











.")



")





























")





















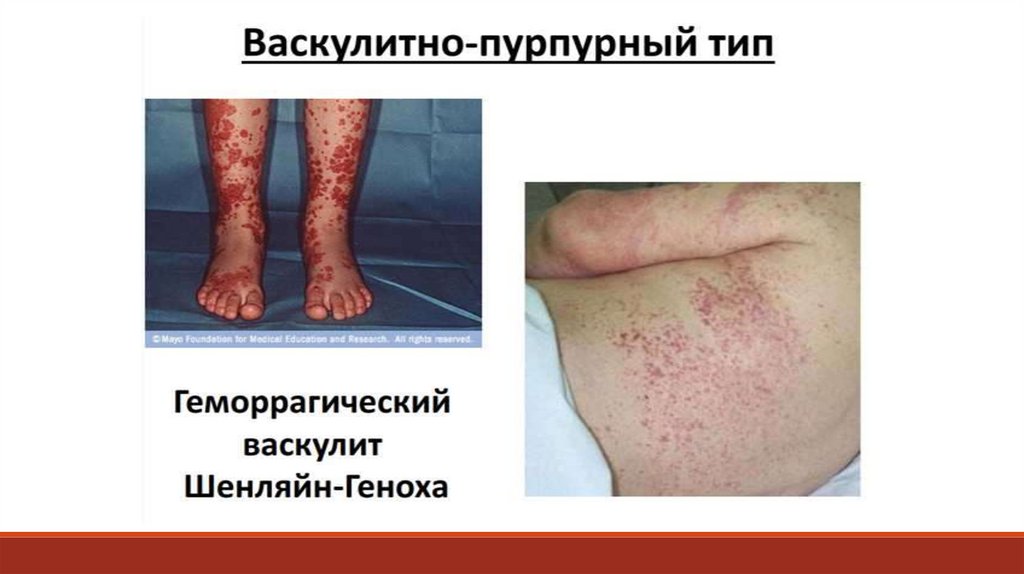
 – системное сосудистое заболевание с IgA-депозитами с преимущественным")

;-")

1. Пальпируемая геморрагическая сыпь.Слегка возвышающаяся над поверхностью")


;- антиагреганты (дипиридамол 300 мг/сут., пентоксифиллин 600 мг/сут.);- СЗП")











































 Медицина
МедицинаПохожие презентации:










Дифференциальная диагностика и ведение больного с геморрагическим и тромбофилическим синдромами
1. Дифференциальная диагностика и ведение больного с геморрагическим и тромбофилическим синдромами
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА ИВЕДЕНИЕ БОЛЬНОГО
С ГЕМОРРАГИЧЕСКИМ И
ТРОМБОФИЛИЧЕСКИМ СИНДРОМАМИ
КАФЕДРА ВНУТРЕННЕЙ МЕДИЦИНЫ №1
ДОЦЕНТ ЗАХАРЬЯН Е.А.
2.
Геморрагический синдром — это вид нарушениягемостаза, который проявляется совокупностью
симптомов, связанных со склонностью к
кровоточивости.
3.
В обеспечении нормального гемостаза участвуют:- тромбоциты (тромбоцитарный компонент),
- факторы свертывания крови (плазменный
компонент)
- сосудистая стенка (сосудистый компонент).
Фибринолитическая система обеспечивает
растворение избыточных тромботических масс.
4.
5.
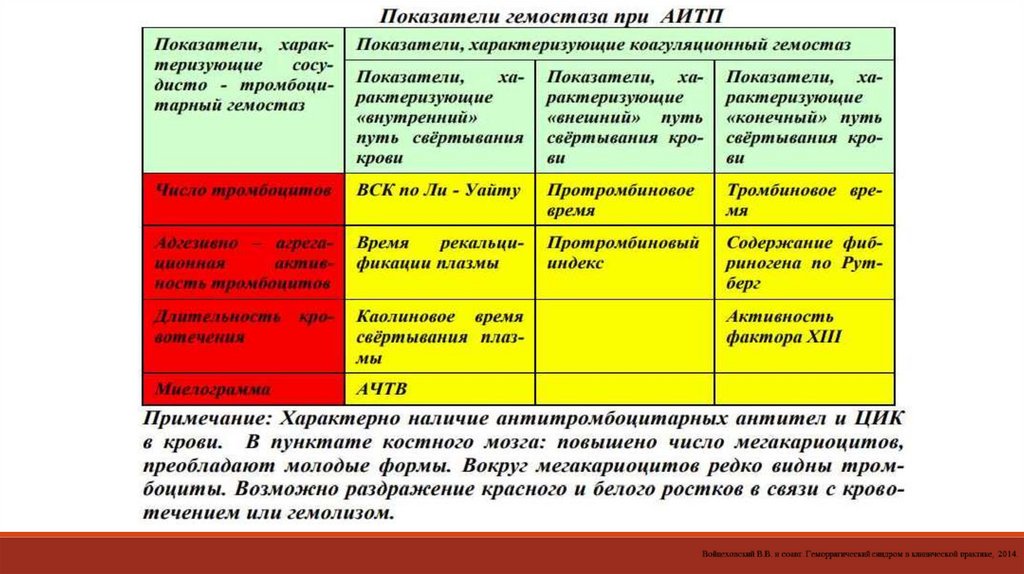
ПАТОЛОГИЯ ТРОМБОЦИТАРНО-СОСУДИСТОГО ГЕМОСТАЗТромбоцитопении
Аутоиммунная (идиопатическая) тромбоцитопеническая пурпура
Тромбоцитопении у беременных
Синдром Фишера- Эванса
Апластическая анемия
Гипоплазия мегакариоцитарного ростка вследствие медикаментозной интоксикации
Гипоплазия мегакариоцитарного ростка при алкогольной интоксикации
Гипопролиферация и гипоплазия мегакариоцитарного ростка инфекционного генеза
Тромбоцитопения при ВИЧ (СПИДе)
Тромботические микроангиопатии
Тромбоцитопения обусловленная нарушением распределения пулов тромбоцитов
Тромбоцитопатии
ПАТОЛОГИЯ КОАГУЛЯЦИОННОГО (ПЛАЗМЕННОГО) ЗВЕНА ГЕМОСТАЗА
Наследственные коагулопатии
Гемофилия А и В
Болезнь Виллебранда
Гемофилия С
Дефицит фактора XIII
Дефицит фактора XII
Наследственный дефицит фактора Х (болезнь Стюарта-Прауэра)
Дефицит фактора VII
Дефицит фактора V (парагемофилия)
Дефицит фактора II
Дефицит фактора I и дисфибриногенемия
Приобретенные коагулопатии
Передозировка прямых антикоагулянтов
Дефицит К-витаминзависимых факторов свертывания
Нарушение гемостаза при лечении препаратами фибринолитического
Коагулопатия при заболеваниях печени
и дефибринизирующего действия
Войцеховский В.В. и соавт. Геморрагический синдром в клинической практике, 2014.
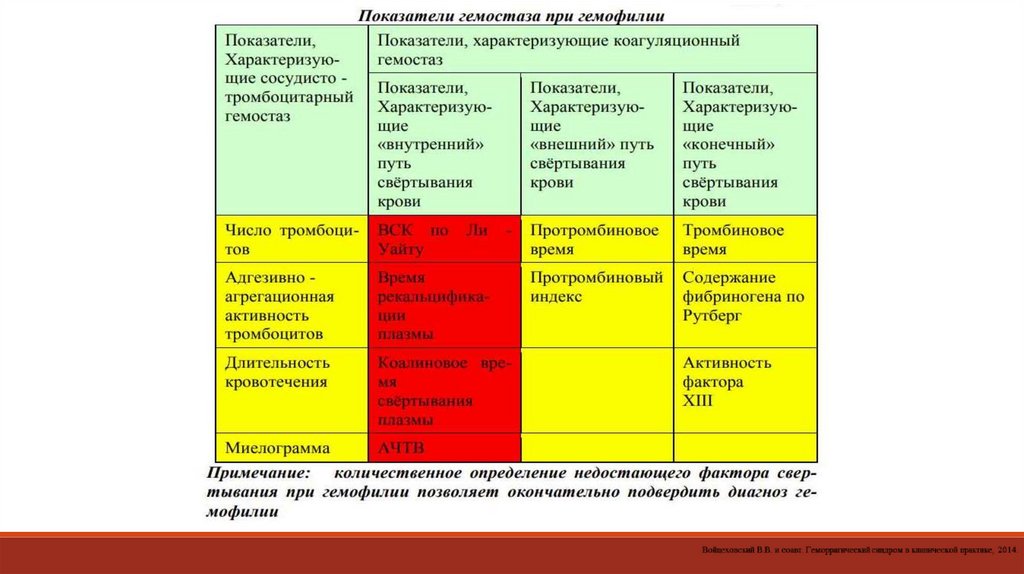
6.
СИНДРОМЫ ДИССЕМИНИРОВАННОГО ВНУТРИСОСУДИСТОГО СВЕРТЫВАНИЯ КРОВИПАТОЛОГИЯ ГЕМОСТАЗА У БОЛЬНЫХ ГЕМОБЛАСТОЗАМИ
Острый промиелоцитарный лейкоз
ВАСКУЛИТЫ
• Геморрагический васкулит
• Криоглобулинемический васкулит
ГЕМОРРАГИЧЕСКАЯ ЛИХОРАДКА С ПОЧЕЧНЫМ СИНДРОМОМ
ГЕМОРРАГИЧЕСКИЕ ЗАБОЛЕВАНИЯ ОБУСЛОВЛЕННЫЕ ПАТОЛОГИЕЙ СОСУДИСТОЙ
СТЕНКИ
• Наследственная геморрагическая телеангиэктазия (Болезнь Рендю-Ослера)
• Синдром Элерса-Данло
• Синдром Казабаха-Меррита
АТРОФИЧЕСКИЕ И ИМИТАЦИОННЫЕ ФОРМЫ ГЕМОРРАГИЧЕСКОГО СИНДРОМА
7. ОБСЛЕДОВАНИЕ БОЛЬНОГО ПРИ ПОДОЗРЕНИИ НА ПАТОЛОГИЮ ГЕМОСТАЗА
1. Анамнез:1) кровоточивость у родственников больного;
2) заболевания, которые могут вызвать геморрагический
синдром (заболевания печени, СЗСТ, заболевания почек,
злокачественные новообразования, тиреотоксикоз, алкоголизм и
т.д.);
3) лекарственные препараты (антикоагулянты, дезагреганты,
сульфаниламиды, НПВС, бета-блокаторы, антибиотики,
транквилизаторы, нитрофураны и т.д.).
8. 2. Осмотр
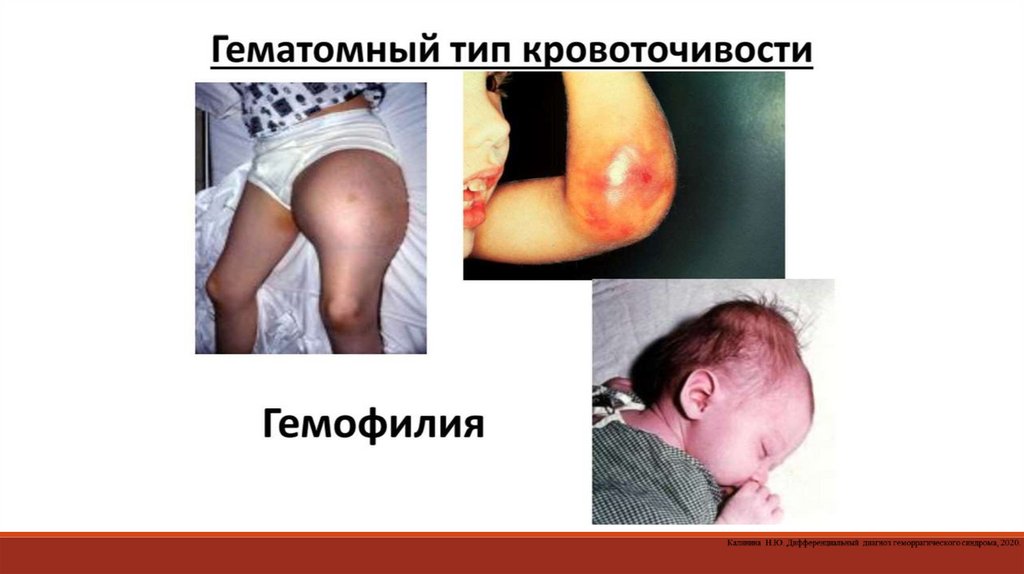
2.1 Определение типа кровоточивости:1. Гематомный тип
2. Микроциркуляторный (петехиально-пятнистый,
синячковый) тип
3. Смешанный (микроциркуляторно-гематомный) тип
4. Васкулитно-пурпурный тип
5. Ангиоматозный тип
9.
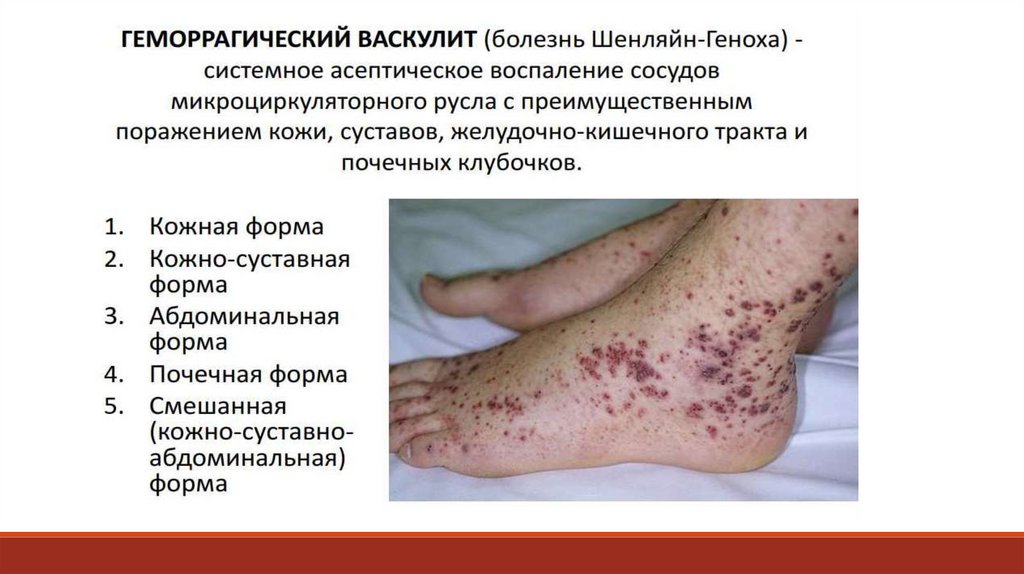
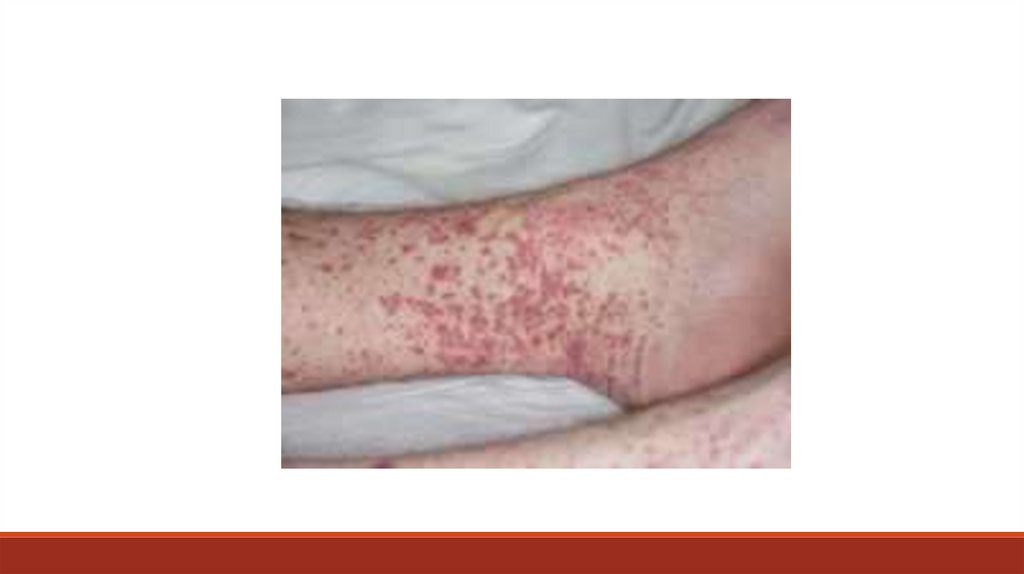
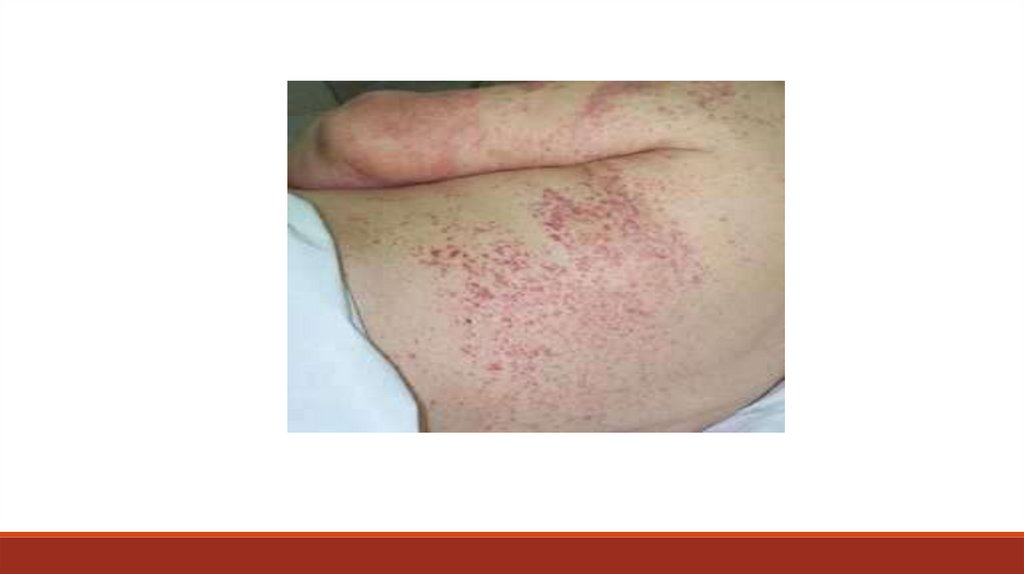
Дополнительное обследование больного:- тесты, оценивающие функциональное состояние системы гемостаза
(рутинные пробы, позволяющие выявить общую коагуляционную
направленность крови);
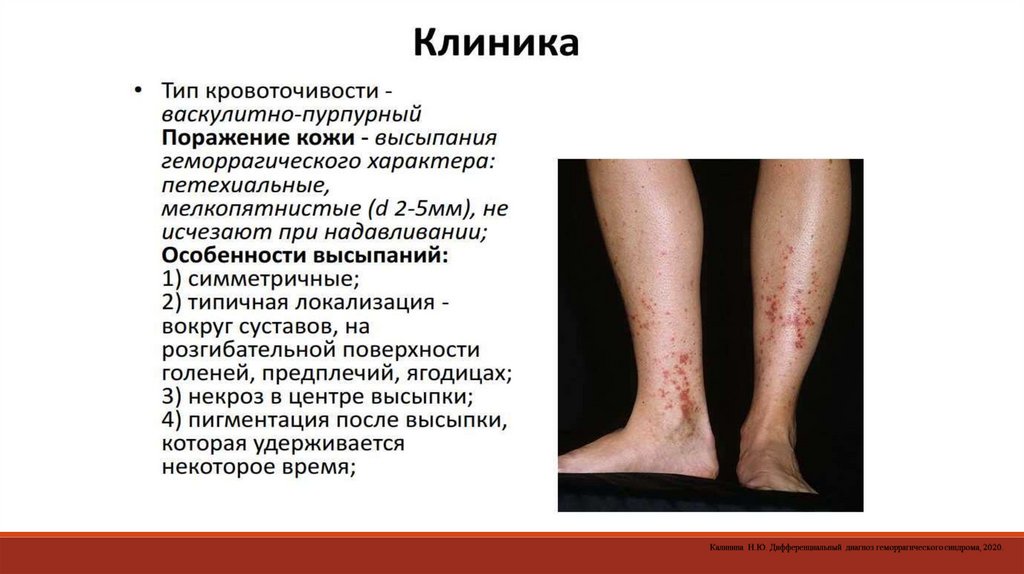
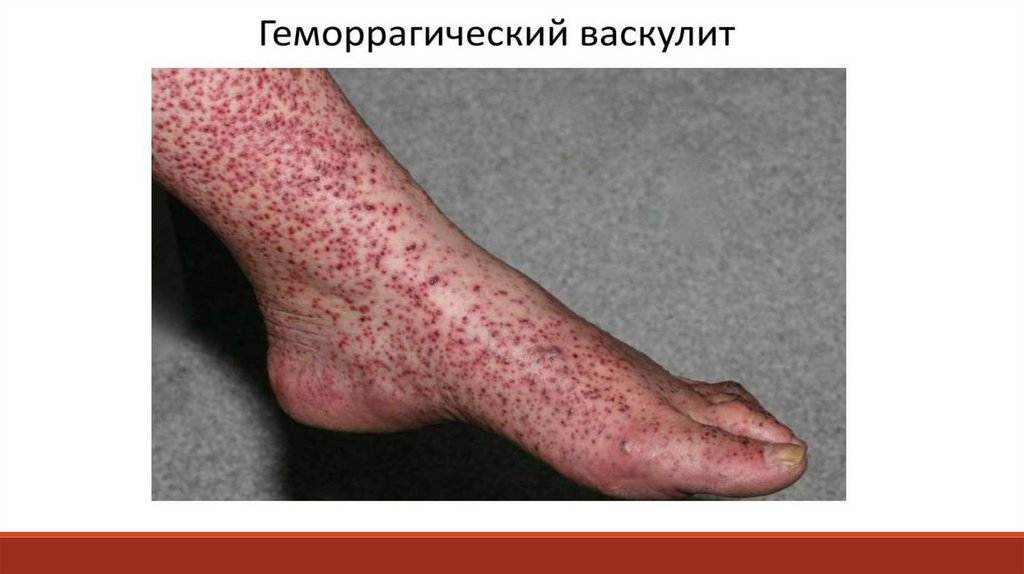
- дифференциальные пробы, которые обнаруживают дефицит отдельных
факторов свертывания, оценивают функциональное состояние
тромбоцитарного звена, свертывающей и противосвертывающей систем
гемостаза).
10.
Калинина Н.Ю. Дифференциальный диагноз геморрагического синдрома, 2020.11.
12.
Исследование сосудисто-тромбоцитарного гемостаза- определение ломкости микрососудов с помощью
пробы манжеточной компрессии, пробы щипка,
баночной пробы и т.д.,
- определение времени кровотечения из микрососудов
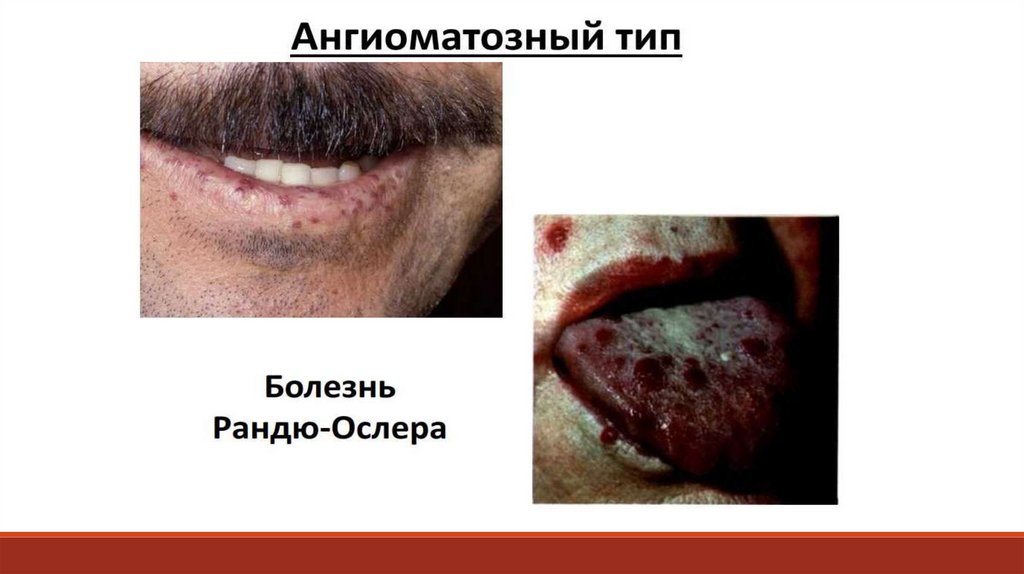
(пробы Дьюка, Квика, Айви, Борхгревика),
- подсчет количества тромбоцитов в крови, определение
их размеров и структуры, адгезивно-агрегационной
функции,
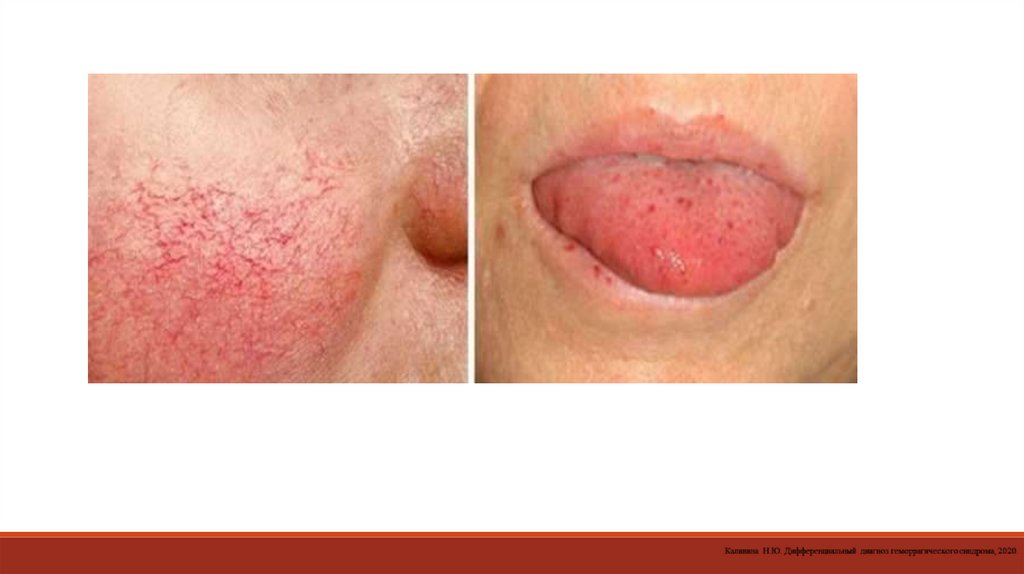
- определение антитромбоцитарных антител.
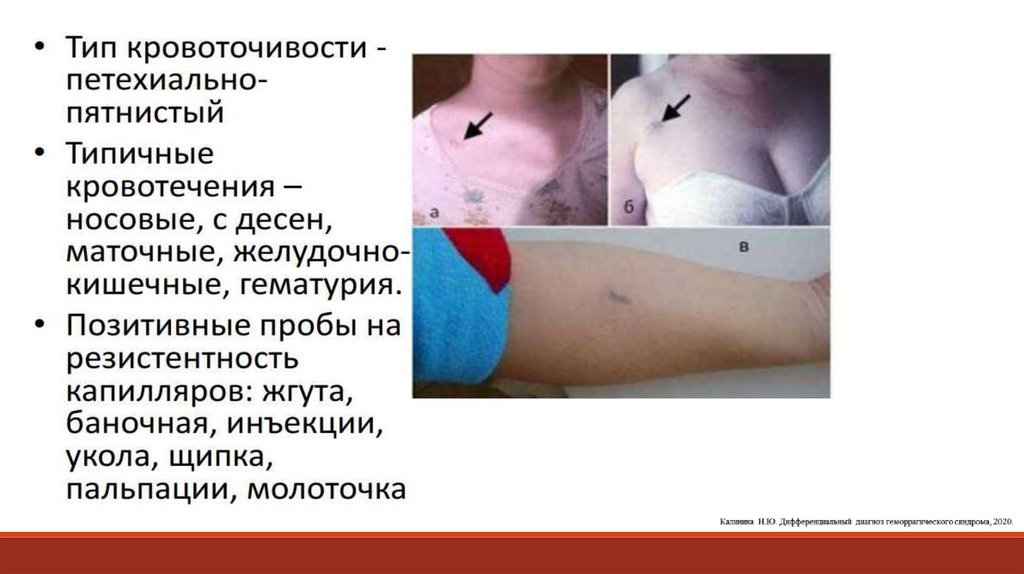
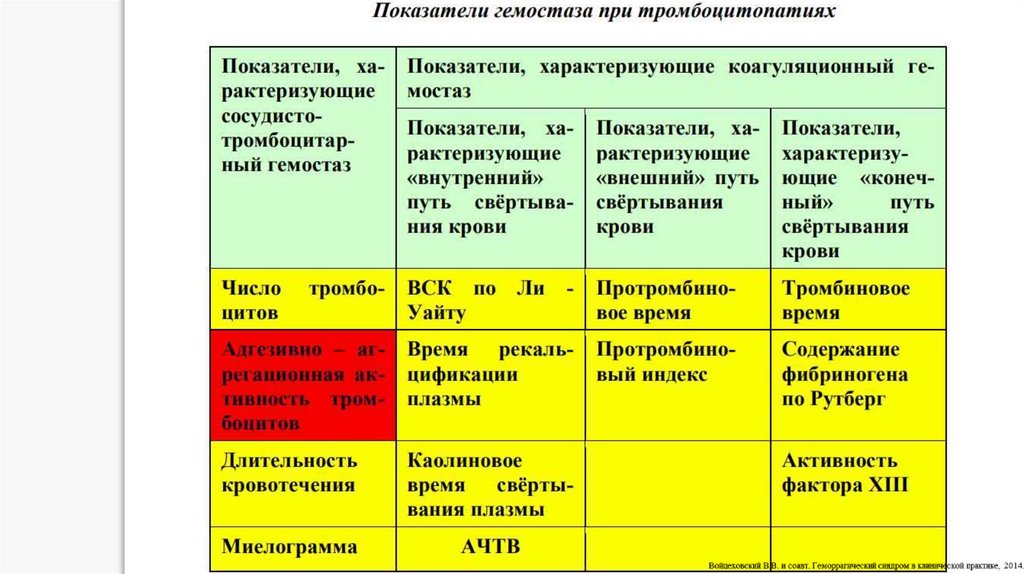
13. При диагностике патологии тромбоцитарно-сосудистого гемостаза исследуют сосудистый компонент (капиллярные пробы).
При диагностике патологии тромбоцитарнососудистого гемостаза исследуют сосудистыйкомпонент (капиллярные пробы).
1. Проба щипка. Врач собирает над ключицей кожу и делает щипок. При снижении резистентности
капилляров на месте щипка появляются петехии или кровоподтеки, особенно через 24 часа.
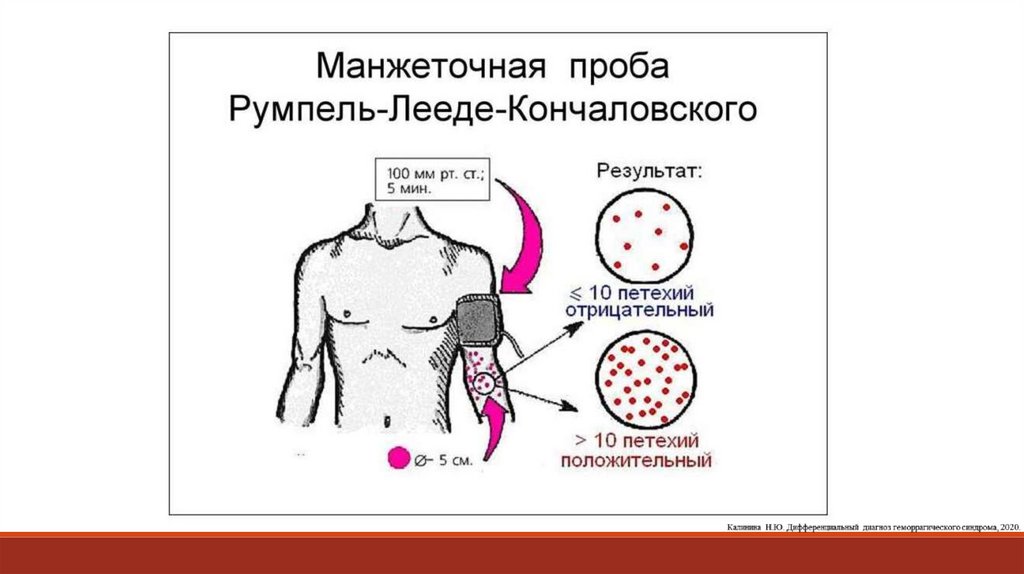
2. Проба жгута, манжеточная проба. На плечо накладывают манжету тонометра, соединяют с
манометром и поддерживают давление на уровне 90–100 мм рт. ст. в течение 5 мин. Количество петехий
более 10 свидетельствует о снижении резистентности стенки капилляров, что может быть связано с
нарушением функционального состояния тромбоцитов, снижением их количества и ангиотрофической
функции, а также с повышением проницаемости капилляров при различных патологических процессах
(васкулиты, вазопатии).
3. Баночная проба основана на определении резистентности капилляров по отношению к
отрицательному давлению, получаемому с помощью приставляемой к поверхности груди или предплечья
присасывающей банки, соединенной с манометром. Положительной проба считается при появлении
многочисленных петехий. Единичные петехии (до 10), как и отсутствие петехий, расценивается как
отрицательный результат пробы.
14. Методы лабораторного исследования тромбоцитаро-сосудистого гемостаза
1. Классическая проба Дьюка.2. проба Борхгревинка-Ваалера
3. исследование по А.С. Шитиковой
4. тест по Г.Н. Сушкевичу
5. Подсчет количества тромбоцитов в крови
6. Подсчет количества тромбоцитов по Фонио
7. Изучение размеров тромбоцитов в мазке
(тромбоцитометрия)
8. Ретракция кровяного сгустка
9. Исследование адгезивно-агрегационной
функции тромбоцитов
10. Методы ретенции тромбоцитов на
стекле/фильтрах.
11. Методы исследования агрегационной
функции тромбоцитов.
12. Визуальный микрометод определения
агрегации тромбоцитов.
13. Графическая регистрация процесса
агрегации.
14. визуальное или графическое исследование
агрегации тромбоцитов под влиянием
ристомицина
15. тесты, отражающие спонтанную
агрегацию тромбоцитов (Wu-Hoak, Н.И.
Тарасовой)
15.
При необходимости проводят:- исследование количества мегакариоцитов в миелограмме и трепанате
костного мозга с изучением морфологии этих клеток,
- электронно-микроскопическое изучение ультраструктуры тромбоцитов,
- определение антитромбоцитарных антител путем
иммунофлюоресцентного исследования в суспензии тромбоцитов
иммуноглобулина, связанного с этими клетками - метод Диксона,
- определение продолжительности жизни меченых аутологичных
тромбоцитов,
- количественное определение содержания в плазме крови до и после
агрегации тромбоцитарных факторов,
- определение маркеров неполноценности эндотелия и активации
сосудисто-тромбоцитарного гемостаза и другие специфические методы
исследования.
16.
Тесты коагуляционного звена системы гемостаза«Локальные» тесты – характеризуют состояние отдельных факторов или звеньев
каскадной реакции.
- активированное частично тромбопластиновое время (АЧТВ),
- протромбиновое время (ПВ),
- протромбиновый индекс (ПТИ),
- международное нормализованное отношение (МНО),
- фибриноген,
- D-димер,
- антитромбин III,
- протеин С,
- фактор VIII,
- концентрация и активность некоторых других факторов.
«Глобальные» коагулогические тесты, результаты которых позволяют оценить работу
системы гемостаза в целом (тромбоэластография/метрия, тест генерации тромбина и
тромбодинамика).
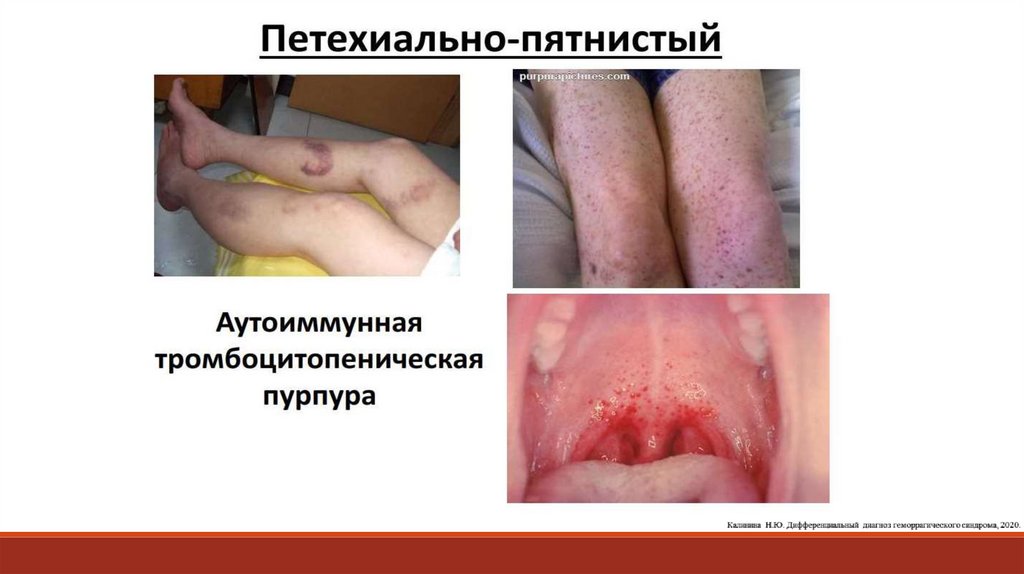
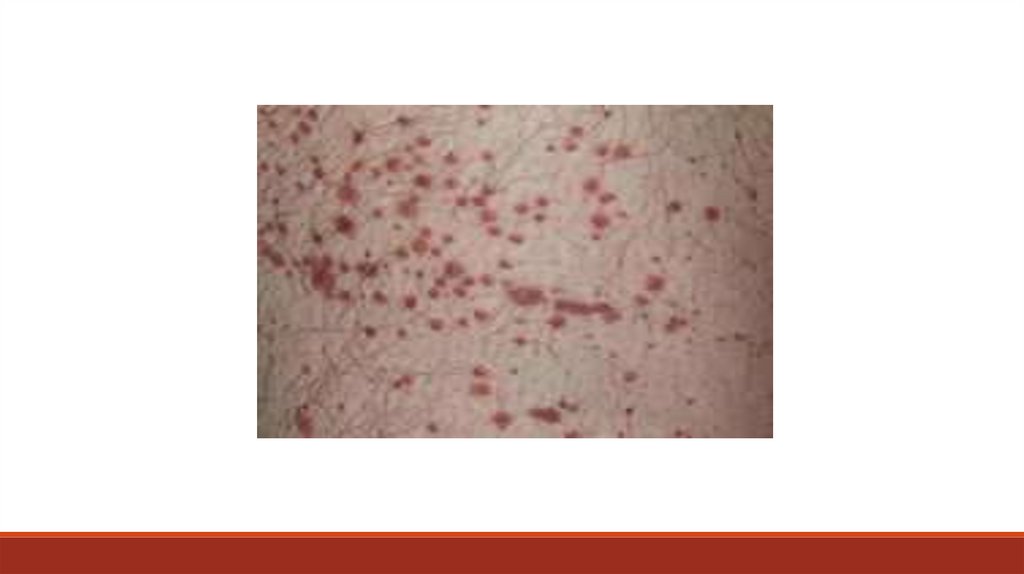
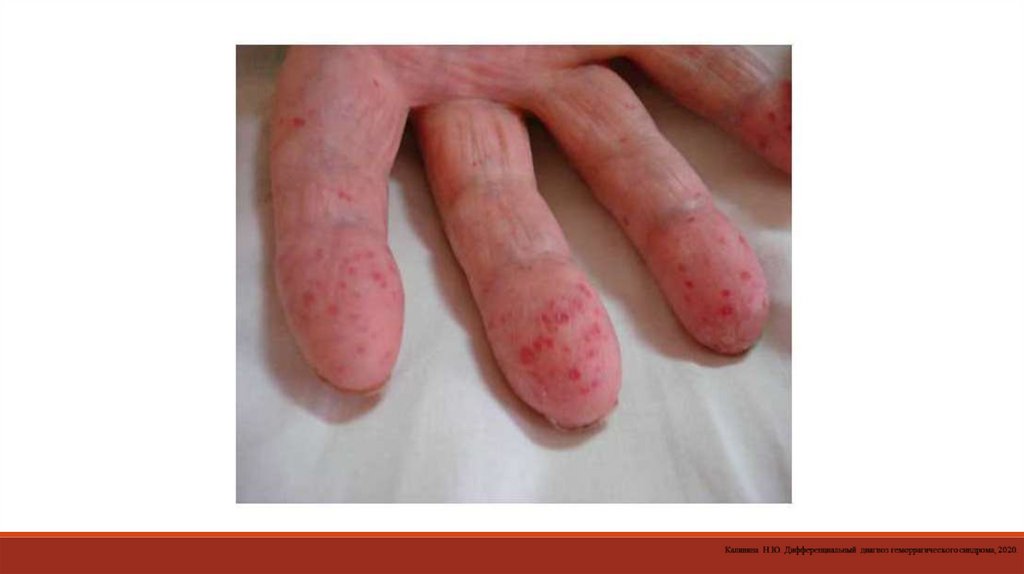
17. Петехиально-пятнистый (синячковый, микроциркуляторный)
18.
19. ПАТОЛОГИЯ ТРОМБОЦИТАРНО-СОСУДИСТОГО ГЕМОСТАЗА
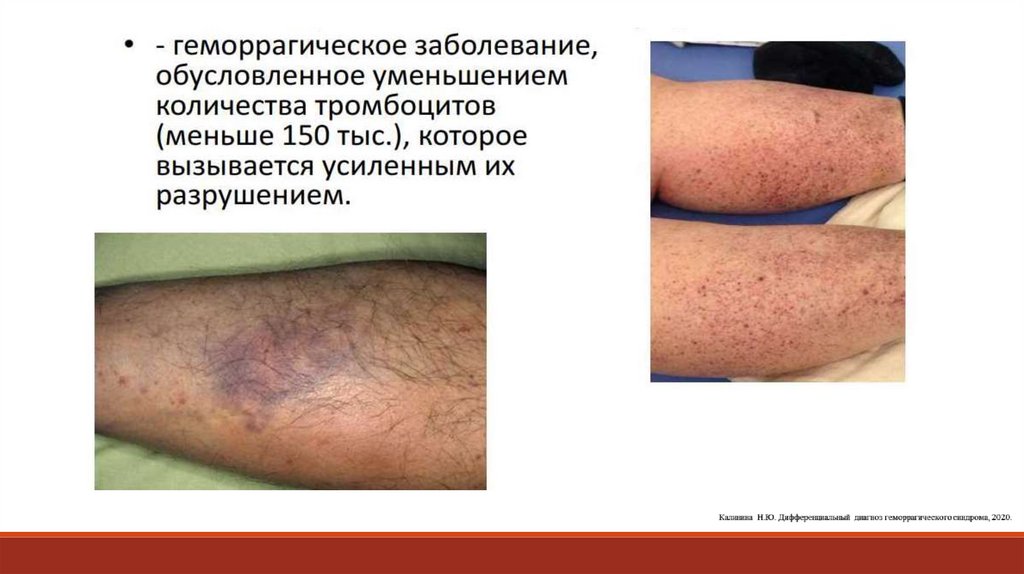
Тромбоцитопении (снижение количества тромбоцитов в периферической крови)- Аутоиммунная (идиопатическая) тромбоцитопеническая пурпура
- Тромбоцитопении у беременных
- Синдром Фишера- Эванса
- Апластическая анемия
- Гипоплазия мегакариоцитарного ростка вследствие медикаментозной интоксикации
- Гипоплазия мегакариоцитарного ростка при алкогольной интоксикации
- Гипопролиферация и гипоплазия мегакариоцитарного ростка инфекционного генеза
- Тромбоцитопения при ВИЧ (СПИДе)
- Тромботические микроангиопатии
- Тромбоцитопения обусловленная нарушением распределения пулов тромбоцитов
Тромбоцитопатии (нарушение функционального состояния тромбоцитов при их достаточном
количестве в крови)
20.
21.
Приобретенные тромбоцитопенииПовышенная деструкция тромбоцитов
Нарушение продукции тромбоцитов
1) Вторичная иммунная тромбоцитопения
при следующих заболеваниях:
- аутоиммунный тиреоидит,
- СКВ, АФЛС и другие коллагенозы,
- Лекарственно-опосредованная тромбоцитопения;
- ЛПЗ,
- лекарственно-опосредованная,
- Тромбоцитопения при инфекционных заболеваниях;
- вирусного генеза (герпесвирусы, хронические
- Токсическая тромбоцитопения (алкоголь и др.);
вирусные гепатиты, ВИЧ),
- постинфекционная и др.;
- Метастатическое поражение костного мозга при
2) Посттрансфузионная пурпура;
3) Синдром диссеминированного внутрисосудистого неоплазмах;
свертывания, фаза истощения;
- Заболевания системы кроветворения (острый лейкоз,
4) Сердечно-сосудистые аномалии и заболевания
апластическая анемия, миелодиспластический
(аневризмы, пороки сердца, стентирование);
5) Гестационная тромбоцитопения;
синдром, лимфопролиферативные заболевания и др.).
6) Тромботическая тромбоцитопеническая
пурпура;
7) Гемолитико-уремический синдром;
8) Тромботическая микроангиопатия и др.
22.
Наследственные тромбоцитопении• тромбастения Гланцмана;
• синдром Бернара-Сулье;
• синдром серых тромбоцитов;
• синдром Вискотта-Олдрича;
• врожденная амегакариоцитарная тромбоцитопения;
анемия Фанкони и другие
Псевдотромбоцитопении
23.
24.
Первичнаяиммунная
тромбоцитопения
–
идиопатическая
тромбоцитопеническая пурпура (ИТП, болензь Верльгофа) – аутоиммунное
заболевание, обусловленное выработкой антител к структурам мембраны
тромбоцитов и их предшественников – мегакариоцитов, что вызывает не только
повышенную деструкцию тромбоцитов, но и неадекватный тромбоцитопоэз,
характеризующийся изолированной тромбоцитопенией ниже 100,0 × 10⁹/л и
наличием/отсутствием геморрагического синдрома различной степени
выраженности.
Вторичная иммунная тромбоцитопения – иммунная тромбоцитопения,
являющаяся симптомом других аутоиммунных заболеваний: системной красной
волчанки (СКВ), антифосфолипидного синдрома (АФЛС), ревматоидного
артрита (РА) и др.
25.
ИТП не имеет географических особенностей.Мужчины болеют в 2-3 раза реже женщин
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ.
Этиология ИТП не известна. Аутоиммунное заболевание, характеризующимся выработкой
антител подкласса IgG1 к гликопротеинам мембраны тромбоцитов, в основном к ГП IIb/IIIa,
реже к ГП Ib-IX-V, Ia/IIa, IV или VI и формированием комплекса антиген-антитело,
фиксирующегося своим Fc-фрагментом иммуноглобулина к Fcy-рецепторам макрофагов,
дендритических
клеток
ретикулоэндотелиальной
системы,
МКЦ.
Помимо
антителообразования большую роль в патогенезе ИТП играют субпопуляции Т-лимфоцитов,
дисбаланс Т-хелперов (Th) 1-го и 2-го типов, подтипов цитотоксических CD8+-клеток и
регуляторных Т-лимфоцитов, цитокинов.
26.
27.
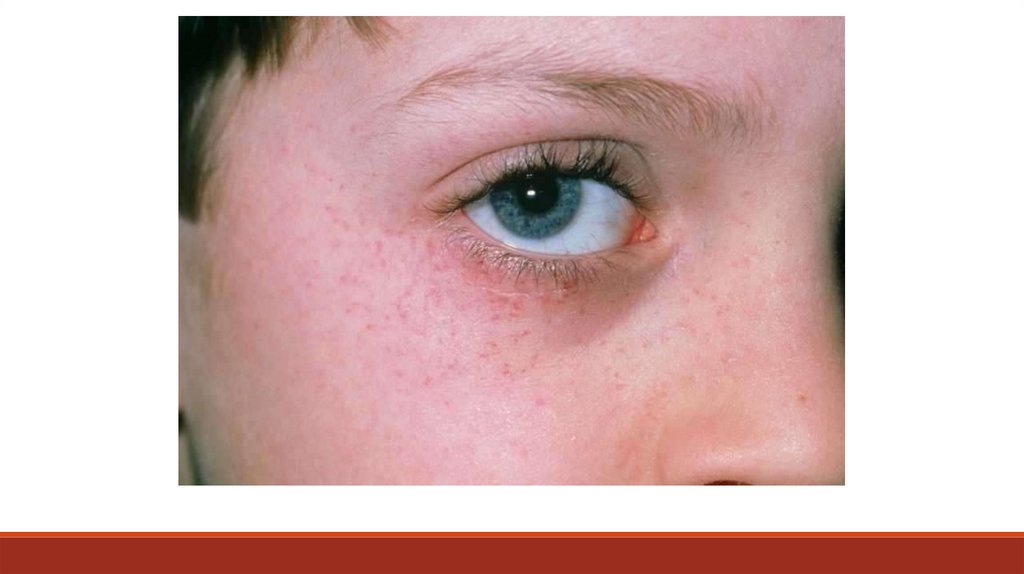
Клинически ИТП проявляется геморрагическим синдромом:- спонтанным или посттравматическим кожным (единичная или
генерализованная петехиальная сыпь и экхимозы),
- петехиями и экхимозами на слизистых,
- носовыми и десневыми кровотечениями,
- мено- и метроррагиями,
- реже – желудочно-кишечными кровотечениями и гематурией,
жизнеугрожающими субарахноидальныим кровоизлияниями (менее 0,5%).
Тяжелый геморрагический синдром, развивающийся при количестве
тромбоцитов более 30,0 × 10⁹/л, требует поиска дополнительных причин
кровоточивости (коагулопатия, патология сосудов и др.).
28.
Диагноз устанавливается на основании следующих критериев:1. изолированная тромбоцитопения менее 100,0 × 10⁹/л как минимум в двух последовательных
анализах крови;
2. отсутствие морфологических и функциональных аномалий тромбоцитов;
3. отсутствие патологии лимфоцитов, гранулоцитов и эритроцитов;
4. нормальные показатели гемоглобина, эритроцитов и ретикулоцитов, если не было существенной
кровопотери;
5. повышенное или нормальное количество МКЦ в миелограмме;
6. нормальные размеры селезенки;
7. отсутствие других патологических состояний, вызывающих тромбоцитопению;
8. наличие тромбоцит-ассоциированных антител в высоком титре (нормальный титр не исключает
ИТП).
29.
30.
31.
32.
33. Лабораторные тесты для диагностики ИТП
Обязательные тестыОАК + ретикулоциты + подсчет тромбоцитов по Фонио
Потенциально полезные тесты
Антитела к гликопротеинам (тромбоцитассоциированные)
Мазок периферической крови
Биохимический анализ крови
Антинуклеарный фактор
Антитела к нативной (двуспиральной) ДНК
Коагулограмма, агрегация тромбоцитов с АДФ, коллагеном, Волчаночный антикоагулянт
ристомицином, адреналином , фибринолиз, антитромбин III, Dдимер
Иммунохимическое
исследование
(количественный анализ)
ВИЧ
сыворотки
крови Антитела к кардиолипину (IgG и IgM) и другим фосфолипидам
Антитела к бета-2-гликопротеину 1 (IgG и IgM)
Вирусы гепатитов В и С
Антитела к ТПО и гормоны щитовидной железы
Helicobacter pilori
Тест на беременность у женщин детородного возраста
Исследование костного мозга (цитологическое и гистологическое)
Герпес-вирусы (антитела и ПЦР)
Прямая проба Кумбса
Маркеры тромбофилии
Коагулограмма и агрегация тромбоцитов с АДФ, коллагеном,
ристомицином, адреналином
34.
35.
36.
ЛЕЧЕНИЕ ИТПОсновные принципы терапии
Современными методами терапии полного излечения от ИТП достигнуть не
удается, возможно получение ремиссии различной длительности или
состояния клинической компенсации. Основная цель терапии ИТП –
достижение безопасного купирующего геморрагический синдром количества
тромбоцитов (не менее 50 × 10⁹/л), а не коррекция их числа до нормальных
показателей.
37.
Схема 1. Терапия больных с впервые диагностированной ИТП(длительность болезни до 3 мес.) с низким уровнем тромбоцитов
Тромбоциты менее 30-50,0 × 10⁹/л
геморрагический синдром есть ( + )
1-я линия
терапии
ГКС:
преднизолон
метилпреднизолон
дексаметазон
2-я линия
терапии
Спленэктомия
геморрагического синдрома нет ( - )
ВВИГ
Динамическое наблюдение
Лечение при подготовке к
операциям и при работе
пациента,
связанной
с
повышенным травматизмом
38.
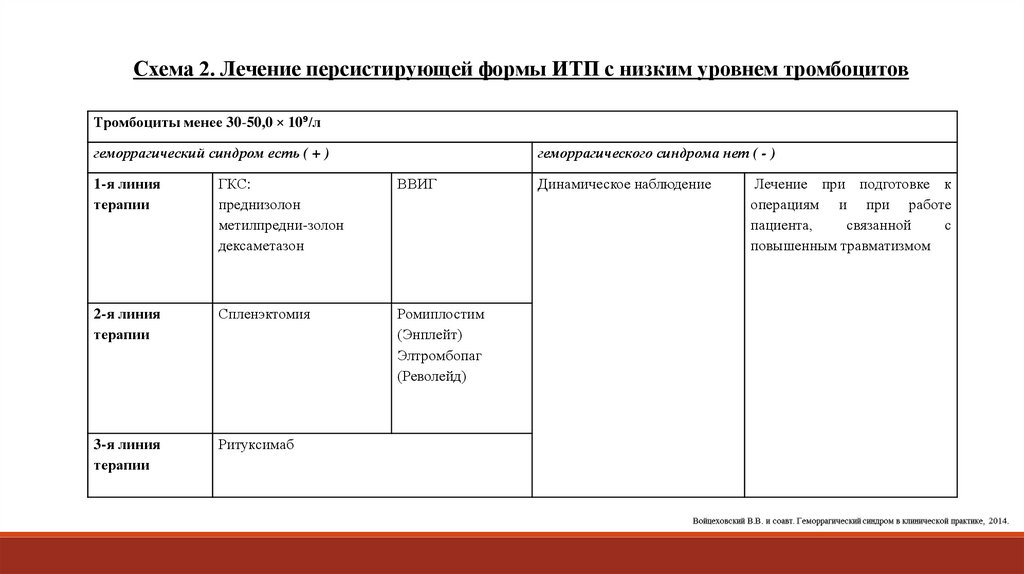
Схема 2. Лечение персистирующей формы ИТП с низким уровнем тромбоцитовТромбоциты менее 30-50,0 × 10⁹/л
геморрагический синдром есть ( + )
геморрагического синдрома нет ( - )
1-я линия
терапии
ГКС:
преднизолон
метилпредни-золон
дексаметазон
ВВИГ
2-я линия
терапии
Спленэктомия
Ромиплостим
(Энплейт)
Элтромбопаг
(Револейд)
3-я линия
терапии
Ритуксимаб
Динамическое наблюдение
Лечение при подготовке к
операциям и при работе
пациента,
связанной
с
повышенным травматизмом
39.
Агонисты тромбопоэтиновых рецепторов, или миметики тромбопоэтина – препаратынеиммуносупрессивного действия, стимулирующие выработку тромбоцитов за счет
активации рецептора к тромбопоэтину на тромбоцитах и МКЦ. Препараты этой группы
используют при хронических рецидивирующих и рефрактерных формах болезни.
Показаниями к лечению миметиками тромбопоэтина являются:
• неэффективность терапии 1-й линии;
• осложнения от ГКС и ВВИГ;
• противопоказания к спленэктомии;
• отказ больного от спленэктомии;
• необходимость уменьшения риска кровотечения перед плановым хирургическом
вмешательстве.
40.
Ромиплостим (Энплейт) – препарат для подкожного введения 1 раз внеделю – в среднем эффективен у 95% больных, у 88-97% пациентов до
и у 79-90% после спленэктомии.
Начальная доза - 1 мкг/кг массы тела, максимальная - 10 мкг/кг массы
тела.
Время ожидаемого ответа - минимум 2 недели. До определения
индивидуальной дозы необходим еженедельный контроль ОАК, затем 1 раз в месяц.
Введение препарата больной может осуществлять самостоятельно.
41.
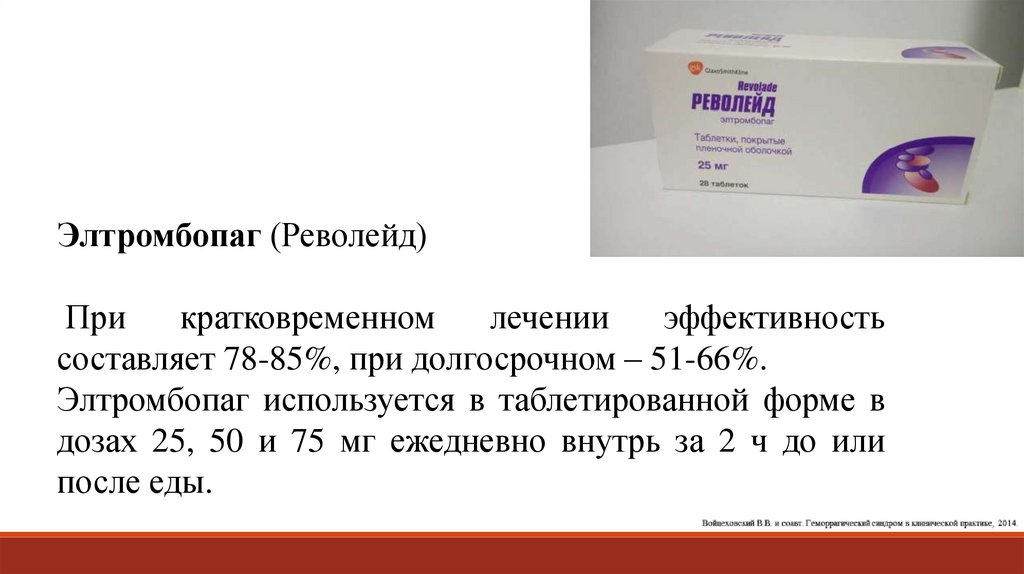
Элтромбопаг (Револейд)При
кратковременном
лечении
эффективность
составляет 78-85%, при долгосрочном – 51-66%.
Элтромбопаг используется в таблетированной форме в
дозах 25, 50 и 75 мг ежедневно внутрь за 2 ч до или
после еды.
42.
К препаратам терапии 3 линии при персистирующей ИТП относится ритуксимаб.Показаниями к назначению ритуксимаба являются:
• неэффективность предыдущих методов лечения;
• противопоказания/осложнения терапии 1 и 2 линий;
• отказ пациента от спленэктомии.
Противопоказания: хронический вирусный гепатит В.
Разовая доза составляет 375 мг/м2 (в среднем 500 мг в/в). Частота введения – 1 раз в неделю;
длительность курса – 4 введения. Возможно поддерживающее лечение – 1 раз месяц при контроле
количества CD19+ лимфоцитов.
43.
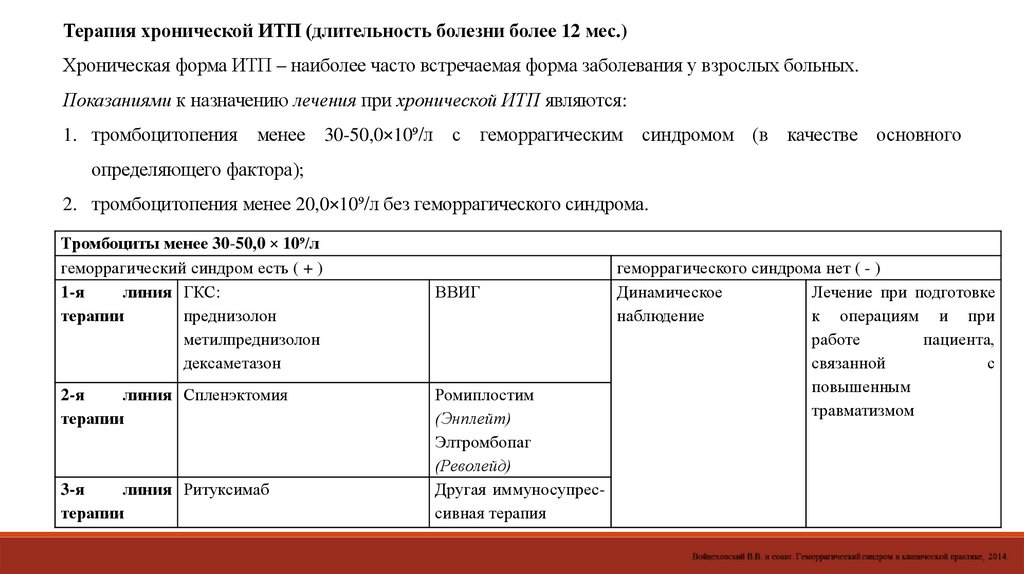
Терапия хронической ИТП (длительность болезни более 12 мес.)Хроническая форма ИТП – наиболее часто встречаемая форма заболевания у взрослых больных.
Показаниями к назначению лечения при хронической ИТП являются:
1. тромбоцитопения менее 30-50,0×10⁹/л с геморрагическим синдромом (в качестве основного
определяющего фактора);
2. тромбоцитопения менее 20,0×10⁹/л без геморрагического синдрома.
Тромбоциты менее 30-50,0 × 10⁹/л
геморрагический синдром есть ( + )
1-я
линия ГКС:
терапии
преднизолон
метилпреднизолон
дексаметазон
2-я
линия Спленэктомия
терапии
3-я
линия Ритуксимаб
терапии
ВВИГ
Ромиплостим
(Энплейт)
Элтромбопаг
(Револейд)
Другая иммуносупрессивная терапия
геморрагического синдрома нет ( - )
Динамическое
Лечение при подготовке
наблюдение
к операциям и при
работе
пациента,
связанной
с
повышенным
травматизмом
44.
Cиндром Фишера-Эванса – сочетание аутоиммунной гемолитическойанемии и аутоиммунной тромбоцитопенической пурпуры. Выделяют
симптоматическую и идиопатическую формы синдрома Фишера-Эванса.
Симптоматическая форма наблюдается при хроническом гепатите,
системной красной волчанке, ревматоидном артрите, хроническом
лимфолейкозе, лимфомах.
Клиническая картина этой патологии состоит из гемолитической желтухи,
анемического синдрома различной степени тяжести, геморрагического
синдрома по петехиально-пятнистому типу. Лабораторные проявления
характеризуются
высоким
ретикулоцитозом
и
непрямой
гипербилирубинемией, сочетающимися с тромбоцитопенией.
45.
Тромбоцитопения при ВИЧ (СПИДе)1. ВИЧ инфицирует мегакариоциты, следовательно, может привести к нехватке тромбоцитов.
2. Некоторые препараты против ВИЧ могут повредить красный костный мозг (например,
некоторые нуклеозидные ингибиторы обратной транскриптазы). То же относится и к
некоторым заболеванием на стадии СПИДа (например, лимфоме или МАК).
3. Иммунная система способна вырабатывать антитела, которые направлены против здоровых
тромбоцитов организма, то есть развивается АИТП. Аутоантитела подают сигнал селезенке,
которая уничтожает и удаляет тромбоциты из организма.
Наиболее эффективное лечение для тромбоцитопении у людей с ВИЧ - это высокоактивная
антиретровирусная терапия (ВААРТ).
В том случае, если ВААРТ оказывается неэффективной, то лечение проводится по программе
АИТП.
46.
47.
Классификация тромбоцитопатий и дисфункций тромбоцитов. (З.С.Баркаган 1988, 2005 гг)А. Наследственные и врожденные формы.
1. Связанные с мембранными аномалиями (все варианты тромбастении Гланцмана, эссенциальная атромбия, аномалия Бернара-Сулье и др.)
2. Внутриклеточные аномалии: а) болезни недостаточного пула хранения: -дефицит плотных (безбелковых) гранул (болезнь Хержманского-Пудлака,
ТАР-синдром и др.); -дефицит α-гранул (белковых) – синдром серых тромбоцитов и др. б) нарушение реакции высвобождения гранул и их
компонентов: -дефицит циклооксигеназы; -дефицит тромбоксан-синтетазы; -другие патогенетические формы.
3. Смешанные тромбоцитарные нарушения (аномалии Мея-Хеглина, Вискотта-Олдрича и др.).
4. Дисфункции плазменного генеза и при сосудистых дисплазиях: а) болезнь Виллебранда; б) аномалии коллагена – болезнь Элерса – Данло и
другие мезенхимальные дисплазии.
Б. Приобретенные тромбоцитопатии
1. При гемобластозах: а) дезагрегационные гипорегенераторные; б) формы потребления (при развитии
ДВС-синдрома); в) смешанного типа.
2. При миелопролиферативных заболеваниях и эссенциальной тромбоцитемии.
3. При В12-дефицитной анемии.
4. При уремии (нарушение ААФТ, доступности фактора 3, реже - ретракции сгустка).
5. При ДВС-синдроме и активации фибринолиза – быстрое потребление тромбоцитов и блокада их функции продуктами расщепления фибриногена.
6. При циррозах, опухолях и паразитарных заболеваниях печени (нарушение ААФТ вследствие метаболических нарушений, секвестрации
тромбоцитов в портальной системе, потреблении их при развитии ДВС-синдрома).
7. Блокада тромбоцитов макро- и парапротеинами (миеломная болезнь, болезнь Вальденстрема, моно- и поликлоновые гаммапатии и др.).
8. При цинге (нарушение АДФ-агрегации).
9. При гормональных нарушениях (гипо- и дистиреозах, гипоэстрогении и др.)
10. Лекарственные и токсигенные (при лечении нестероидными противовоспалительными препаратами, ацетилсалициловой кислотой,
пиразолоновыми производными, бутазолидинами, бруфеном, индометацином, β – адреноблокаторами, дипиридомолом, большими дозами папаверина и
некоторыми антибиотиками (карбеннциллином, пенициллином), транквилизаторами, мочегонными препаратами, нитрофуранами, антигистаминами,
цитостатиками и другими средствами), после приема алкоголя.
11. При лучевой болезни.
12. При массивных гемотрансфузиях, инфузиях реополиглюкина.
13. При больших тромбозах.
48.
Клинические проявления тромбоцитопатий и ихдиагностика.
Заподозрить тромбоцитопатию следует у больных с геморрагиями петехиально –
пятнистого типа, меноррагиями неясного генеза, десневыми и носовыми
кровотечениями, продолжительным подтеканием крови после удаления
зубов, небольших порезов.
При этом количество тромбоцитов в пределах нормы или слегка снижено, может
быть удлинено время кровотечения.
Отмечается патология при исследовании адгезивно-агрегационной функции
тромбоцитов.
49.
50.
51.
52.
53.
54.
Гемофилия — наследственное заболевание свертывающей системы крови, возникающее врезультате дефицита фактора свертывания крови VIII (FVIII) — гемофилия А, или фактора
свертывания крови IX (FIX) — гемофилия B.
Передается по X-сцепленному рецессивному пути наследования. Примерно у 70% больных
имеется положительный семейный анамнез по заболеванию. Причиной гемофилии являются
мутации гена, кодирующего FVIII (Xq28), или гена, кодирующего FIX (Xq27). В 30-35% случаев
возможны спорадические мутации без наличия семейного анамнеза заболевания.
55.
Королева Виктория (1819 – 1901).Через ее детей гемофилия попала
в царствующие семьи Германии,
Испании и России.
Цесаревич Алексей родился летом 1904 года
(через 10 лет после венчания Николая
Александровича и Александры Федоровны). Не
прошло и шести недель, как выяснилось
ужасное: у цесаревича гемофилия, против
которой в то время медицина была бессильна.
56.
57.
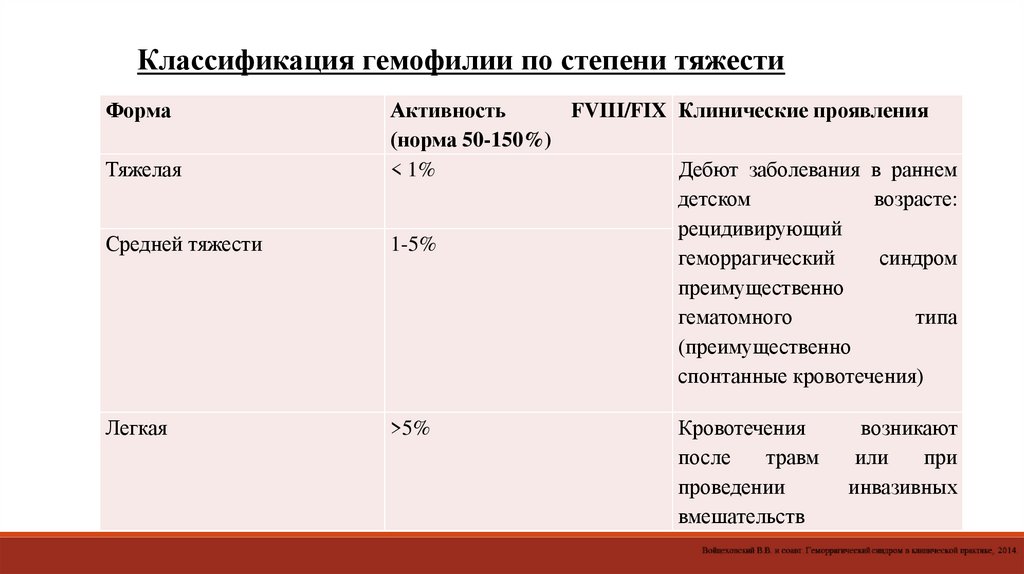
Классификация гемофилии по степени тяжестиФорма
Средней тяжести
Активность
FVIII/FIX Клинические проявления
(норма 50-150%)
< 1%
Дебют заболевания в раннем
детском
возрасте:
рецидивирующий
1-5%
геморрагический
синдром
преимущественно
гематомного
типа
(преимущественно
спонтанные кровотечения)
Легкая
>5%
Тяжелая
Кровотечения
после
травм
проведении
вмешательств
возникают
или
при
инвазивных
58.
59.
Клинические признакиОсновное проявление гемофилии – кровотечения и кровоизлияния, возникающие
спонтанно или вследствие травмы. ГА и ГВ имеют схожую клиническую картину.
60. Плазменные нарушения гемостаза (гемофилии)
Дефектный факторНазвание болезни
Синонимы заболевания
I (фибриноген)
Афибриногенемия, гипофибриногенемия,
дисфибриногенемия
Дефицит I фактора
II (протромбин)
V (проакцелирин)
Гипопротромбинемия
Дефицит V фактора
Дефицит II фактора
Парагемофилия, болезнь Оврена
VII (проконвертин)
VIII (антигемофилический глобулин)
Дефицит VII фактора
Гемофилия А
Гипопроконвертинемия
Классическая гемофилия, дефицит VIII фактора
IX (фактор Кристмаса)
Болезнь Виллебранда
Гемофилия В
Ангиогемофилия
Болезнь Кристмаса, дефицит IX фактора
Х (фактор Стюарта – Прауэр)
XI (предшественник плазменного
тромбопластина)
Дефицит Х фактора
Дефицит XI фактора
Болезнь Стюарта – Прауэра
Гемофилия С
XII* (фактор Хагемана)
XIII (фибринстабилизирующий фактор, фактор
Лаки-Лорана, фибриназа)
Дефицит XII фактора
Дефицит XIII фактора
Симптом Хагемана
–
XIV**(фактор Флетчера, прекалликреин)
Дефицит прекалликреина
Дефицит фактора Флетчера, дефицит XIV
фактора
XV** (кининоген высокой молекулярной массы
– КВММ, фактор Фитцжеральда, Вильямса,
Фложак)
Дефицит кининогена ВММ
Болезнь Фитцжеральда, Вильямса, Фложак
* Дефицит XII, XIV и XV факторов свертывания крови не проявляется геморрагиями, хотя лабораторное обследование обнаруживает у этих больных нарушение контактной активации (удлинение АЧТВ).** Название
факторов XIX и XV не принято в Международной номенклатуре.
61.
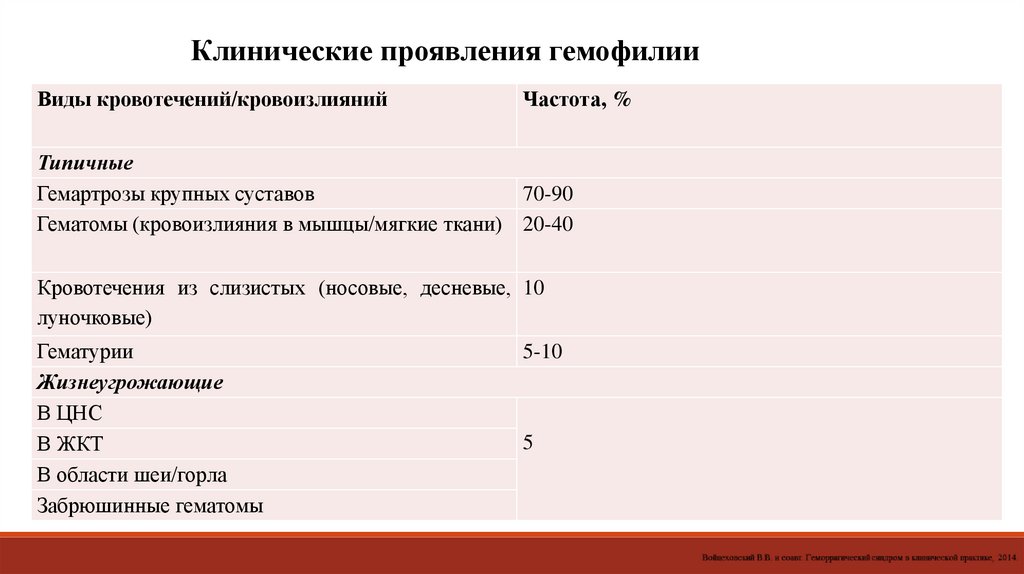
Клинические проявления гемофилииВиды кровотечений/кровоизлияний
Частота, %
Типичные
Гемартрозы крупных суставов
70-90
Гематомы (кровоизлияния в мышцы/мягкие ткани) 20-40
Кровотечения из слизистых (носовые, десневые, 10
луночковые)
Гематурии
Жизнеугрожающие
В ЦНС
В ЖКТ
В области шеи/горла
Забрюшинные гематомы
5-10
5
62.
Диагностика1. Выявление наличия геморрагического синдрома в анамнезе у пациента и членов
семьи.
2. Физикальный
осмотр:
(кожный
геморрагический
синдром
различной
выраженности в виде множественных экхимозов и гематом; признаки поражения
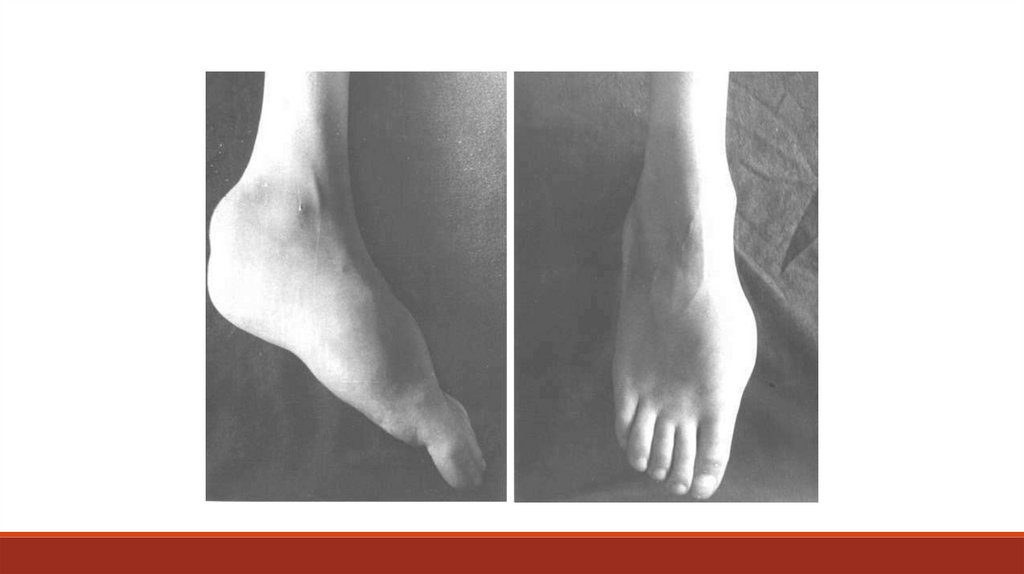
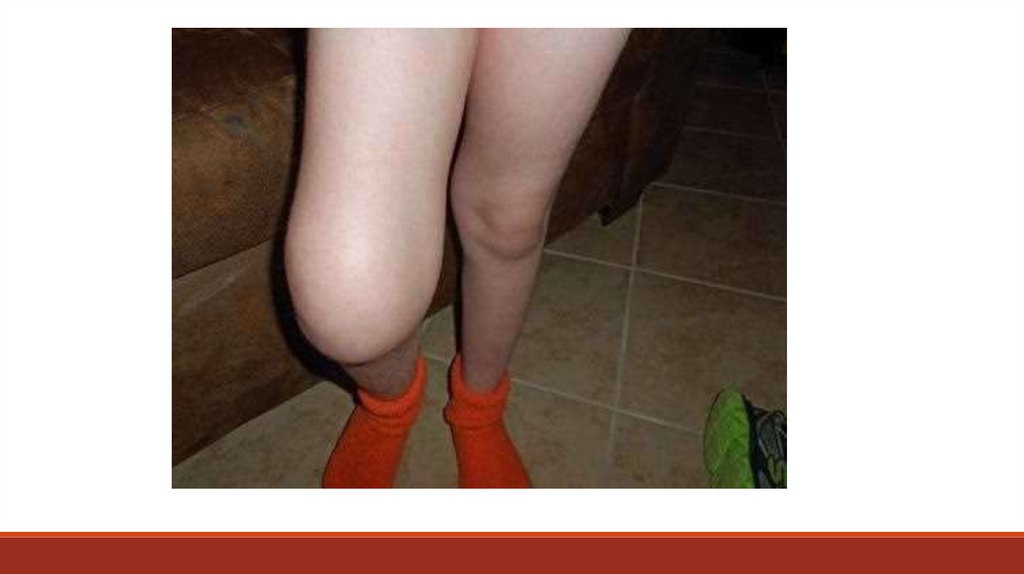
суставов в виде деформации, отека и локального повышения температуры кожи
(острый гемартроз) и/или признаков нарушения подвижности, объема движений
суставов, гипотрофии мышц конечности на стороне пораженного сустава,
нарушение походки (деформирующая артропатия).
63.
64.
65.
В качестве первого этапа рекомендуется проводить коагулологический скрининг:активированное частичное тромбопластиновое время (АЧТВ);
протромбиновое время (ПВ);
тромбиновое время (ТВ);
концентрацию фибриногена (по Клауссу);
время кровотечения стандартизованным методом (например, по Айви, или с помощью анализаторов
функции тромбоцитов PFA-100 и PFA-200);
подсчет количества тромбоцитов по Фонио.
Второй этап диагностики рекомендуется выполнять при выявлении изолированного удлинения АЧТВ, либо при
отсутствии изменений в ходе скрининга у пациентов с клиническими признаками легкой формы гемофилии. На
данном этапе определяют активность факторов плазменного гемостаза — FVIII, FIX, фактора фон
Виллебранда (vWF), факторов свертывания крови XI и XII.
66.
При выявленном снижении активности FVIII или FIX третьим этапом диагностики рекомендуется выполнятьопределение специфического ингибитора к сниженному фактору. При снижении активности нескольких
факторов свертывания крови и/или удлинении фосфолипид-зависимых тестов (АЧТВ с чувствительными
реактивами) проводится определение неспецифического ингибитора (волчаночного антикоагулянта).
Для исключения приобретенных дефицитов FVIII, FIX рекомендуется молекулярно-генетическая диагностика
нарушений FVIII, FIX.
Критерии диагноза гемофилии (при наличии геморрагического синдрома в анамнезе больного или
семейном анамнезе):
отсутствие приобретенных коагулопатий;
снижение активности FVIII/FIX ниже 50%;
наличие мутаций генов FVIII или FIX.
Диагноз устанавливается при наличии как минимум 2 из 3 критериев.
67.
68.
ЛечениеОсновным принципом лечения гемофилии является специфическая заместительная терапия концентратами
факторов свертывания.
- очищенные, вирус-инактивированные препараты, изготовленные из донорской плазмы человека (концентрат
FVIII, концентрат FIX, концентрат FVIII + фактор фон Виллебранда, антиингибиторный коагулянтный
комплекс (АИКК))
- рекомбинантные концентраты факторов свертывания (Октоког альфа, Мороктоког альфа, Нонаког альфа,
Эптаког альфа (активированный), Симоктоког альфа, Туроктоког альфа)
Концентраты факторов свертывания крови вводятся внутривенно. Чаще всего используется болюсная инфузия со
скоростью, рекомендованной производителем. В редких случаях - непрерывная инфузия. Современная терапия
гемофилии базируется на принципе «домашнего лечения». Обязательными условиями для проведения «домашнего
лечения» являются: наличие у пациента гемостатических препаратов (препарат находится там же, где пациент),
решение о применении гемостатического препарата принимает пациент или его родственники в соответствии с
рекомендациями гематолога, пациент и/или его родственники обучены правилам хранения и использования
препаратов.
69.
70.
71.
72.
ПРИОБРЕТЕННЫЕ КОАГУЛОПАТИИДефицит К-витаминзависимых факторов свертывания
Витамин К относится к группе липофильных (жирорастворимых) и гидрофобных витаминов,
необходимых для синтеза белков, обеспечивающих достаточный уровень коагуляции.
Причины развития дефицита витамина К:
- нарушения обмена;
- нарушения синтеза;
- недостаточное поступление с пищей;
- действие препаратов, связывающих желчные кислоты;
- заболевания желчных путей, препятствующие адсорбции жирорастворимых витаминов;
- заболевания кишечника, сопровождающиеся нарушением всасывания вит. К,
- выработка ингибиторов к факторам свертывания крови;
- заболевания печени;
- ДВС-синдром,
- прием антикоагулянтов непрямого действия;
- отравление родентицидами, содержащими непрямые антикоагулянты.
(Баркаган З.С., 2005; И.В. Колосова и соавт., 2012; Chung K.S., et al., 1981)
73.
Недостатки приема варфарина:- необходимость частого лабораторного контроля в связи с высоким риском кровотечений,
- диарея,
- повышение активности печеночных трансаминаз,
- остеопороз,
- экзема,
- некроз кожи,
- васкулиты,
- алопеция
- у части больных - высокая резистентность к кумаринам (нарушение в печени сродства клеточных
рецепторов к кумаринам).
(Баркаган З.С., и соавт., 2001; Бокарев И.Н., и соавт., 2009; Стойко Ю.М., Замятин М.Н., 2007).
74.
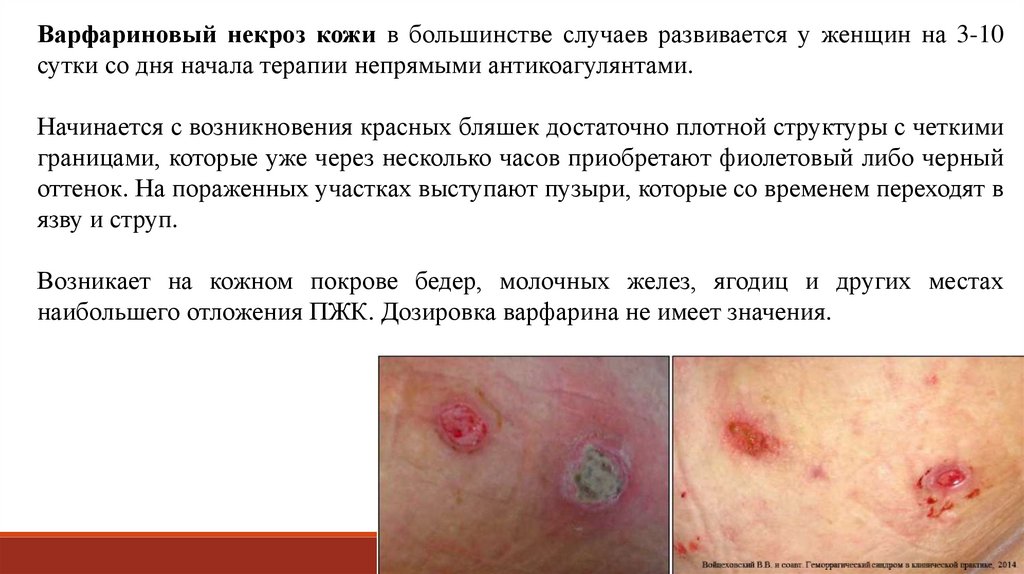
Варфариновый некроз кожи в большинстве случаев развивается у женщин на 3-10сутки со дня начала терапии непрямыми антикоагулянтами.
Начинается с возникновения красных бляшек достаточно плотной структуры с четкими
границами, которые уже через несколько часов приобретают фиолетовый либо черный
оттенок. На пораженных участках выступают пузыри, которые со временем переходят в
язву и струп.
Возникает на кожном покрове бедер, молочных желез, ягодиц и других местах
наибольшего отложения ПЖК. Дозировка варфарина не имеет значения.
75.
Если при повышенных цифрах МНО у больного отмечаются серьезныегеморрагические проявления, то он надлежит немедленной временной
госпитализации, отмене приема варфарина, введениям внутривенно
или внутрь витамина К. При больших кровотечениях – выполняются
также трансфузии свежезамороженной плазмы по 500 – 800 мл/сут.
76.
Передозировка прямых антикоагулянтовАнтикоагулянты прямого действия – лекарственные препараты, непосредственно влияющие на факторы
свертывания крови в сосудистом русле: гепарин; низкомолекулярные гепарины – эноксапарин-натрий
(клексан), надропарин кальций (фраксипарин), дельтапарин натрий (фрагмин) и др.; гепариноиды.
Побочные эффекты:
- кровотечения, особенно при почечной недостаточности, в связи с передозировкой (гематурия,
гемартрозы, кровотечения из ЖКТ (при язвенной болезни желудка и двенадцатиперстной кишки), при
внутримышечных инъекциях и введении под кожу – гематомы)
- аллергические реакции в виде крапивницы, затрудненного дыхания, отека слизистой оболочки носа.
- иммунная тромбоцитопения (на гепарин влияет антигепариновый фактор тромбоцитов (фактор IV),
образуя комплекс гепарин-фактор IV, что может вызвать гепариновую иммунную тромбоцитопению
вследствие образования антител к этому комплексу (наиболее опасная форма тромбоза).
При передозировке гепарина: внутривенно протамина сульфат до 5 мл 1 %-го раствора медленно под
контролем пробы на скорость свертывания крови. На каждые 100 ЕД гепарина, которые необходимо
нейтрализовать, вводят 1 мг протамина сульфата.
77.
78. Смешанный тип кровоточивости
79.
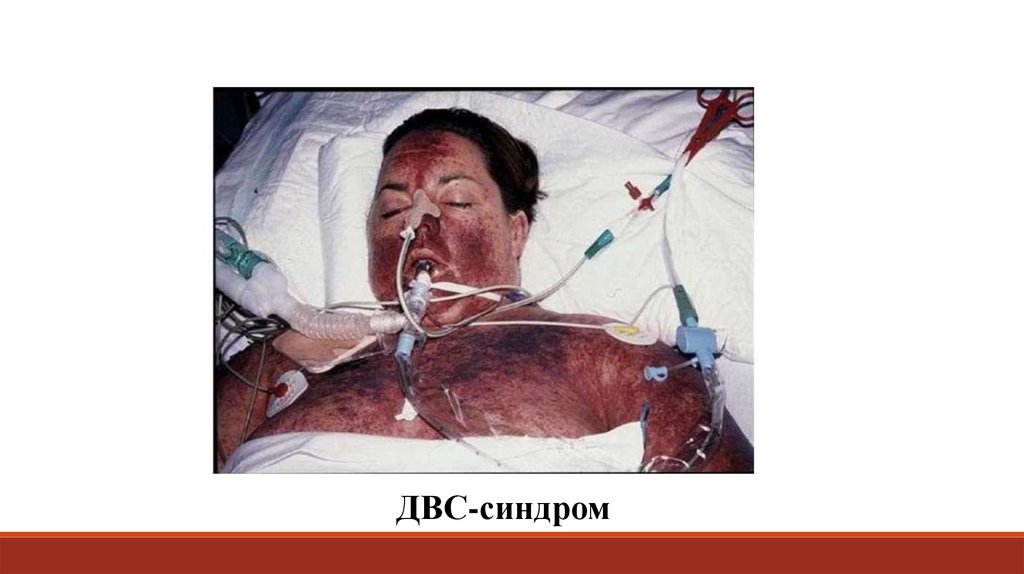
ДВС-синдром80.
Болезнь Виллебранда (БВ) - наиболее распространенная наследственнаякоагулопатия, обусловленная снижением количества или нарушением функции
фактора Виллебранда (vWF).
БВ – фенотипически гетерогенная коагулопатия с аутосомно-рецессивным или
аутосомно-доминантным типом наследования. Высокая вариабельность не только
клинических проявлений со значительным количеством легких и малосимптомных
форм, но и лабораторных показателей (FVIII, RIPA, vWF, vWF:Ag, vWF:СВ) при
БВ обусловлена генетическими мутациями в гене vWF. Поскольку природа
генетических дефектов разнообразна, заболевание характеризуется широким
клиническим полиморфизмом.
81.
82.
Классификация болезни ВиллебрандаТип заболевания
Частота, %
Характеристика
Тип 1
70
Частичный дефицит vWF; уровень FVIII нормальный или
сниженный пропорционально степени снижения vWF
Тип 2
2A
25
Качественные дефекты vWF
Снижение количества высокомолекулярных мультимеров
vWF вследствие повышенной чувствительности к протеазе
ADAMTS13
2B
Повышение аффинности vWF к рецепторам GPIb тромбоцитов, избыточное связывание vWF с тромбоцитами
2M
Снижение ристоцетин-кофакторной активности vWF при
нормальном содержании антигена vWF; нормальная
мультимерная структура vWF
2N
Тип 3
Cнижение аффинности к FVIII
Практически полное отсутствие vWF с низкой активностью FVIII
5
83.
Клинические признакиОсновное проявление БВ – геморрагический синдром микроциркуляторного/смешанного типов
спонтанного или посттравматического характера.
При БВ 1 и 2 типов преобладает микроциркуляторный тип кровоточивости: экхимозы,
кровотечения из слизистых (десневые, носовые, луночковые), меноррагии, кровотечения при
проведении хирургических вмешательств и инвазивных диагностических процедур.
При типах БВ, характеризующихся выраженным снижением уровня FVIII (2A, 2N, 3), часто
наблюдается смешанный (микроциркуляторно-гематомный) тип геморрагического синдрома.
БВ 3 типа по своим проявлениям схожа с тяжелой формой гемофилии A ввиду почти полного
отсутствия vWF и, как следствие, FVIII. Несмотря на идентичность клинических проявлений, у
пациентов с БВ симптоматика менее выражена, чем у больных гемофилией, и всегда сочетается с
кровотечениями по микроциркуляторному типу.
Жизнеугрожающие кровотечения более характерны для БВ 3 типа.
84.
85.
Первым этапом лабораторной диагностики рекомендуется проведение коагулологического скрининга,включающего следующие показатели:
общий анализ крови с подсчетом количества тромбоцитов по Фонио;
исследование агрегации тромбоцитов с основными индукторами - ристоцетином, коллагеном, АДФ,
тромбином, адреналином;
определение АЧТВ, ПТИ, ТВ и фибриногена.
Для верификации диагноза БВ рекомендуется проведение второго этапа диагностики - специфических
коагулологических исследований:
соотношение vWF:RСo/vWF:Ag (обязательно);
FVIII:C (обязательно),
уровень RIPA в двух концентрациях ристомицина (для уточнения типа БВ);
анализ мультимеров vWF (в неясных случаях).
Специфические тесты могут быть выполнены в специализированных центрах по лечению коагулопатий.
86.
87.
Для лечения и профилактики БВ рекомендовано применение вирусинактивированных концентратов FVIII, содержащих vWF и/или FVIII+vWF сраспределением мультимеров, максимально приближенным к таковому в
нормальной плазме человека.
88. Васкулитно-пурпурный тип
89.
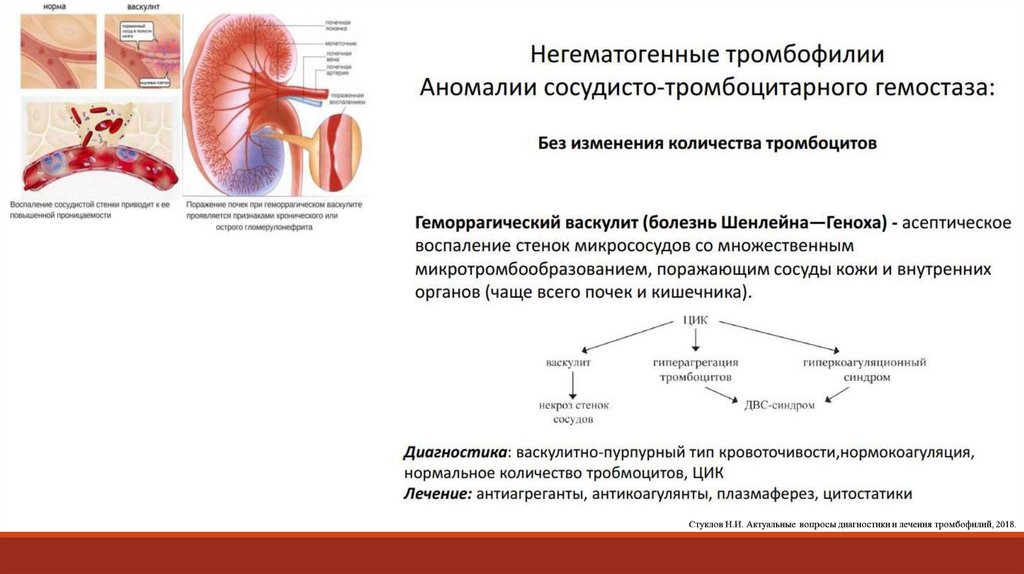
90. Геморрагический васкулит(пурпура Шенляйна-Геноха) – системное сосудистое заболевание с IgA-депозитами с преимущественным
ГЕМОРРАГИЧЕСКИЙ ВАСКУЛИТ(ПУРПУРА ШЕНЛЯЙНА-ГЕНОХА)
– системное сосудистое заболевание с IgA-депозитами с преимущественным
поражением капилляров, артериол и венул кожи, суставов, брюшной полости и
почек.
Болеют дети и подростки до 14 лет. Развивается после инфекции (стрептококк),
введения вакцин и сывороток, в связи с лекарственной непереносимостью
и переохлаждением.
91.
92.
93. Клиника:- пурпура (геморрагические высыпания на коже конечностей, ягодиц, реже на туловище; при надавливании не исчезает);-
Клиника:- пурпура (геморрагические высыпания на коже конечностей, ягодиц, реже
на туловище; при надавливании не исчезает);
- артралгии и/или артрит (симметричное поражение крупных суставов);
- абдоминальный синдром (изъязвления, боли, рвота и диарея кровью,
перфорации, перитонит).
94.
Кожный синдром проявляется симметрично расположенной папулезно-геморрагической сыпью на нижнихконечностях, ягодицах, реже туловище, которая, в тяжелых случаях, осложняется центральными некрозами. При
надавливании элементы сыпи не исчезают.
Суставной синдром возникает вместе с кожным синдромом или спустя несколько часов или дней после него в
виде болей в крупных суставах (коленных, локтевых и др.).
Абдоминалный синдром чаще наблюдается в детском возрасте. Основной признак – сильные боли в животе,
постоянные или схваткообразные. Эти боли могут сочетаться с кровавой рвотой, миленой или свежей кровью в
кале, а также ложными позывами с частым стулом или, наоборот, с его задержкой. При обильных кровотечениях
развиваются коллапс и острая постгеморрагическая анемия. Дифференцировать абдоминальный синдром следует с
острой кишечной непроходимостью, аппендицитом, прободной язвой кишечника, перекрутом кисты яичника.
Почечный синдром чаще развивается по типу острого или хронического гломерулонефрита – с микро- или
макрогематурией,
протеинурией, цилиндрурией. Возможен нефротический синдром. Значительно реже
выявляются сосудистое поражение легких, дающее иногда смертельное легочное кровотечение, и церебральная
форма болезни с головными болями, менингеальными симптомами, эпилептиформными припадками.
95.
96.
97.
98.
99.
100. Диагностические критерии, предложенные ACR (1990)1. Пальпируемая геморрагическая сыпь.Слегка возвышающаяся над поверхностью
Диагностические критерии, предложенные ACR (1990)1. Пальпируемая геморрагическая сыпь.
Слегка возвышающаяся над поверхностью кожи, пальпируемая геморрагическая сыпь, не связанная с
тромбоцитопенией.
2. Начало болезни в возрасте до 20 лет.
3. Ишемия кишечника.
Диффузные боли в животе, усиливающиеся после еды, диарея геморрагического характера.
4. Обнаружение скоплений гранулоцитов в стенках артериол и венул.
При наличии двух из четырех вышеперечисленных критериев можно поставить диагноз
геморрагический васкулит Шенляйн-Геноха.
101.
102.
Лечение.1. Госпитализация и соблюдение постельного режима не менее 3 недель
обязательны.
2. Избегать охлаждения, физ. нагрузок, стресса.
3. Из рациона исключают индивидуально непереносимые виды пищи, а также
какао, кофе, шоколад, цитрусовые, свежие ягоды (земляника, клубника и
др.) и блюда из них.
4. Отказ от приема антибиотиков, устранение контакта с аллергенами и
медикаментами. Противопоказаны пробы с бактериальными антигенами –
туберкулиновые, Бюрне и др.
5. Больные подлежат диспансеризации в гематологическом кабинете.
103. - антикоагулянты (гепарин 25-30 тыс ЕД/сут.);- антиагреганты (дипиридамол 300 мг/сут., пентоксифиллин 600 мг/сут.);- СЗП
- антикоагулянты (гепарин 25-30 тыс ЕД/сут.);- антиагреганты (дипиридамол 300 мг/сут., пентоксифиллин 600 мг/сут.);
- СЗП (300-400 мл е/д + гепарин – 3-4 дня);
- ГКС (преднизолон 1,5-2 мг/кг/сут. при абдоминальном синдроме);
- цитостатики при нефротическом варианте гломерулонефрита (азатиоприн,
циклофосфан
1-2 мг/кг/сут);
- НПВС (при артралгиях и поражении кожи – диклофенак 150 мг/сут.);
- аминохинолоновые производные (при легких формах - делагил – 0,25-0,5 г/сут.);
- экстракорпоральная терапия (плазмаферез).
104. Ангиоматозный тип
105.
106.
ГЕМОРРАГИЧЕСКИЕ ЗАБОЛЕВАНИЯ, ОБУСЛОВЛЕННЫЕ ПАТОЛОГИЕЙ СОСУДИСТОЙСТЕНКИ
Наследственная геморрагическая телеангиэктазия (болезнь Рандю-Ослера)
Характерно очаговое истончение стенок малых кровеносных сосудов и аневризматическое
расширение их просвета с образованием мелких узловатых или паукообразных сосудистых
высыпаний, которые легко кровоточат. В аномальных сосудах нарушен локальный гемостаз, в
частности адгезия и агрегация тромбоцитов и образование стабилизированного фибрина, в
результате чего наблюдаются обильные кровотечения, которые трудно поддаются остановке.
Патогенез. Наследуется болезнь по аутосомно-доминантному типу. Кровоточивость связана
как с малой резистентностью и легкой ранимостью сосудистой стенки в локусах ангиэктазии,
так и с очень слабой стимуляцией в этих участках адгезии и агрегации тромбоцитов, а также
локального свертывания крови.
107.
108.
У. Ослер выделял три типа телеангиэктазий:1. Ранний в виде небольших неправильной формы мелких
сосудистых образований-пятнышек.
2. Промежуточный в виде небольших сосудистых «паучков».
3. Узловатый тип, имеющий вид ярко красных круглых или
овальных возвышающихся узелков диаметром 3-7 мм.
109.
Клиника.Телеангиэктазы могут образовываться и кровить на всех слизистых оболочках – в зеве, гортани, бронхах и на
всем протяжении ЖКТ, а также в мочевых путях и во влагалище.
Чаще всего геморрагические явления начинаются с обильных и рецидивирующих носовых кровотечений.
Легочное, желудочно-кишечное кровотечение и др. при болезни Рандю-Ослера требуют исключения всех других
причин кровотечений.
Возможно развитие кровоизлияния в головной и спинной мозг, его оболочки и во внутренние органы. В этих
случаях врожденная неполноценность сосудов чаще всего проявляется развитием в органах артериовенозных
аневризм, которые чаще всего локализуются в легких и протекают с одышкой, цианотически-красным цветом
лица, инъекцией сосудов склер, снижением оксигенации крови. Реже артериовенозные аневризмы выявляются
при болезни Ослера в печени, почках, селезенке. Длительно существующий ангиоматоз ведет к развитию тяжелых
изменений в органах – к легочно-сердечной недостаточности, циррозу печени, хронической печеночной
недостаточности и др. Наиболее частым осложнением болезни Рандю-Ослера является железодефицитная анемия.
110.
111.
112.
113.
114.
Диагностика болезни Рандю-Ослера основана на выявлении телеангиэктазов на коже и слизистыхоболочках и артериовенозных шунтов с помощью всех доступных физикальных и инструментальных
методик. Важен учет семейного анамнеза.
Лечение. Больным болезнью Рандю-Ослера противопоказан прием аспирина и других нестероидных
противовоспалительных
препаратов,
нарушающих
функцию
тромбоцитов
и
усиливающих
кровоточивость. Усиление кровоточивости наблюдается также при приеме острой пищи, алкоголя.
Для остановки носовых кровотечений используются орошения полости носа 5–8% аминокапроновой
кислотой (в смеси с тромбином или без него).
К хирургическому лечению – иссечению участков слизистой оболочки с расположенными в них
телеангиэктазами прибегают при частых и очень обильных желудочно-кишечных, носовых, бронхолегочных и лоханочных кровотечениях, но эффект таких вмешательств бывает временным.
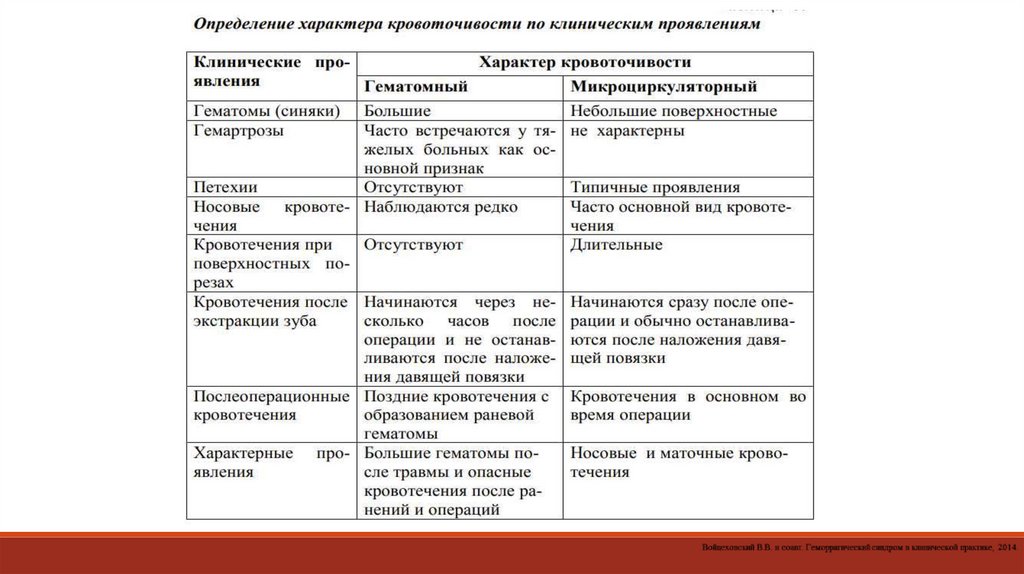
115. Дифференциально-диагностическое значение кровоточивости
Вид кровоточивостиВероятность ГД
Другие наиболее частые причины повышенной кровоточивости
Спонтанные геморрагии
Носовые кровотечения
±
Местный дефект (ринит, дефект сосудов сплетения Киссельбаха), АГ
Десневые кровотечения
±
Пародонтоз
Меноррагии
±
Полипы, эрозии, опухоли гениталий
Гематурия
±
Местное повреждение урологического тракта (камни, опухоли,
полипы)
Желудочно-кишечные кровотечения
±
Язвенные поражения слизистой, опухоли желудочно-кишечного тракта
Кровохарканье
±
Тромбоэмболия легочной артерии, рак легких или туберкулез
Реакция на травму
Петехии, экхимозы
++
Глубокие подкожные гематомы ("синяки")
++
Гемартрозы
++
Длительные или обильные кровотечения:из порезов
++
при удалении зубов
++
при тонзиллэктомии
++
во время или после операции
++
Пупочные кровотечения (при рождении)
++
Примечание. ± – ГД мало вероятен; ++ – ГД вероятен.
116. Наиболее частые причины геморрагий при отдельных патологических состояниях
Вид патологииНаиболее вероятные причины кровоточивости
Опухоли
ДВС-синдром, тромбоцитопения (метастатическое поражение костного мозга - КМ), прорастание сосудов
Инфекционные заболевания
ДВС-синдром, тромбоцитопения (угнетение КМ; аутоиммунное поражение тромбоцитов)
Острый лейкоз
ДВС-синдром, тромбоцитопения (поражение КМ)
Шоковое состояние
ДВС-синдром
Состояние после экстракорпорального кровообращения и оксигенации
Тромбоцитопения (отложение тромбоцитов на диализных мембранах)
Побочная реакция на прием лекарства
Васкулиты (гиперчувствительность), тромбоцитопения (угнетение КМ, повышенное разрушение
тромбоцитов иммунными механизмами), тромбоцитопатия
Хронический алкоголизм
Тромбоцитопения
Заболевания печени, сопровождающиеся печеночно-клеточной недостаточностью Снижение синтеза факторов свертывания крови в гепатоцитах, тромбоцитопения (при гиперспленизме)
Обтурационная желтуха
Снижение синтеза факторов протромбинового комплекса (II, VII, IX, X) из-за дефицита витамина К
Хронический миелопролиферативный синдром (болезнь Вакеза, хр. миелолейкоз)
Тромбоцитемия
Миеломная болезнь
Сосудистые нарушения, тромбоцитопатия, тромбоцитопения
Макроглобулинемия Вальденстрема
Сосудистые нарушения, тромбоцитопатия, тромбоцитопения
Криоглобулинемия
Сосудистые нарушения, тромбоцитопатия, тромбоцитопения
Амилоидоз
Сосудистые нарушения, тромбоцитопатия, тромбоцитопения
Гипотиреоз
Тромбоцитопения (гипоплазия КМ)
Уремия
Тромбоцитопения (гипоплазия КМ), тромбоцитопатия
Гемотрансфузии
Тромбоцитопения в результате иммунной аллергической реакции, при разведении большим количеством
"старой" крови, не содержащей тромбоцитов, ДВС-синдром
Коллагенозы (системная красная волчанка, ревматоидный артрит, дерматомиозит
и др.)
Тромбоцитопения (повышенное разрушение в результате иммунных механизмов), ингибиторная
гемофилия(антитела к какому-либо фактору свертывания), васкулит
117. Тромбофилический синдром
118.
119.
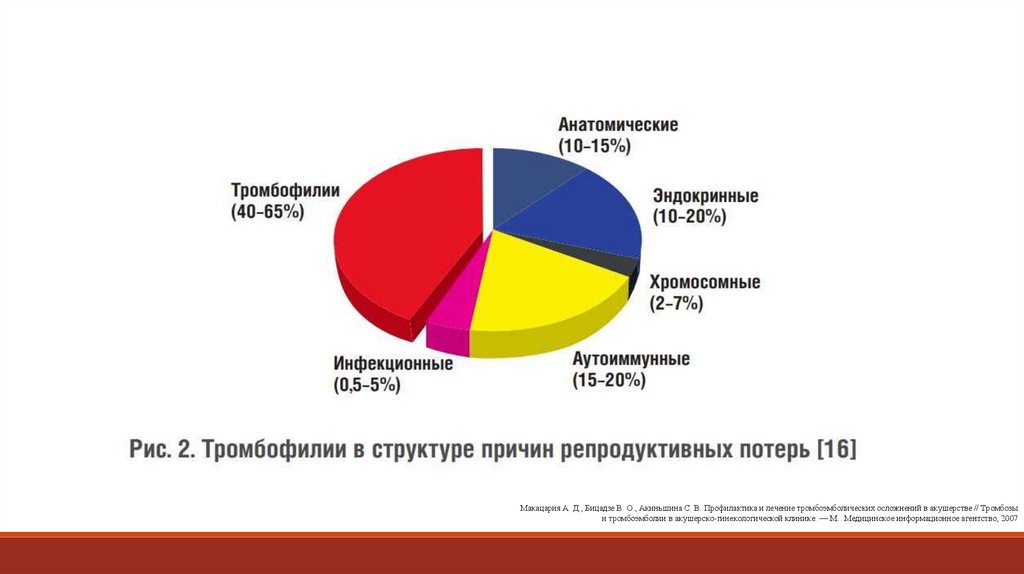
Стуклов Н.И. Актуальные вопросы диагностики и лечения тромбофилий, 2018.120.
Макацария А. Д., Бицадзе В. О., Акиньшина С. В. Профилактика и лечение тромбоэмболических осложнений в акушерстве // Тромбозыи тромбоэмболии в акушерско-гинекологической клинике. — М.: Медицинское информационное агентство, 2007
121.
122.
Самойленко В.В. Тромбофилия: диагностика и лечение. Место и роль лабораторной диагностики, 2018.123.
Зотова И.В., Затейщиков Д.А. Наследственная тромбофилия и венозные тромбоэмболические осложнения: правила тестирования вклинической практике. Российский кардиологический журнал. 2020
124.
125.
126.
127.
128.
129.
130.
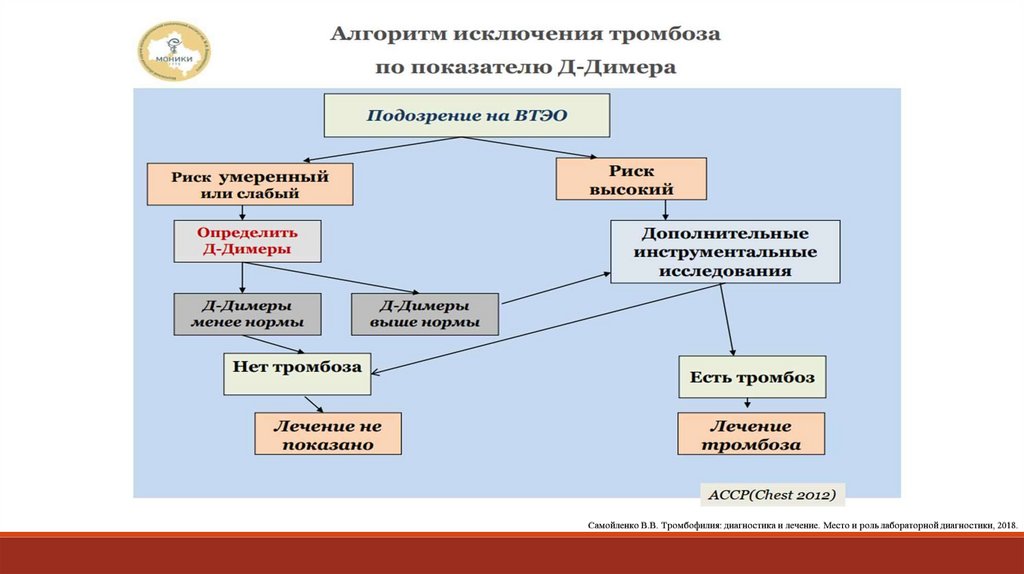
131. Причины повышения D-димера:
Онкопатология;Воспалительные процессы, инфекция, некроз тканей;
Заболевания печени;
Беременность;
Фибрилляция предсердий;
Послеоперационный период;
Травмы, ожоги, гематомы;
Старческий возраст (>80 лет);
Расслоение аорты;
Инсульт;
Венозный тромбоз;
Длительное нахождение в стационаре.
132.
133.
134.
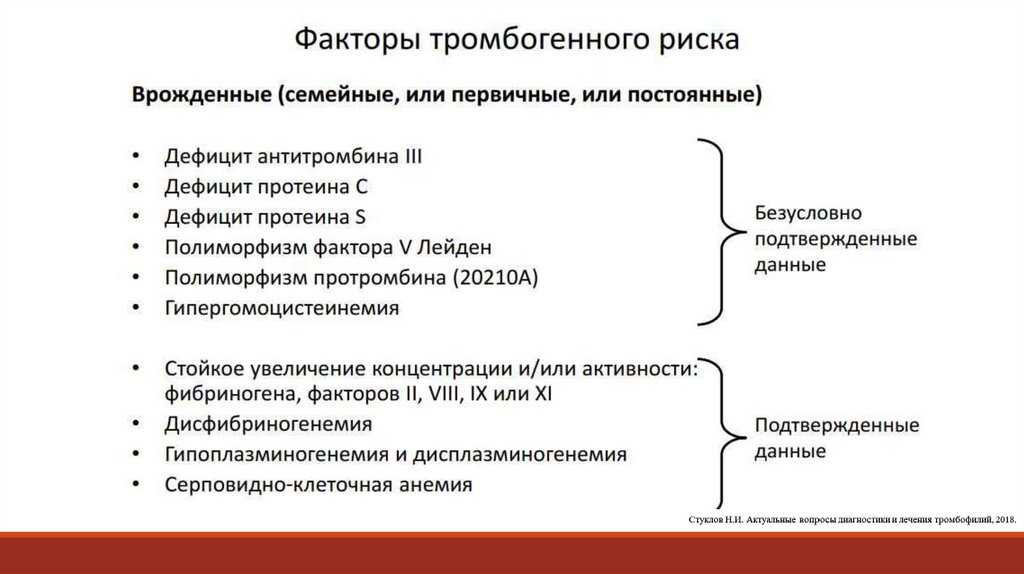
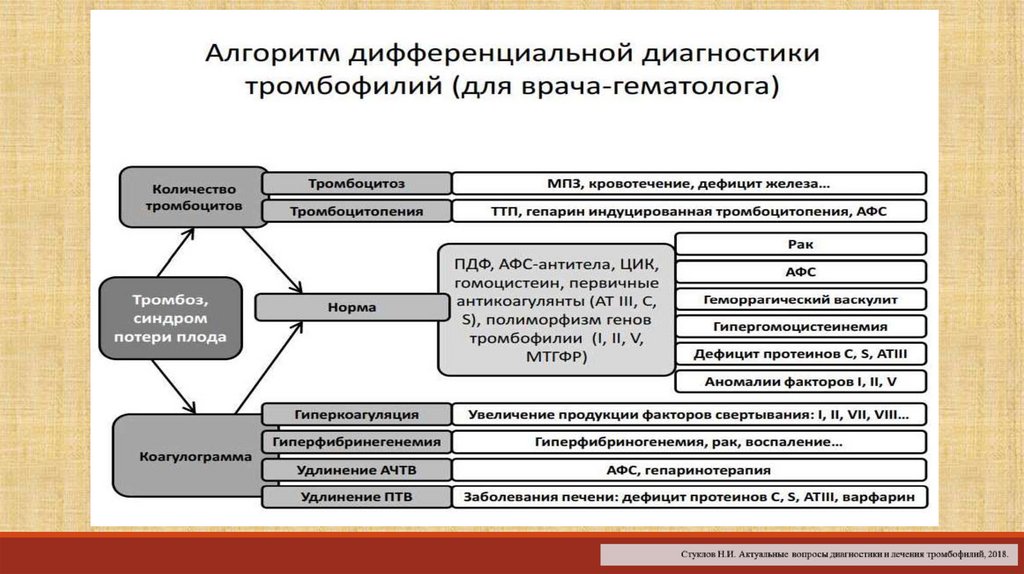
Показания для проведения генетического исследования с целью верификациинаследственной тромбофилии:
1. пациенты с ТЭЛА, независимо от пола или возраста;
2. мужчины< 70 лет с рецидивирующими тромбозами глубоких вен;
3. женщины< 50 лет с рецидивирующими тромбозами глубоких вен;
4. женщины детородного возраста с семейным анамнезом тромбозов или тромбофилии;
5. пациенты с недавно диагностированным антифосфолипидным с венозным или артериальным тромбозом;
6. возраст ≤ 50 лет с тромбозом;
7. тромбоз с нетипичной локализацией (мезентериальный, селезѐночный, портальный, печеночный,
мозговой)
8. родственник первой степени с тромбозом (особенно, если в возрасте ≤ 50 лет)
9. тромбоз во время беременности
10.тромбоз на фоне приема пероральных противозачаточных препаратов
http://www.hematology.ru/general/hemostasis/publication/002.pdf
135.
Перечень обязательных исследований:1. Общий анализ крови;
2. АЧТВ (АЧТВ+АФ ПВК);
3. Тромбиновое время;
4. Протромбиновый комплекс (протромбин+МНО);
5. Фибриноген;
6. Антитромбин II;
7. I Хагеман-зависимый фибринолиз (ХЗФ);
8. Д-димер;
9. Агрегация тромбоцитов (с универсальным индуктором агрегации);
10. Протеин С;
11. Определение волчаночного антикоагулянта.