенностей структуры исследуемого участка
ДНК вплоть до расшифровки первичной последовательности
оснований.
Все методы начинаются с выделения из образцов (чаще всего
крови) ДНК или РНК, которые в дальнейшем используют для
анализа.
В пробирку с предварительно добавленными цитратом натрия и
ЭДТА набирают 5 мл крови (соотношение консерванта и крови
1:4) и хорошо перемешивают.
Хранение при +40С не более двух недель.
Обязательные данные при сборе материала: ФИО, пол, дата
рождения, место жительства и место рождения, национальность.
45.
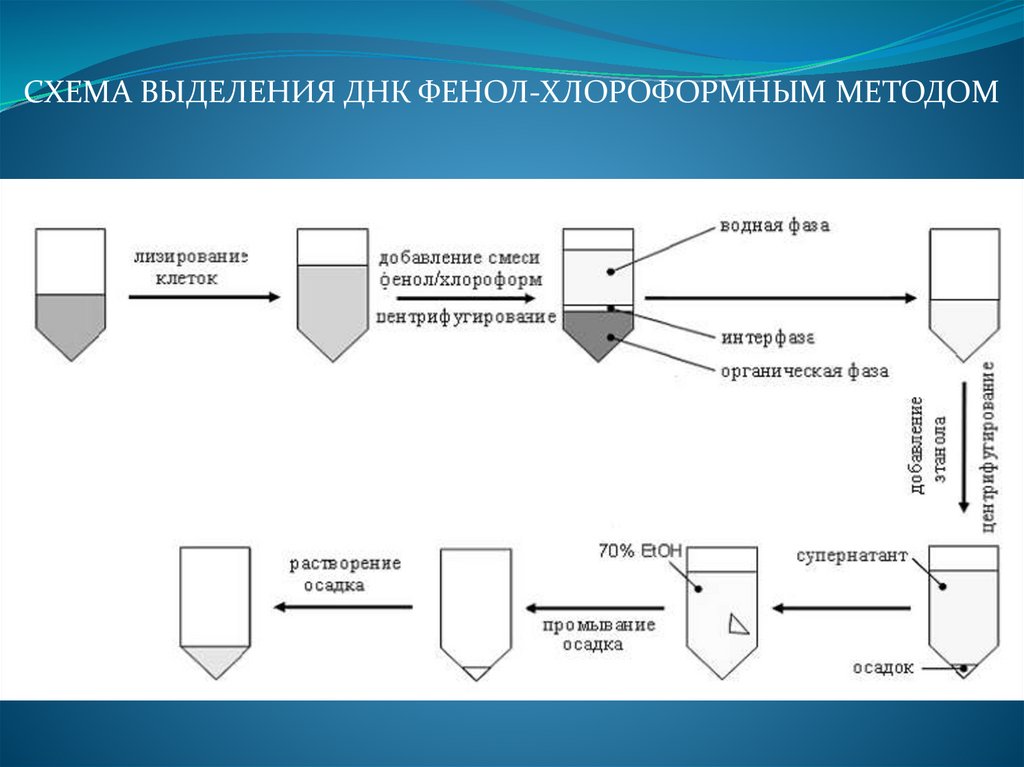
Фенол-хлороформная экстракция ДНК из кровиСуть метода: выделить ДНК из клеток путем лизиса их оболочек, далее
осадить ДНК центрифугированием и очистить фенол-хлороформной
смесью.
Применение фенола и хлороформа при веделении ДНК способствует
созданию двухфазной системы. Фенол отделяет белки от ДНК.
Хлороформ денатурирует белок и липиды и помогает поддерживать
разделение органической и водной фазы, а также делает ДНК менее
растворимой в феноле.
ОСНОВНЫЕ ЭТАПЫ:
ПЕРВЫЙ ДЕНЬ:
1) К 5 мл крови добавляют 30 мл холодного лизирующего буфера
(таблица 6). Точно уравновешивают пробирки, стоящие в центрифуге
друг против друга!
Полученную смесь центрифугируют при +40С со скоростью 4000
оборотов/минуту в течение 20 минут.
2) Супернатант сливают, к осадку добавляют 20 мл холодного
лизирующего буфера, перемешивают и центрифугируют при 4000
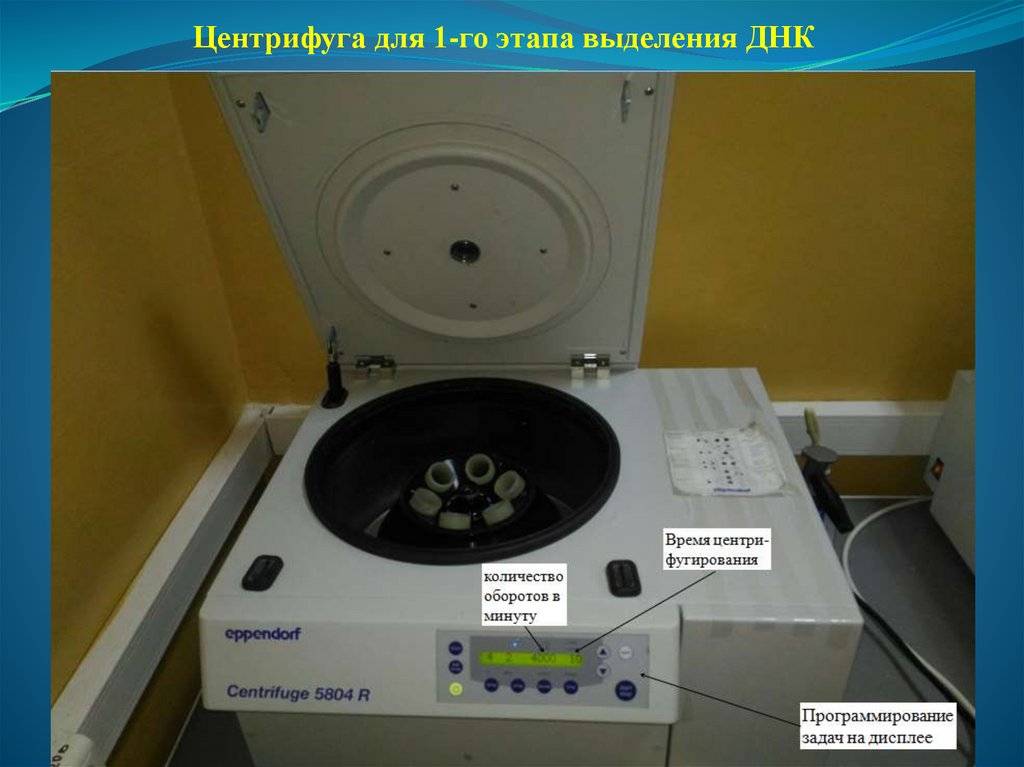
об/мин, при +40С в течение 10 минут.
46.
Центрифуга для 1-го этапа выделения ДНК47.
3) Надосадочную жидкость слить, ресуспензировать осадок в 800 мкл буферадля протеиназы К (Soline ЭДТА).
4) Перенести осадок с Soline EDTA в эппендорф на 2 мл, добавить 80 мкл 10%
SDS (таблица 12) и 20 мкл протеиназы К (10мг/мл), слегка перемешать и
добавить Soline EDTA до 1 мл.
5) Готовую смесь хорошо перемешать на ротаторе (рисунок ), добавить Soline
EDTA до 1 мл и инкубировать в термостате при t=370С 12 часов.
48.
ВТОРОЙ ДЕНЬ:1) В эппендорф с инкубированной смесью добавляют 800 мкл
забуференного фенола (на 50 мл фенола-Трис-НCl 200 мкл
меркаптоэтанола). Перемешивать на ротаторе в течение 10 минут
до однородной массы.
2) Центрифугировать при 5000 об/мин в течение 10 минут.
3) Супернатант перенести в новый эппендорф, добавить 500 мкл
забуференного фенола и 500 мкл хлороформ-изоамилового
спирта (24 мл хлороформа + 1 мл изоамилового спирта).
Перемешивать на шейкере в течение 10 минут до однородной
массы.
После центрифугирования раствор разделится на 2 слоя –
для следующего этапа осторожно берем верхний слой,
стараясь не захватить нижнего, иначе очистку придется
повторять!
49.
4) Центрифугировать при 5000 об/мин в течение 10 минут.5) Супернатант перенести в новый эппендорф, добавить 800 мкл
хлороформ-изоамилового спирта. Перемешивать в течение 5
минут на шейкере до однородной массы.
6) Центрифугировать при 3000 об/мин в течение 3 минут.
7) Супернатант перенести в новый эппендорф, добавить 1000 мкл
охлажденного (при -200С) 96% этанола (2 объема). Как можно
быстрее перемешать, наблюдая преципитацию ДНК. Если
супернатанта, содержащего ДНК, больше 500 мкл, то лучше
разлить по двум эппендорфам и также добавить охлажденный
96% этанол.
8) Центрифугировать при 10000 об/мин в течение 5 минут.
9) Осторожно слить спирт, промыть осадок 2 мл 70% этанола до
удаления солей, тщательно встряхивая пробирку.
10) Полностью слить спирт и высушить осадок при комнатной
температуре о полного испарения спирта.
11) Высушить ДНК при комнатной температуре и растворить в
дистиллированной воде.
50.
СХЕМА ВЫДЕЛЕНИЯ ДНК ФЕНОЛ-ХЛОРОФОРМНЫМ МЕТОДОМ51.
Для определения концентрации и чистоты (наличие примеси белка)раствора ДНК применяют аппарат NanoDrop. Необходимо обязательно
провести оценку качества экстрагированной ДНК путем измерения
оптической плотности раствора ДНК в области белкового и
нуклеинового спектров поглощения. В чистых образцах ДНК
соотношение А(260)/А(280)> 1,8; где А(260) и А(280) – оптическая
плотность раствора при длине волны 260 и 280 нм. В противном случае
процедуру очистки необходимо повторять.
52.
МИКРОПИПЕТКА ДЛЯ РАСКАПЫВАНИЯ РЕАГЕНТОВ53. Методы детекции мутаций
I. Поиск неизвестных мутацийконформационный полиморфизм одноцепочечных
фрагментов -SSCP
электрофорез в денатурирующем гралиентом геле DGGE
химическое расщепление некомлементарных сайтов-CMC
анализ гетеродуплексов -HA
прямое секвенирование -DS
II. Идентификация известных мутаций
Рестрикционный анализ, ПДРФ
Блот - гибридизация по Саузерну (1975)
Полимеразная цепная реакция -ПЦР (1985)
ПЦР-ПДРФ
Диагностика с помощью биочипов ( 2 000)
54.
ЧТО ДАДУТ МЕДИЦИНЕ ИССЛЕДОВАНИЯГЕНОМА В "ПОСТГЕНОМНУЮ" ЭРУ ?
• Идентификацию генов и генных сетей частых
мультифакториальных заболеваний;
• Диагностику заболеваний по экспрессионным профилям тысяч
генов;
• Диагностику наследственной предрасположенности по
тестированию аллельных вариантов "смысловых" SNPs;
• Внедрение предиктивного генетического тестирования семей
с высоким риском частых мультифакториальных болезней;
• Создание индивидуальных, семейных и специализированных
баз ДНК-данных (генетический паспорт беременной,
спортсмена, призывника и пр.);
• Создание новых лекарств направленного действия для
индивидуальной молекулярной терапии.
55. Молекулярно-генетические методы
Дляполучения
достаточного
количества
фрагментов
ДНК
используется полимеразная цепная
реакция (ПЦР) - метод селективной
амплификации отдельных локусов
ДНК посредством имитации in vitro
репликации ДНК.
Для проведения ПЦР необходимо
наличия праймеров (прямых «Forward» и обратных - «Revers»),
Taq-полимеразы,
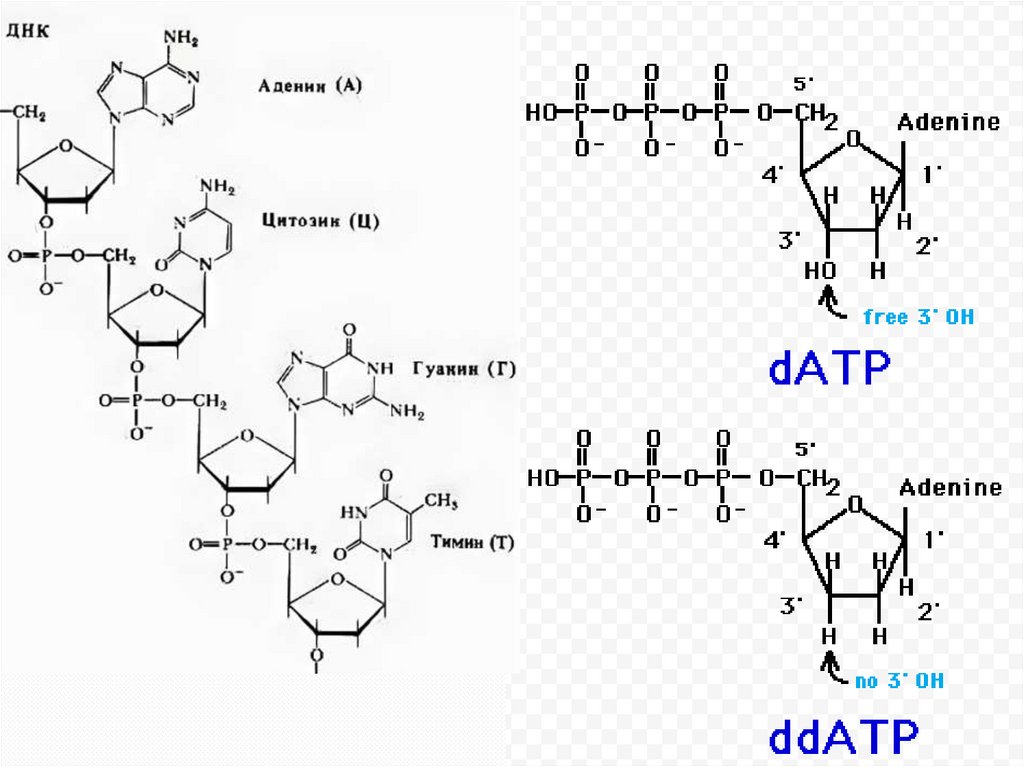
смеси дезоксинуклеотидтрифосфатов
(dATP, dTTP, dGTP, dCTP),
буфер и
определяемый образец ДНК.
56. Метод ПЦР
Методом ПЦР можно синтезировать фрагмент ДНК invitro и получить его как химически чистое вещество.
Для синтеза используются короткие синтетические
отрезки ДНК, называемые праймерами (затравка для
синтеза).
С 3’-конца праймера начинается синтез фрагмента ДНК
по матричной нити, на которую он отжигается
(прилипает при комплементарном взаимодействии
между нуклеотидами праймера и матрицы).
За один цикл достройки ДНК из двух нитей ДНК
получают 4.
В следующем цикле из 4 нитей получится уже 8 и т.д.
Каждый цикл занимает несколько минут.
За 30 циклов ПЦР нужный фрагмент размножится в 1
миллиард раз, что позволяет наблюдать фрагмент
(после окраски).
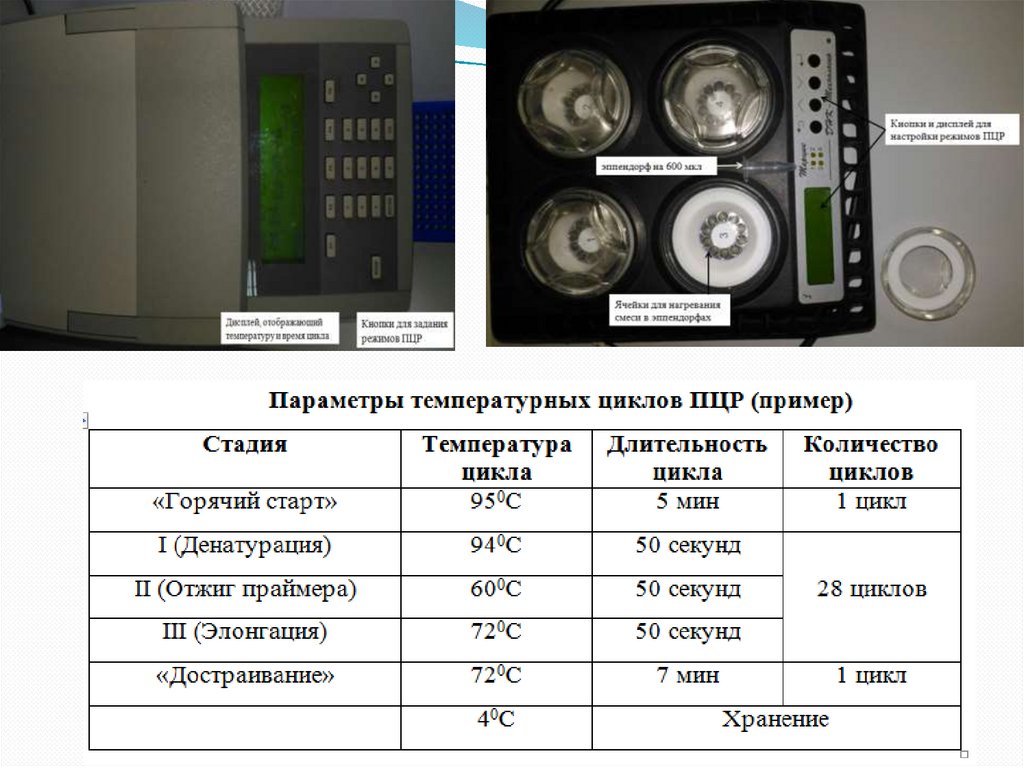
57. Метод ПЦР
58.
59. Ламинарный бокс для приготовления смесей для ПЦР
60. Эппендорфы (А) и стрипы (В)
61. Термоциклер для эппендорф 600 мкл
62. Термоциклер для эппендорф 200 мкл
63. Метод гель-электрофореза
Сканирующая электронная микроскопия 7,5% ПААГна поверхности (слева) и изнутри (справа)
64. Приготовление ПААГ-геля
65. Электрофорез ПЦР-продукта в ПААГ-геле
66. Окраска ПААГ-геля бромистым этидием
Окраска ПААГ-геля нитратом серебра67. ПЦР В РЕАЛЬНОМ ВРЕМЕНИ
Метод основан на одновременнойамплификации и количественном
определении специфической ДНКпоследовательности.
Один из вариантов метода
заключается в добавлении в смесь
интеркалирующих красителей, при
помощи которых затем определяют
количество ДНК в смеси. Флюорофор
приобретает способность к свечению
только после того, как встраивается в
ДНК-дуплекс. Чем больше
синтезировано ПЦР-продукта, тем
более интенсивна флюоресценция,
благодаря чему можно определить
количество продукта амплификации и
исходную концентрацию ДНК. Метод
отличается относительной простотой
дизайна праймеров и дешевизной, но
характеризуется низкой
специфичностью.
68. ПЦР В РЕАЛЬНОМ ВРЕМЕНИ
Второй вариант метода (TaqMan)заключается в том, что флюорофор
начинает светиться при
вытеснении его полимеразой в
процессе амплификации. Метод
отличается высокой
специфичностью и дает
возможность проводить
мультиплексный анализ до пяти
целевых генов в одной пробирке.
Для Real-Time PCR используются
те же реагенты, что и для ПЦР, но
дополнительно добавляется
интеркалирующий краситель
SYBR Green (I вариант) или
флуоресцентно меченые ДНКзонды (II вариант).
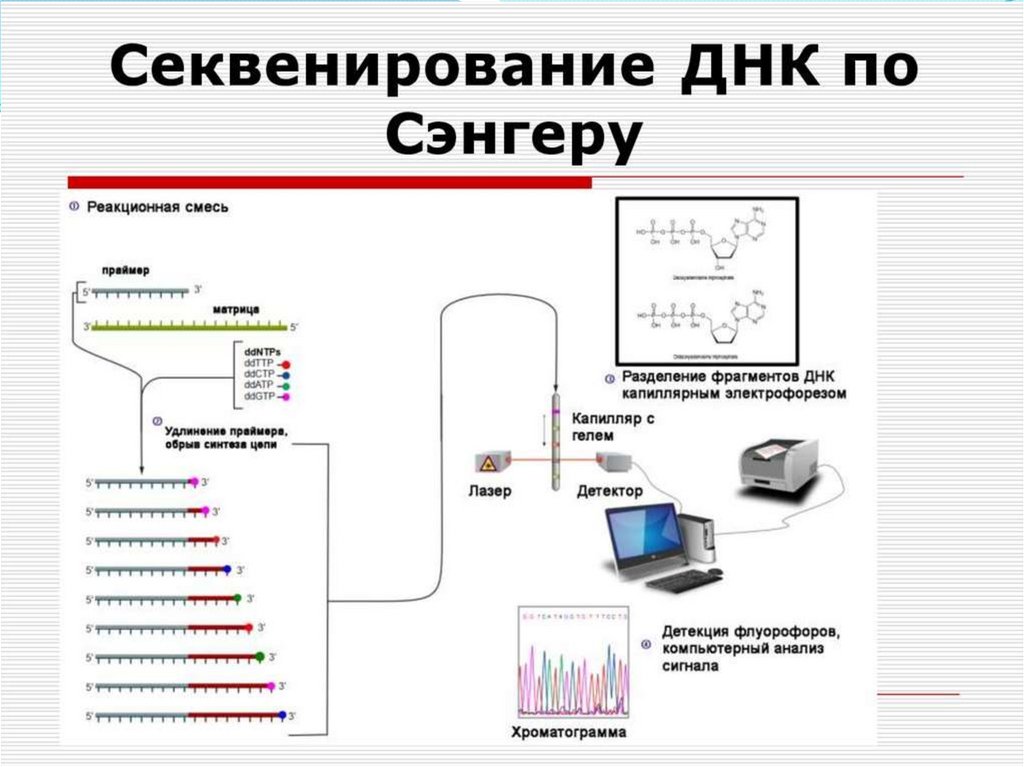
69. СЕКВЕНИРОВАНИЕ
Секвенирование (англ. sequence – последовательность) - методопределения последовательности нуклеотидов исследуемой
ДНК.
К первому поколению методов секвенирования относятся:
1) Химическое расщепление ДНК по одному основанию
Максама-Гильберта (открыт в 1977 году). В связи со
сложностью и дороговизной химический метод МаксамаГильберта в настоящее время практически не используется.
2) Дидезоксисеквенирование по Сэнгеру (открыт в 1975 году).
Данный метод определения последовательностей нуклеотидов
ДНК путем получения комплементарных молекул ДНК,
различающихся по длине на одно основание с использованием
дидезоксинуклеотидов и радиоизотопов или флюорохромных
красителей.
Исторически Сэнгер проводил секвенирование с
использованием радиоизотопов. Суть метода заключалась в
следующем:
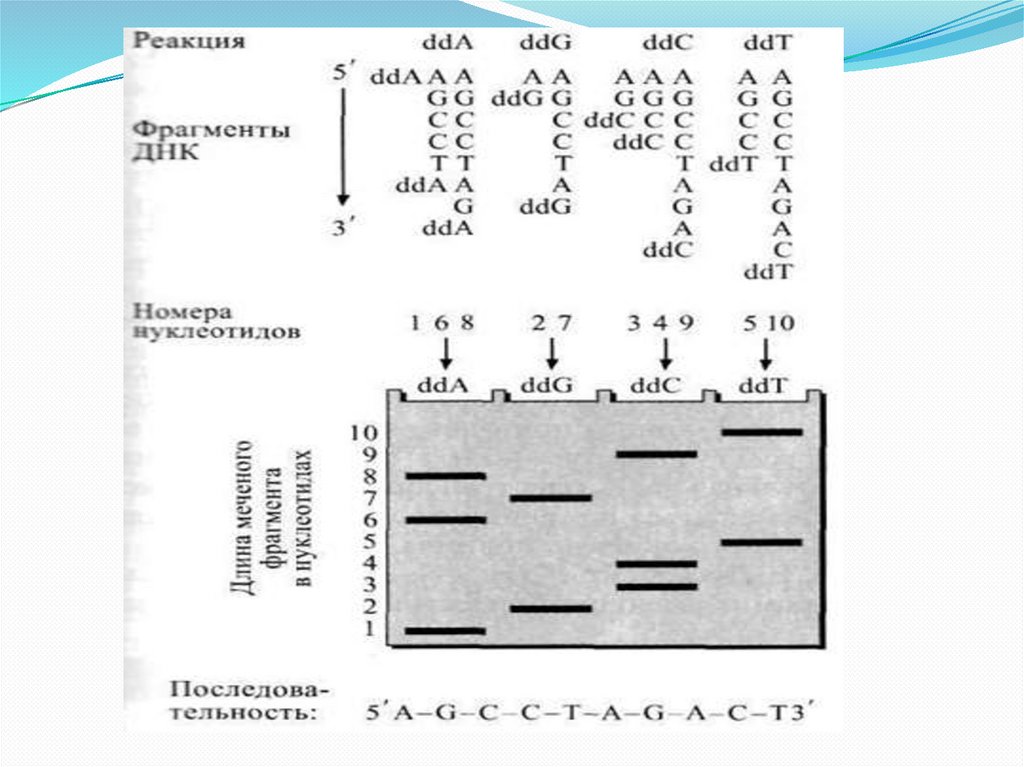
70.
Вначале ДНК денатурировали. Затем добавлялисеквенирующий праймер, проводили отжиг праймера и
инициирование синтеза ДНК, добавляя в реакционную смесь
ДНК-полимеразу и дезоксинуклеотидтрифосфаты – dATP,
dCTP, dGTP, dTTP, один из которых радиоактивен. Синтез
вели в четырех параллельных пробирках, в каждую из
которых добавляли один из специфических
дидезоксинуклеотидтрифосфатов, или терминаторов– ddNTP.
При встраивании ddNTP на место соответствующего
нуклеотида синтез ДНК прекращался.
Таким образом, в каждой из пробирок получали набор
различающихся по длине радиоактивно меченых фрагментов
ДНК с одним и тем же специфическим для данной пробирки
дидезокситерминатором на конце молекулы.
После одновременного электрофоретического разделения
этих фрагментов на четырех соседних дорожках и
радиоавтографии размер синтезированных фрагментов может
быть определен, а значит, определена и локализация ddNTP, и
порядок соответствующих им нуклеотидов в исходной
молекуле ДНК (рисунок).
71.
72.
73.
74.
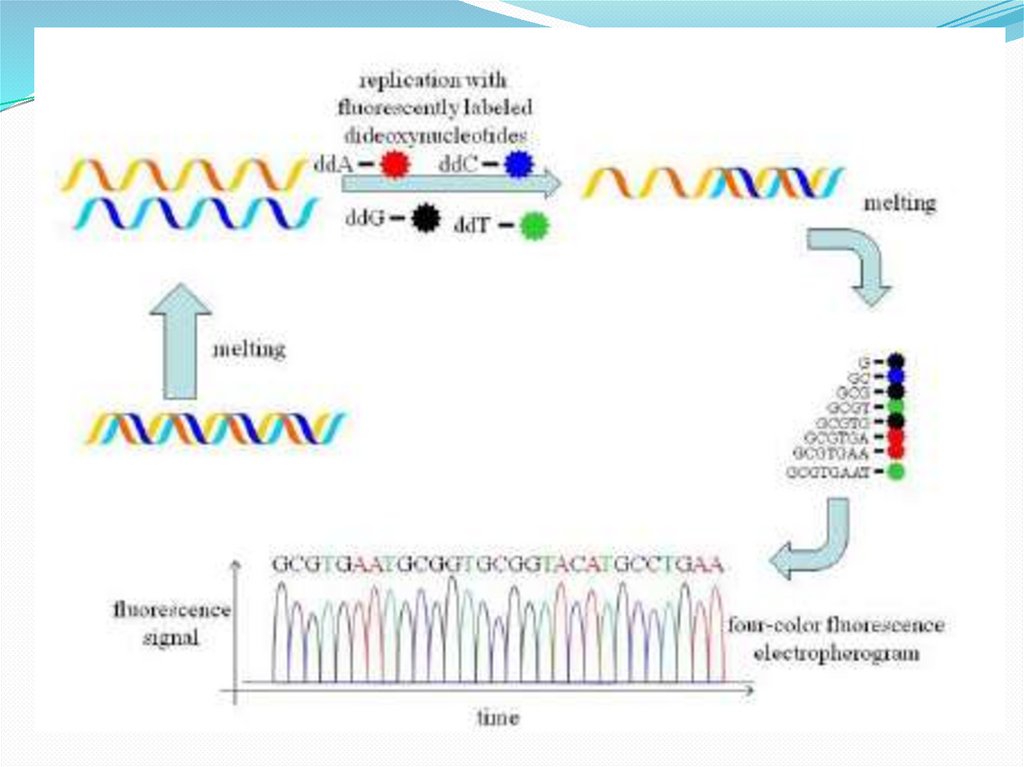
В настоящее время метод секвенирования сиспользованием радионуклидов не применяется.
Используется метод автоматического
секвенирования при помощи присоединенных к
каждому из ddNTP четырех разных флуоресцентных
маркеров (рисунок). Синтез ДНК проводят в одном
сосуде, после чего продукты флюоресцентной
реакции подвергают электрофорезу в тонком
стеклянном капилляре (капиллярный
электрофорез). Проходя через отверстие в
капилляре, флюоресцентные метки возбуждаются
лазерным лучом и начинают испускать свечение,
которое регистрируется цифровой камерой,
превращается в электрический сигнал и выводится в
виде графика.
75.
76.
77. Рестрикционный анализ
Рестриктазы – это ферменты бактерий, специфическиразрезающие ДНК бактериофагов в определенных сайтах. У
самих бактерий ДНК в сайтах рестрикции модифицирована
метилированием. Для обозначения рестриктаз используют
три первые буквы из названия вида бактерии (например,
BglII – из Bacillus globigi, EcoRI – из E. coli), после которых
курсивом следуют определенный буквенный символ,
обозначающий генетическую линию или штамм, и римская
цифра.
Известно три основных типа рестриктаз: I тип – узнают НП
и разрезают ДНК неподалеку от нее, но само место разреза
не строго специфично; II тип – узнают НП и разрезают ДНК
в определенной фиксированной точке внутри этой
последовательности; III тип – узнают НП и разрезают ДНК,
отступив определенное число нуклеотидных пар от ее
конца.
78.
Для выявления специфических фрагментов ДНКиспользуется метод блот-гибридизации по Саузерну.
Эта методика состоит из следующих этапов:
1. После окончания электрофореза гели помещают в
щелочной раствор для денатурации фрагментов ДНК получают одноцепочечные ДНК.
2. Одноцепочечные ДНК вымывают из геля на
нитроцеллюлозный или нейлоновый фильтры
перпендикулярным поверхности геля током буфера.
3. Одноцепочечные фрагменты ДНК фиксируют на
фильтре.
79. Блот-гибридизация по Саузерну
4. Для визуального выявлениянужных фрагментов проводят
гибридизацию исследуемого
образца со специфическим по
нуклеотидной последовательности
меченным радиоактивно или
флюоресцентной меткой
олигонуклеотидным
синтетическим зондом.
5. Радиоактивно меченные участки
выявляют путем экспонирования
фильтра с рентгеновской пленкой
(авторадиография).
6. Флюоресцентные метки
выявляют в люминесцентном
микроскопе.
Этот метод позволяет обнаружить
единственный ген среди десятков тысяч.
(из obi.img.ras.ru/humbio/har)
80. Биохимические методы
Наследственные заболевания, которые обусловлены генными мутациями, изменяющимиструктуру или скорость синтеза белков, обычно сопровождаются нарушением углеводного,
белкового, липидного и других типов обмена веществ. Наследственные дефекты обмена можно
диагностировать посредством определения структуры измененного белка или его количества,
выявления дефектных ферментов или обнаружения промежуточных продуктов обмена веществ
во внеклеточных жидкостях организма (крови, моче, поте и т.д.)
Кроме выявления гомозиготных носителей мутантных генов существуют методы выявления
гетерозиготных носителей некоторых рецессивных генов, что особенно важно при медикогенетическом консультировании.
Так, у фенотипически нормальных гетерозигот по фенилкетонурии после приема фенилаланина
обнаруживается повышенное его содержание в крови.
При гемофилии гетерозиготное носительство мутантного гена может быть установлено с
помощью определения активности фермента, измененного в результате мутации.
81. СЕМИОТИКА НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ (НБ) Семиотика - раздел медицинской генетики, изучающий симптомы наследственных болезней,
Клинические проявления НБ• семейный характер
• хроническое, рецидивирующее течение
• резистентность к лекарственной терапии
• специфические «маркерные» симптомы
• системные поражения органов и тканей
82. Семиотика наследственных болезней
Термины «синдром» и «болезнь» для наследственнойпатологии равнозначны. Для некоторых нозологических
форм применяются оба термина (синдром/болезнь Дауна =
трисомия 21).
Семиотика – методическая основа объективного врачебного
осмотра, направленного на идентификацию специфической
наследственной патологии у пробанда на основании
определения характерных знаков патологического процесса и
динамики клинических проявлений. С этой целью
используются различные предметно-образные знаки для
обозначения признаков НБ.
83.
Характерные «знаки» (симптомы) НБ«лицо эльфа» при синдроме Вильямса (АД НБ, 1:10 000)
84.
85. «Птицеголовая карликовость» (синдром Секкеля – АР НБ )
86.
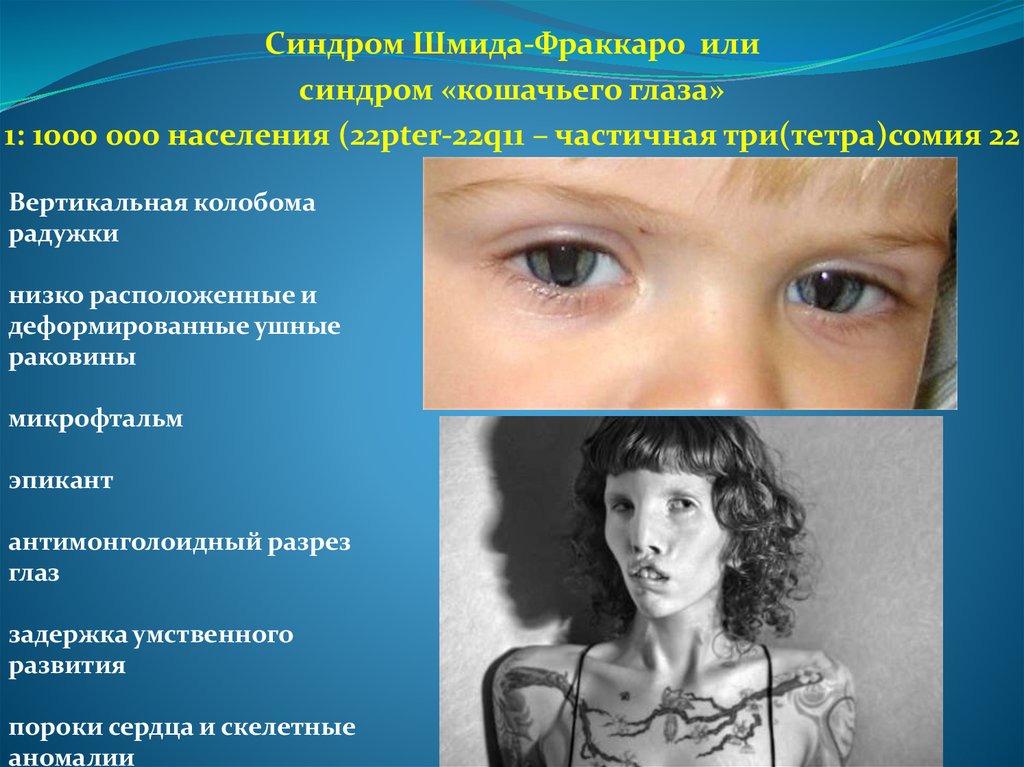
Синдром Шмида-Фраккаро илисиндром «кошачьего глаза»
1: 1000 000 населения (22pter-22q11 – частичная три(тетра)сомия 22
Вертикальная колобома
радужки
низко расположенные и
деформированные ушные
раковины
микрофтальм
эпикант
антимонголоидный разрез
глаз
задержка умственного
развития
пороки сердца и скелетные
аномалии
87. Классификация наследственных болезней
I. Генетическая• Генные НБ (моногенные)
• Мультифакториальные НБ (полигенные или болезни
с наследственной предрасположенностью)
• Хромосомные НБ
• Генетические болезни соматических клеток
• Митохондриальные болезни
• Эпигенетические болезни (болезни импринтинга)
• Болезни экспансии (динамические мутации)
88. Классификация наследственных болезней
II По типу наследования : III По клиническому проявлению:• Аутосомно-доминантные
Классификация по органно• Аутосомно-рецессивные
системному принципу:
• Сцепленные с полом
нервные, мышечные, глазные,
• Х-сцепленные доминантные
болезни опорно-двигательного
• Х-сцепленные рецессивные
аппарата, др.
• Y-сцепленные
•Нетрадиционного типа наследования
IV По патогенезу :
Нарушения обмена веществ (1)
Аномалии морфогенеза
(2)
Комбинированные (1) и (2)
89. Особенности клинического проявления
Клинический полиморфизм (КП)• первичный КП - многообразие проявления
признаков НБ связанных с первичным дефектом,
• вторичный КП - осложнения вследствие
первичного дефекта
• пенетрантность - частота признака среди больных
• экспрессивность - выраженность клинического
признака
Генетический полиморфизм - молекулярная основа
КП
• разные мутации одного гена - разная клиника, разные
заболевания
• мутации разных генов - одно заболевание
90.
Специфические симптомы НБПодвывих или вывих хрусталика (синдром Марфана, синдром Вейля-Марчезани,
гомоцистинурия)
Голубые склеры (несовершенный остеогенез)
Потемнение мочи (алкаптонурия)
Мышиный запах (фенилкетонурия)
Кровоточивость (болезнь Виллебранда, гемофилия)
Грубые черты лица (мукополисахардиоз)
Астеническое телосложение и деформация грудной клетки (синдром Марфана)
Низкий рост, непропорциональные конечности и лицевой череп (ахондроплазия)
91. МНОЖЕСТВЕННОСТЬ ПОРАЖЕНИЙ
Обусловлено плейотропным действием гена (влияние одного генана развитие нескольких признаков).
Любая моногенная НБ всегда проявляется комплексом нарушений
различных органов и систем.
Вторичная плейотропия связана с осложнениями первичных
патологических процессов :
Муковисцедоз (мутация трансмембранного белка ионного транспорта в
клетках >
сгущение слизи бронхов и поджелудочной железы >
вторичные легочные инфекции и нарушения пищеварения)
Талассемия (мутация гена гемоглобина >
усиленное кроветворение >
гемосидероз органов >
утолщение костей черепа и гепатолиенальный синдром
92.
Первичная плейотропияСимптомы НБ обусловлены эффектом действия первичных продуктов
мутантных аллелей гена.
Фенилкетонурия
мутация гена фенилаланин-4-гидроксилазы,
который превращает фенилаланин в тирозин
не синтезируется
тирозин
накопление фенилпировиноградной
кислоты в организме
снижается образование
меланина
нарушение развития нервной системы
гипопигментация кожи,
волос, радужки
слабоумие, судорожные припадки,
тремор, повышенная возбудимость
93.
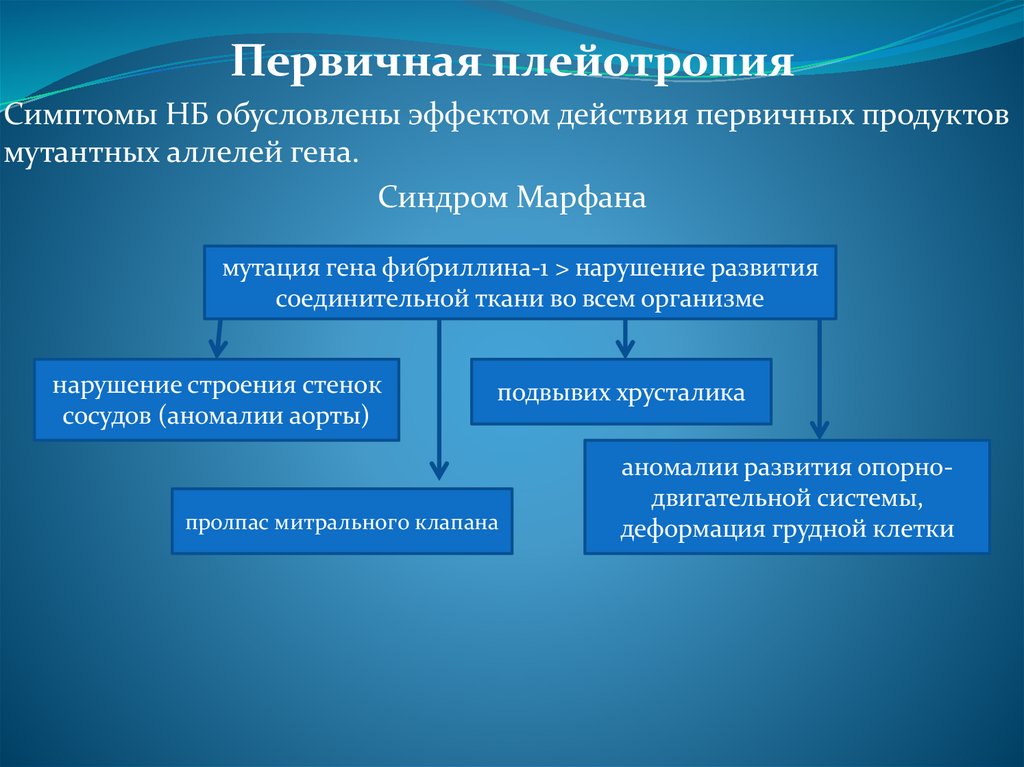
Первичная плейотропияСимптомы НБ обусловлены эффектом действия первичных продуктов
мутантных аллелей гена.
Синдром Марфана
мутация гена фибриллина-1 > нарушение развития
соединительной ткани во всем организме
нарушение строения стенок
сосудов (аномалии аорты)
подвывих хрусталика
пролпас митрального клапана
аномалии развития опорнодвигательной системы,
деформация грудной клетки
94.
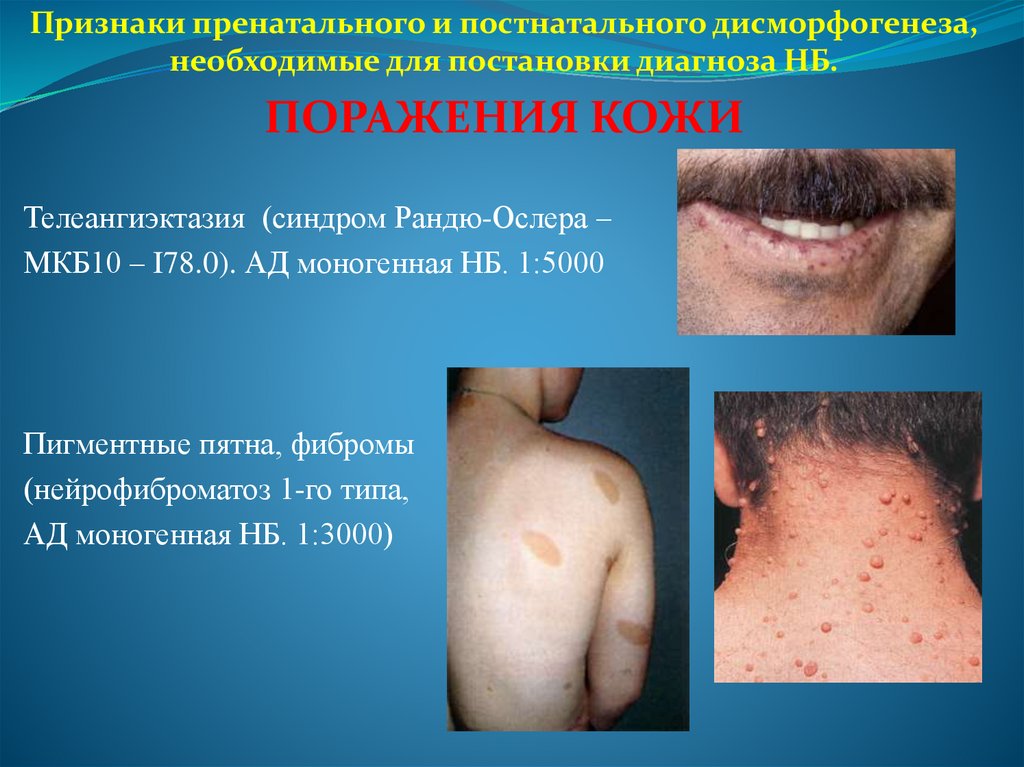
Признаки пренатального и постнатального дисморфогенеза,необходимые для постановки диагноза НБ.
ПОРАЖЕНИЯ КОЖИ
Телеангиэктазия (синдром Рандю-Ослера –
МКБ10 – I78.0). АД моногенная НБ. 1:5000
Пигментные пятна, фибромы
(нейрофиброматоз 1-го типа,
АД моногенная НБ. 1:3000)
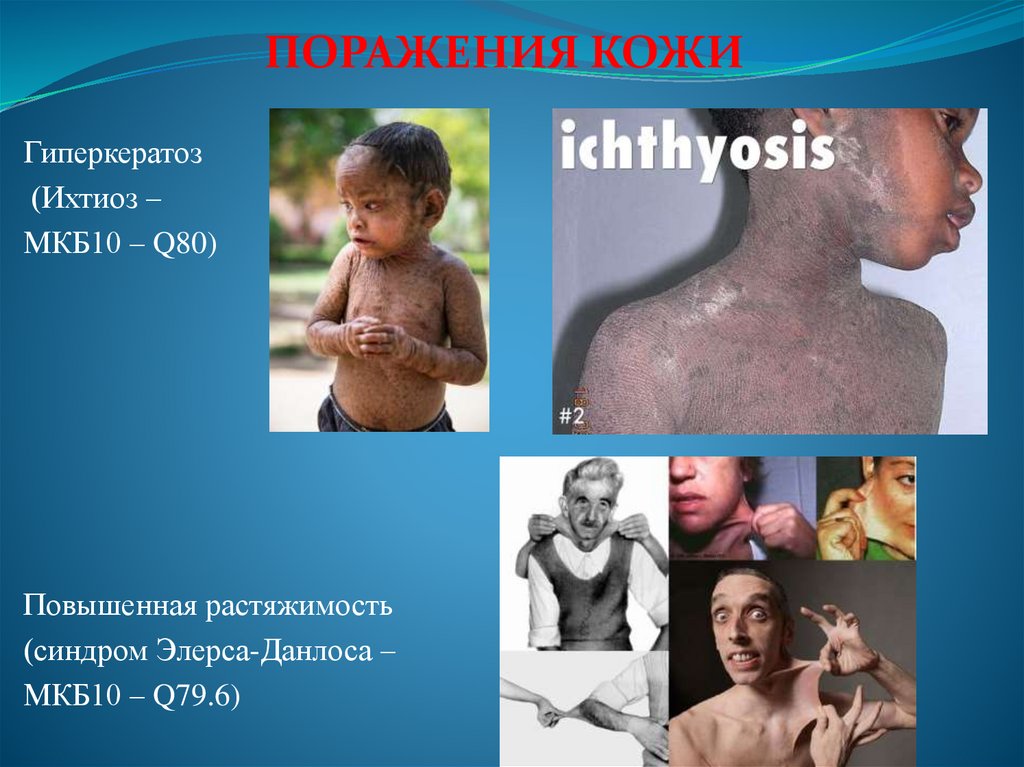
95.
ПОРАЖЕНИЯ КОЖИГиперкератоз
(Ихтиоз –
МКБ10 – Q80)
Повышенная растяжимость
(синдром Элерса-Данлоса –
МКБ10 – Q79.6)
96. ПОРАЖЕНИЯ ЧЕРЕПА
гидроцефалиятрингоцефалия
микроцефалия
акрооцефалия
долихоцефалия
97.
АНОМАЛИИ УШНЫХ РАКОВИНАнотия (отсутствие ушных раковин)
Микротия (недоразвитие ушных раковин)
Макротия (чрезмерный размер ушных раковин)
Деформация , низкое расположение, оттопыревание,
отклонение назад,
завитки со сглаженным упрощенным рисунком,
предушные фистулы,
пердушные папилломы
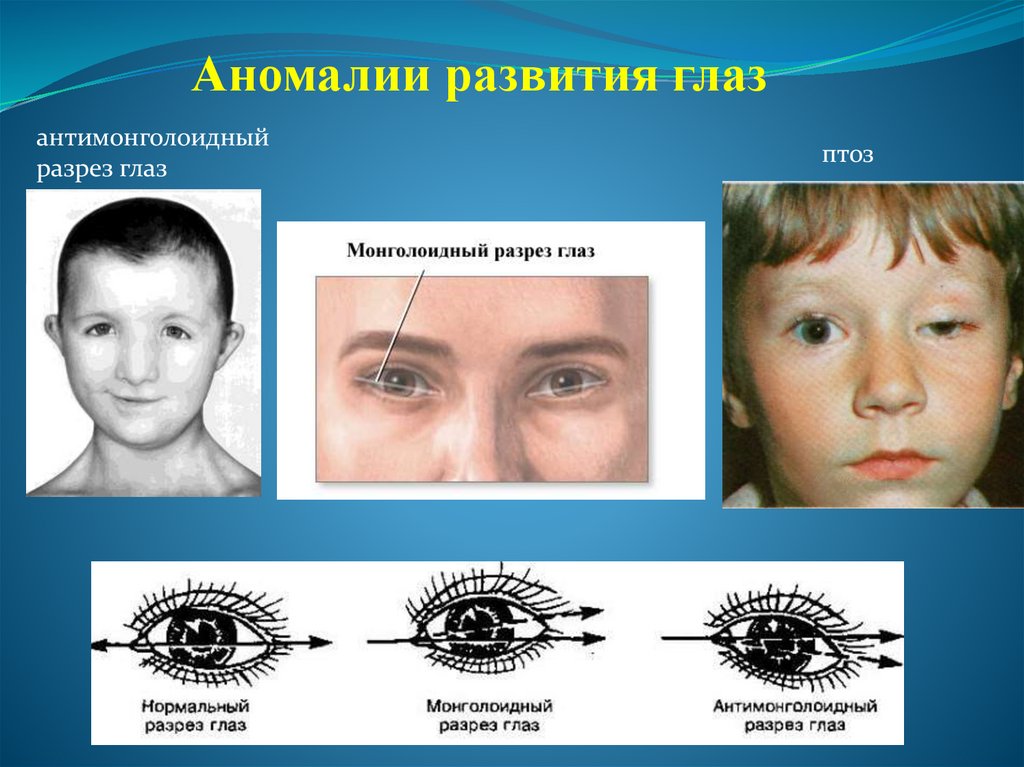
98.
Аномалии развития глазантимонголоидный
разрез глаз
птоз
99.
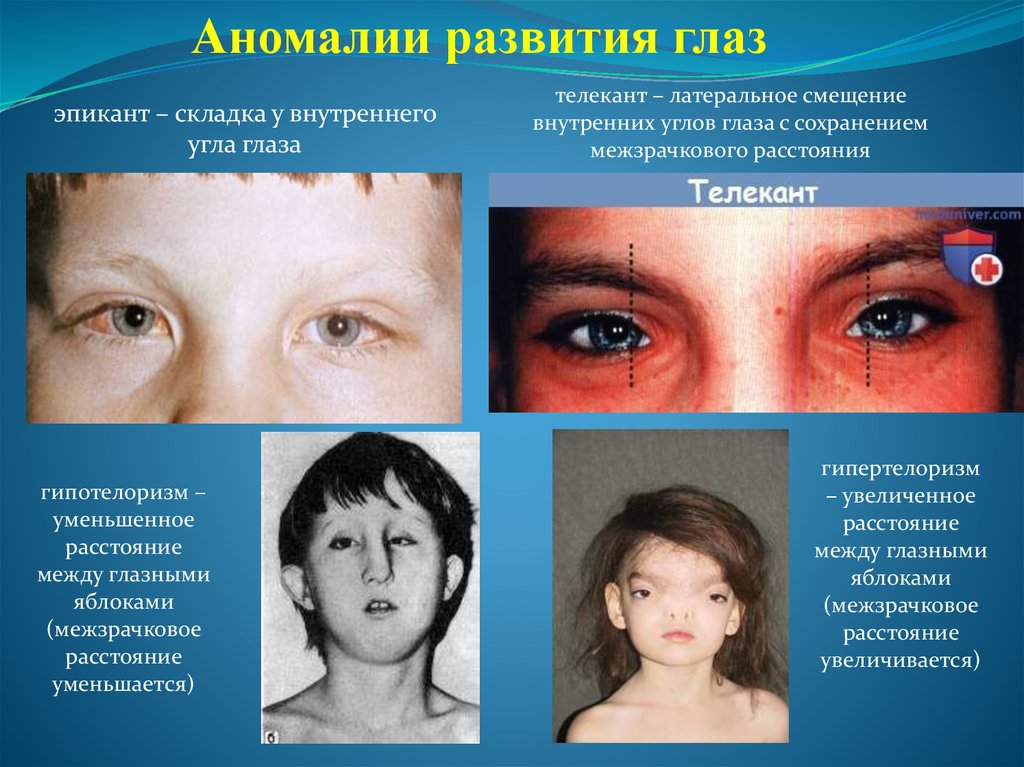
Аномалии развития глазэпикант – складка у внутреннего
угла глаза
гипотелоризм –
уменьшенное
расстояние
между глазными
яблоками
(межзрачковое
расстояние
уменьшается)
телекант – латеральное смещение
внутренних углов глаза с сохранением
межзрачкового расстояния
гипертелоризм
– увеличенное
расстояние
между глазными
яблоками
(межзрачковое
расстояние
увеличивается)
100.
аномалии развития носакороткий
клювовидный
седловидная переносица
широкая плоская
переносица
плоские крылья носа
открытые вперед ноздри
101.
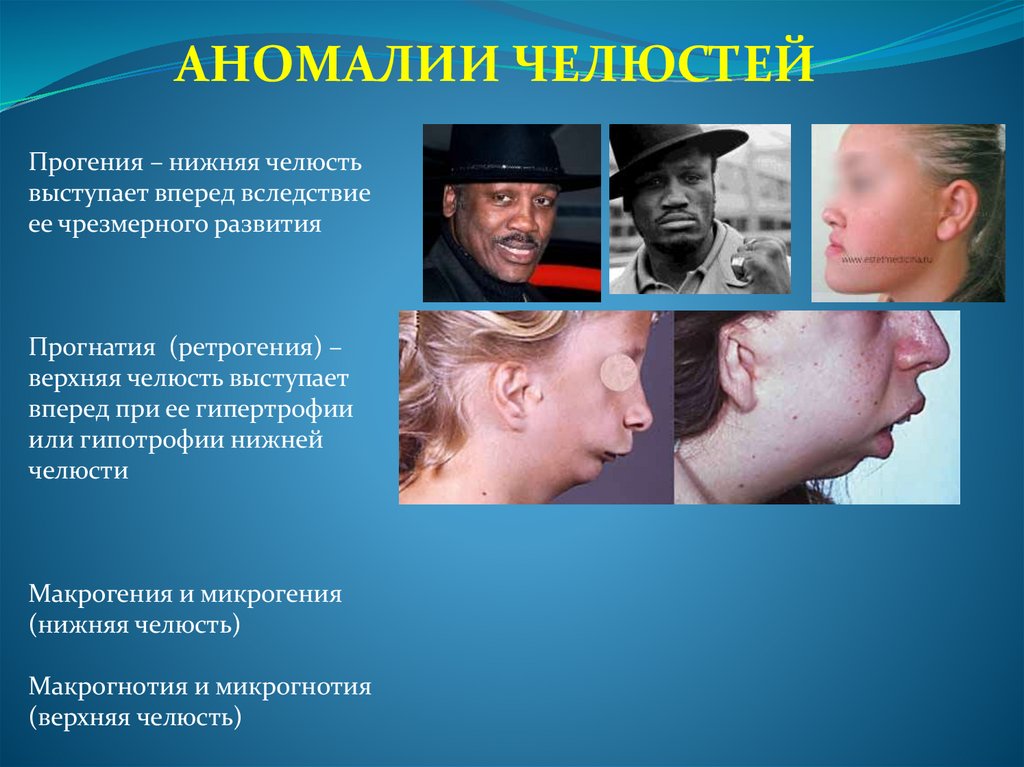
АНОМАЛИИ ЧЕЛЮСТЕЙПрогения – нижняя челюсть
выступает вперед вследствие
ее чрезмерного развития
Прогнатия (ретрогения) –
верхняя челюсть выступает
вперед при ее гипертрофии
или гипотрофии нижней
челюсти
Макрогения и микрогения
(нижняя челюсть)
Макрогнотия и микрогнотия
(верхняя челюсть)
102.
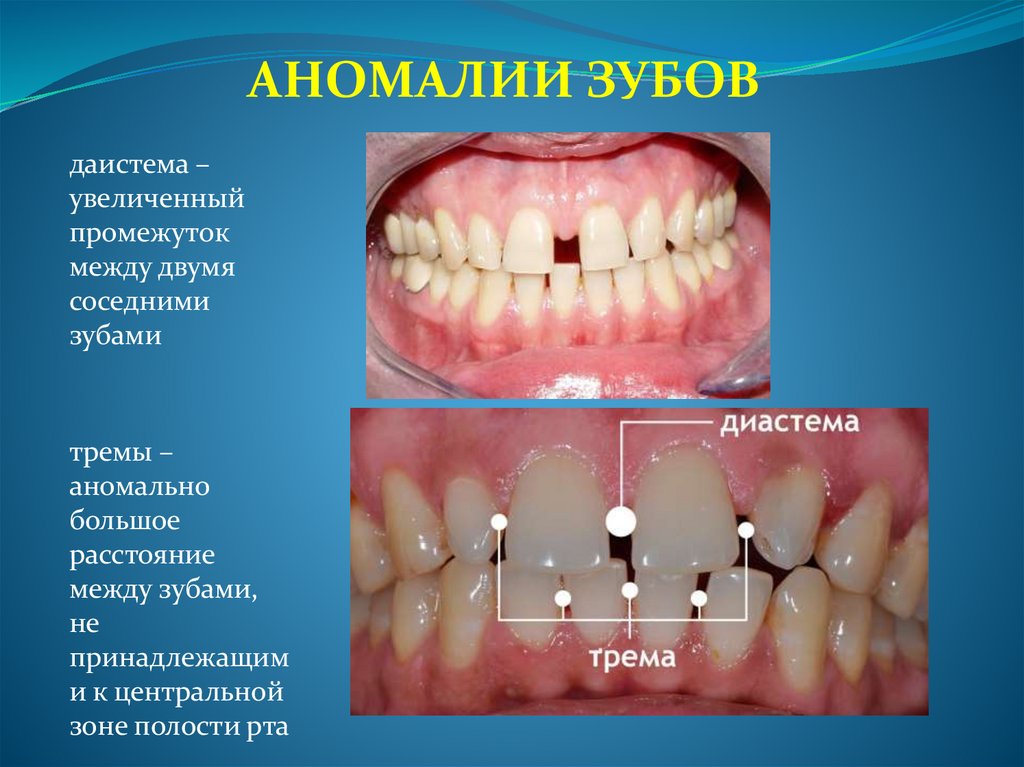
АНОМАЛИИ ЗУБОВдаистема –
увеличенный
промежуток
между двумя
соседними
зубами
тремы –
аномально
большое
расстояние
между зубами,
не
принадлежащим
и к центральной
зоне полости рта
103.
АНОМАЛИИ ШЕИкороткая
длинная
низкая линия роста волос
крыловидные складки
АНОМАЛИИ ГРУДНОЙ КЛЕТКИ
долихостеномелия (dolicho – удлиненный, stenos – узкий,
melos – часть тела)
воронкообразная
килевидная
полителия (добавочные соски)
гипертелоризм сосков
сколиоз
пилонидальная ямка (в области копчика и крестца)
104.
АНОМАЛИИ КОНЕЧНОСТЕЙвальгусная деформация
варусная деформация
удлинение
укорочение
поли-, олиго-, брахи-, арахно-, син-, кампто- (сгибательня
деформация проксимального межфалангового сустава) –дактилия
поперечная ладнная складка
трехфаланговый 1 палец
полая стопа, конская стопа, косолапость, плоскостопие
АНОМАЛИИ МОЧЕПОЛОВОЙ СИСТЕМЫ
крипторхизм (неопущение яичка в мошонку)
шалевидная мошонка
гипоспадия (смещение вниз отверстия мочеиспускательного
канала)