



 анемии различают:")

:")
:")










:")







 характеризуются:")






:")















")











")


:")


")




 Медицина
МедицинаПохожие презентации:










Дифференциальный диагноз заболеваний крови
1.
Дифференциальный диагноззаболеваний крови
Кафедра внутренних болезней педиатрического факультета
КрасГМУ им. проф. В.Ф.Войно-Ясенецкого
проф. Н.А.Борисенко
2. План лекции:
Основные клинические проявлениязаболеваний крови (анемии, острые и хрон.
лейкозы, миеломная болезнь и др.);
Анемический синдром при заболеваниях
крови;
Данные дополнительных методов
исследований;
Дифференциальная диагностика;
Принципы лечения.
3. Цель лекции:
Оценить клинические проявлениязаболеваний, при которых наблюдается
анемический синдром;
Оценить результаты анализа крови и
данных дополнительных методов
исследований (рентгенологические,
цитохимические, иммунологически и др);
Уметь поставить диагноз и назначить
лечение.
4.
Ведущим клиническим признакомзаболеваний крови является -анемический
синдром, (анемия).
Характеризуется:
снижением гемоглобина (Hb) и эритроцитов в
единице объема крови, что приводит к
кислородному голоданию тканей (гипоксии) и
последующим клиническим проявлениям.
5. По степени насыщения гемоглобином (цветовому показателю) анемии различают:
Гипохромные (ЦП < 0,85)Нормохромные (ЦП 0,9 -1,05)
Гиперхромные (ЦП > 1,1)
По степени тяжести анемии различают:
Легкой степени (Hb 90-110 г/л)
Средней степени (Hb 70-90 г/л)
Тяжелой степени (Hb < 70 г/л)
6. Клинические признаки анемии:
Головная боль, головокружение, снижениепамяти, обморочные состояния;
Парестезии, боли в мышцах, сердце,
тахикардия и др.;
Бледность кожных покровов и слизистых
оболочек и др.
7. Заболевания крови, при которых наблюдается анемический синдром (анемия):
ЖДА, В12-дефицитная, апластическая,гемолитическая анемии;
Острые и хронические лейкозы;
Миеломная болезнь;
Геморрагические диатезы (аутоиммунная
тромбоцитопения, тромбоцитопатия) и др.
8. Наиболее часто встречается железодифицитная анемия (ЖДА):
ЖДА- это патологическое состояние, в основекоторого лежит дефицит Fе в организме.
Норма железа 3-4 гр. Fe связано с белками.
Наиболее важный из них Нb (состоит из гема и
белка глобина).
Основным белком, не имеющим гема, но
содержащим железо является Ферритин.
Другим белком запаса железа является –
Гемосидерин.
9. Причины ЖДА:
У детей:Недоношенных;
При многоплодной беременности;
При несбалансированном питании;
У детей, родившихся от матери с ЖДА;
При недостатке в питании
железосодержащих продуктов.
У женщин:
Обильные маточные кровотечения
(дисфункция яичников, частые аборты и
роды).
10. Клинические признаки ЖДА:
Сидеропенический синдром:Ломкость ногтей, их поперечная,
продольная исчерченность;
Выпадение, ранняя седина волос, потеря их
блеска;
Извращение вкуса, обоняния (нравятся
запахи лаков, красок, бензина, ацетона и
др.);
Сухость кожи, мышечная слабость,
дисфагия.
11. Картина крови при ЖДА
Гипохромный характеранемии, анизоцитоз,
пойкилоцитоз
12. В анализе крови наблюдается:
Снижение Нb, эритроцитов, анизоцитоз,пойкилоцитоз;
Снижение содержания СЖ (норма 12-30
мкмоль/л);
Увеличение ОЖСС (норма 48-68
мкмоль/л);
Снижение ферритина в сыворотке крови.
13. Лечение ЖДА:
Устранение причин дефицита железа.Патогенетическое лечение:
• диета, богатая железом;
• ферротерапия: ферроплекс, фенюльс,
феррамид, ферроцирон (внутрь) совместно с
аскорбиновой кислотой и др.;
В условиях Сибири можно употреблять
Кожановскую минеральную воду (в 1 литре30мг железа);
Гемотрансфузии только по жизненным
показаниям (Нв < 40 г/л).
14. Профилактика ЖДА:
Проводится в группах риска:• беременным
• детям, находящимся на искусственном
вскармливании
• подросткам-девочкам
Устраняются этиологические причины
заболевания;
Назначают препараты железа в малых
дозах: железо вводится в продукты питания
(детские смеси, железистые мин.воды).
15. Дифференциальная диагностика с В12 – дефицитной анемией
Дефицит витамина В12 может наступить при:Атрофическом гастрите (наблюдается снижение или
отсутствие гастромукопротеина - внутреннего фактора
Кастла);
Резекции желудка или тонкой кишки, где происходит
всасывание витамина В12;
Гельминтозах (широкий лентец), при которых имеет
место потребление витамина В12 гельминтами;
Раке желудка, операциях на тонком кишечнике,
энтеритах;
Вегетарианской диете (недостаток поступления
витамина В12 с пищей).
16. Клинические симптомы B12 дефицитной анемии:
Слабость, головокружение, снижение памяти;Атрофический гастрит (ахлоргидрия), язык
Хантера;
Фуникулярный миелоз (парастезии,
неустойчивая походка, повышение коленных
рефлексов, положительный симптом
Бабинского);
Умеренная гепато- и спленомегалия.
17. Картина крови при В12 – дефицитной анемии:
Гиперхромная (ЦП больше 1,0),лейкопения, тромбоцитопения
Макроцитоз, мегалоцитоз,
тельца Жолли, кольца
Кебота (остатки ядерных
образований).
18. Лечение и профилактика:
Этиологическое лечение(дегельминтизация, лечение гастрита и
др.);
Витамин В12 1000 мкг в сутки до
нормализации Нb и эритроцитов;
Поддерживающая терапия витамином В12
пожизненно: 1 раз в две недели по 500 мкг
или 1 раз в месяц по 500 мкг;
Гемотрансфузия – только тяжелым больным;
Профилактическое лечение 1-2 раза в год
по 500 мкг в течение 10-15 дней.
19. Гемолитические анемии (ГА):
Жизнь эритроцитов длится 120 дней. ПриГА продолжительность жизни эритроцитов
укорачивается.
Гемолиз может возникать при
разнообразных патологических процессах
и протекать постоянно или в виде кризов.
Он может быть внутрисосудистым и
внесосудистым.
20. Гемолитические анемии различают:
Наследственные ГА возникают в результате нарушений:Структуры мембраны эритроцита;
Структуры или синтеза гемоглобина;
Дефицита ферментов эритроцита;
Приобретенные ГА:
Аутоиммунные (образование антител к собственным
эритроцитам);
Лекарственные: сульфаниламиды, антибиотики (пенициллин,
цефалоспорины большие дозы, допегид);
Переливание несовместимой (резус или групповой) крови;
Идиопатические (причина не ясна).
21. Клиника наследственной ГА:
Длительно с раннего детства нарушениекостеобразования (деформация челюстей с
неправильным расположением зубов, высокое
небо, выступающий лоб);
Желтуха (за счет увеличения непрямого
билирубина);
Увеличение селезенки и печени (при
прогрессировании заболевания);
Склонность к образованию камней в желчном
пузыре.
22. Картина крови при ГА:
Микросфероцитоз,ретикулоцитоз;
Микросферодитарная
ГА (значительное
уменьшение диаметра
эритроцитов).
23. Наследственная овалоклеточная ГА:
24. Наследственная ГА – акантоцитоз:
25. Наследственная серповидноклеточная ГА:
26. Лечение гемолитической анемии:
Терапию начинают с назначения преднизолона (60 мгв день для взрослых). При прогрессировании доза
может быть увеличена до 100 и более мг;
При неэффективности глюкокортикостероидов –
спленэктомия;
По жизненным показаниям и неэффективности
спленэктомии назначают иммунодепрессанты
(циклофосфан, азатиоприн, метотрексит и др);
При тяжелом анемическом синдроме переливают
эритромассу (отмытые, лучше замороженные
эритроциты). Переливать свежие эритроциты не
рекомендуется, усиливается гемолиз.
27. Гипо- и апластические анемии (ГА и АА) характеризуются:
Депрессией одного или трех ростковкроветворения (эритроидного,
миелоидного, мегакариоцитарного), с
последующим развитием панцитопении.
28. Этиология:
Идиопатическая (причина не известна),Врожденная (наследуется по аутосомно
рецессивному типу);
Приобретенная (лекарственные средства:
левомицетин, макролиды, НПВС, сульфаниламиды,
некоторые противотуберкулезные средства, и др.);
Химические вещества (нитроэмали, лаки,
пестициды, применяемые в быту, с/х);
Инфекции (цитомегаловирусы, постгепатитные),
Ионизирующая радиация.
29. Клиника:
Наследственные протекают незаметно,медленно,
Приобретенные – как острое заболевание:
лихорадка, некротическая ангина,
десневые и носовые кровотечения,
геморрагии на коже, печень и селезенка не
увеличены.
30. Картина крови при ГА и АА:
Панцитопения (различной степенивыраженности);
Анемия (нормохромного типа);
Лейкопения с гранулоцитопенией;
Относительный лимфоцитоз;
Тромбоцитопения (до единичных);
Уменьшено кол-во ретикулоцитов;
СЖ в норме.
31. Апластическая анемия: опустошенный костный мозг, единичные клетки костного мозгового кроветворения
32. Апластическая анемия: полное опустошение костно-мозгового кроветворения
33. Лечение гипо- и апластических анемий:
Заместительная терапия (эритромасса,тромбомасса, свежезамороженная плазма);
ГКС (преднизолон 1-2 мг/кг веса в сутки в
течении 1-2 мес);
Спленэктомия;
Циклоспорин – А (сандиммун) в течение 5-6
мес;
Антилимфоцитарный глобулин,
Пересадка костного мозга от близких
родственников.
34. Дифференциальная диагностика с острым лейкозом (ОЛ):
ОЛ – опухолевое заболевание крови,при котором нарушается
дифференциация клеток крови до
зрелых форм. Их основная масса
представлена бластными клетками
35.
Периферическая кровь при ОЛ:Анемия (снижение Hb, эритроцитов, тромбоцитов);
Бластный клетки (1-5 – 99%);
Лейкемический провал;
Лейкопения (может быть умеренный лейкоцитоз)
менее 3,0·109/л
СОЭ часто ускорена.
В костном мозге:
Бластов > 5% (норма - 2%).
36.
Рис.1Тотальная метаплазия,
острый миелобластный
лейкоз
Рис.2
Острый промиелоцитарный
лейкоз, промиелоциты
разной величины,
имеют нежную структуру
37. Для уточнения варианта ОЛ проводятся исследования бластных клеток:
Цитохимическое(в них определяют ферменты):Миелопироксидазу
Липиды
Нафтилэстеразу
Гликоген
Хлорацетатэстеразу
38.
Цитогенетическое исследование:обнаруживают хромосомные перестройки, характерные
для разных видов ОЛ
Иммунологическое исследование
(фенотипирование):
на патологических клетках выявляют антигены (кластеры),
их обозначают СД (sidi) и номером. Для уточнения
вариантов ОЛ имеются соответствующие кластеры.
39. Клиника острого лейкоза:
Условно выделяют 3 стадии:начальная
развернутая
терминальная
Симптоматика полиморфна. Заболевание
начинается постепенно, иногда выявляется
случайно, протекает под маской других
заболеваний.
40. Симптомы ОЛ в начальной стадии:
Слабость, недомогание, снижениетрудоспособности, ангина, не поддающаяся
традиционному лечению;
Анемия неясной этиологии;
Изменения в полости рта (кровоточивость десен,
кровотечения из лунки зуба после его удаления);
Носовые кровотечения;
Стоматиты, пародонтоз;
Боли в позвоночнике, костях.
41. Клинические синдромы в развернутой стадии:
Гиперпластический (увеличение лимфоузлов,селезенки, печени, миндалин);
Геморрагический (различные кровотечения);
Анемический (снижение Нb, эритроцитов,
тромбоцитов)
Интоксикационный (повышение температуры,
слабость);
Бактериальных осложнений (абсцессы, менингит,
парапроктит, пневмонии).
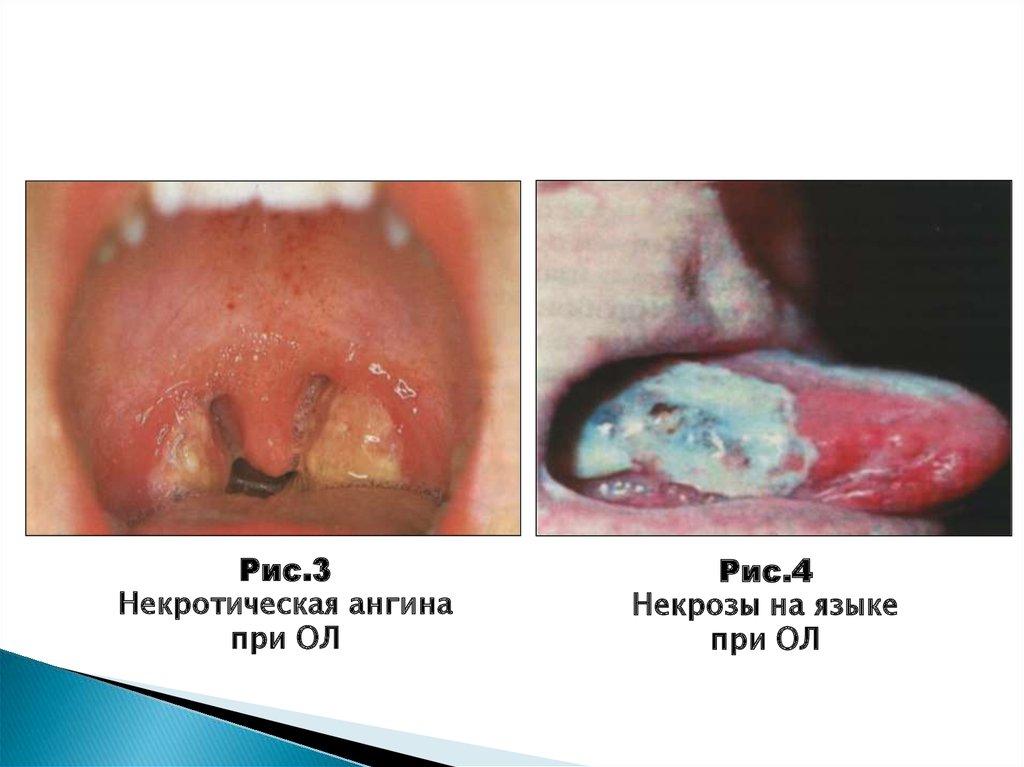
42.
Рис.3Некротическая ангина
при ОЛ
Рис.4
Некрозы на языке
при ОЛ
43.
Рис.5Геморрагии на голенях
при ОЛ
Рис.6
Геморрагии на лице
ребенка с ОЛ
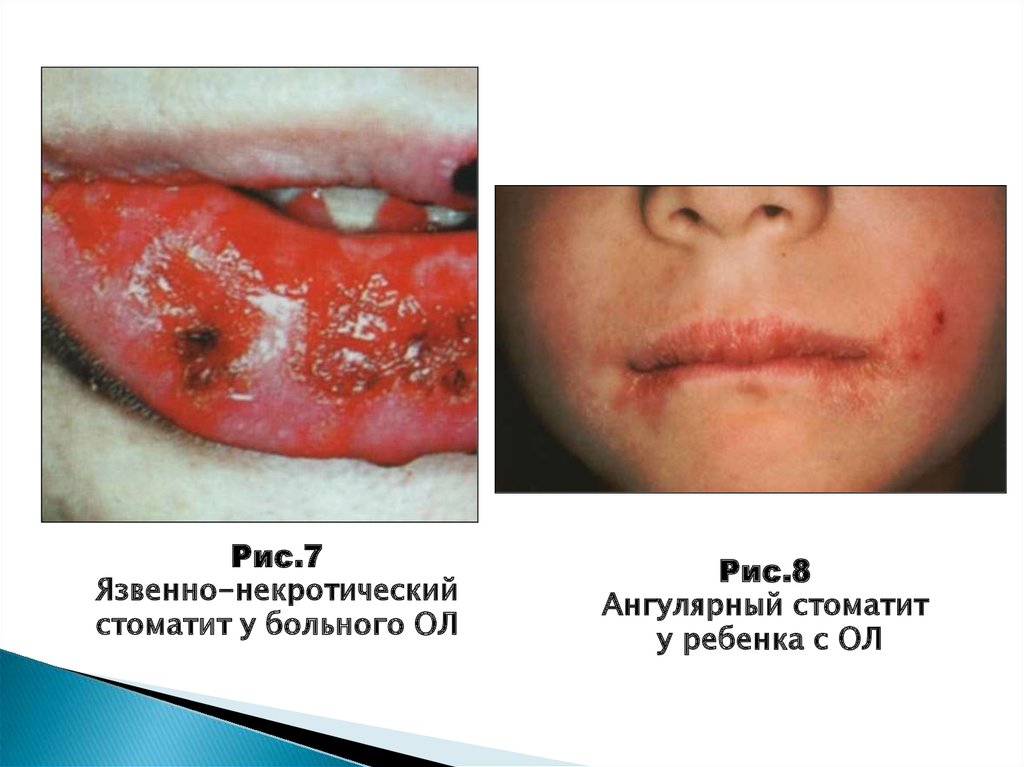
44.
Рис.7Язвенно-некротический
стоматит у больного ОЛ
Рис.8
Ангулярный стоматит
у ребенка с ОЛ
45. Нейролейкемия:
поражение нервной системы:метастазы опухолевых клеток в
мозг, оболочки, вещество мозга,
нервные стволы с соответствующей
клиникой (менингит, парезы,
полиневриты, боли в спине).
46. Принципы лечения:
Лечение начинать только после установленияточного диагноза острого лейкоза.
ПХТ-цитостатики (в виде программ), направлены
на разные этапы деления клеток. Строго
соблюдаются дозы и сроки лечения
Дезинтоксикационная терапия
Заместительная терапия (компоненты крови:
эритромасса, тромбомасса)
Антибиотики
Трансплантация костного мозга
47. Анемический синдром наблюдается при хронических лейкозах:
Анализ крови при хроническоммиелолейкозе (ХМЛ):
Нейтрофильный лейкоцитоз (100-300·109/л) со
сдвигом до миелоцитов, промиелоцитов
Увеличено количество базофилов и эозинофилов
(базофильно-эозинофильная ассоциация)
Выявляется Ph-хромосома («филадельфийская
хромосома»)
48. Клиника ХМЛ
начальнаяразвернутая
терминальная
3 стадии:
ОСНОВНЫЕ СИМПТОМЫ:
Начальная стадия (слабость, утомляемость, снижение
трудоспособности, субфебрильная температура)
Развернутая стадия (увеличение селезенки, печени,
лимфоузлов, геморрагический синдром, боли в
костях)
Терминальная стадия (ухудшение всех симптомов
заболевания, состояние тяжелое)
49.
Рис.9Резкое увеличение
селезенки у больного ХМЛ
Рис.10
Пародонтоз
у больного ХМЛ
50. Рис 11. Геморрагическая сыпь при ХМЛ
51. Лечение ХМЛ:
Гливек (высокоэффективный препарат, хорошо
переносится. Назначают 400-600мг в сутки)
Интерферон (Интрон Л)
Гидреа (гидроксимочевина)
Трансплантация костного мозга
Симптоматическая терапия
Все методы эффективны на ранних стадиях
заболевания.
Средняя продолжительность
жизни 4-6 лет.
52. Хронический лимфолейкоз (ХЛЛ)
- субстратом опухоли являются зрелые лимфоциты, страдаетиммунная система, часто склонность к инфекциям
(пневмонии, ОРВИ).
Анализ крови:
Лейкоцитоз (100-300·109/л)
Лимфоцитоз (60-90·109/л)
Клетки Боткина-Гумпрехта
начальная
развернутая
терминальная
В клинике выделяют 3 стадии:
В развернутой стадии – увеличение лимфоузлов, селезенки,
геморрагический синдром.
53. Лечение ХЛЛ:
В начале заболевания: сдерживающая терапия.При ухудшении состояния:
Лейкеран, Флюдарабин, Циклофосфан, Фотрин,
Дегранол;
Антибиотики (для борьбы с инфекцией);
Лучевая терапия (на лимфоузлы, селезенку).
54. Дифференциальный диагноз с миеломной болезнью:
Миеломная болезнь (множественнаямиелома, болезнь Рустицкого Калера)опухолевое заболевание состоящее из
плазматических (миеломных клеток) разной
степени зрелости.
55. Этиология миеломной болезни:
До конца не изучена;Имеет значение ионизирующая радиация;
Влияние химических факторов (бензол,
бензин, краски, лаки, промышленные яды
и др.).
56. Формы множественной миеломы:
Очаговая;Диффузная;
Диффузно-очаговая.
57. Клиника миеломной болезни полиморфна:
Анемический синдром (бледность кожных покровов ислизистых оболочек);
Гемморагический (кровоточивость десен, носовые и др.);
Болевой (боли в спине по типу остеохондроза);
Интоксикационный (лихорадка), обусловлен распадом
миеломных клеток;
Спонтанные переломы костей;
Компрессия позвонков (параплегии, парезы конечностей,
поражения глазодвигательного и лицевого нервов;
Наследственная ММ протекает медленно;
Приобретенная ММ - как острое заболевание (лихорадка,
десневые и носовые кровотечения, геморрагии на коже,
печень и селезенка увеличены незначительно) .
58. Анализ крови при миеломной болезни:
Анемия нормохромная (ЦП не более1,1 и не менее 0,7);Лейкоциты от 2-16*109 /л;
Тромбоциты (в начале норма,при прогрессировании
болезни их количество снижается);
Умеренная эозинофилия (6-11%);
Моноцитоз (13-20%);
СОЭ ускорена в течение длительного времени (50-80
мм/час);
Плазматические (миеломные клетки) в крови и особенно
в костном мозге (15 и более%).
59. Костный мозг: миеломные клетки крупных размеров, ядра клеток нежной структуры, расположены эксцентрично.
60. Биохимические анализы при ММ:
Увеличение общего белка в сывороткекрови (выявляется М-градиент
парапротеин) продуцируется
злокачественными клетками
(мононуклональный иммуноглобулин),
Увеличение мочевины, креатинина, мочевой
кислоты,
Гиперкальциемия (результат деструкции
костной ткани и ее остеолиза)
61.
Анализ мочи при ММ:Обнаруживается белок Бенс-Джонса (разной
степени выраженности);
Отмечается снижение клубочковой фильтрации.
R-графия позвоночника (обязательно):
компрессия тел позвонков (в отличие от МТS рака
отростки и дужки позвонков в процесс не
вовлекаются).
R-графия трубчатых костей, тазовых костей
ключицы:
дифф. остеопороз, очаги деструкции, переломы
костей
Для уточнения диагноза проводится ЯМР,
биопсия пораженных участков кости
62. R-графия черепа:
Отмечаются дефекты костной ткани (множественные участкиостеолиза):
63. Принципы лечения ММ:
ПХТ: цитостатики (алкеран, вепезид,циклофосфан, винкристин, адриамицин,
доксарубицин) в виде программ с
применением больших доз преднизолона.
Баркезамид (вейкейд) новый препарат
биологического действия обладает
противоопухолевой активностью (8 курсов).
Прогноз ММ в целом неблагоприятный.
64. Дифференциальную диагностику следует проводить с геморрагическими диатезами (ГД)
Связаны с системой гемостаза.Могут быть приобретенными и наследственными
ГД делят на следующие группы:
Болезни сосудов (вазопатии);
Изменение качественного состава тромбоцитов;
Нарушение свертываемости крови (коагулопатии);
ДВС – синдром;
Болезнь Рандю-Ослера.
65. Геморрагический васкулит
Классическая форма – болезнь Шейлейна-Геноха. Воснове лежит множественный
микротромбоваскулит. Поражаются сосуды кожи и
внутренних органов.
Преимущественно поражается эндотелий сосудов
мелкого калибра (капилляры): асептическое
воспаление, деструкция стенки сосудов, их
тромбирование, сосуды становятся
непроходимыми.
66. Геморрагическая сыпь на голенях и тыле кисти
67. Аутоиммунная тромбоцитопения (болезнь Верльгофа):
Наследственная;Приобретенная;
Болеют чаще женщины;
В патогенезе: появляются антитела к собственным
тромбоцитам.
Клиника: выражен геморрагический синдром
(«синяки» на коже разной формы и величины,
кровоточивость десен, носовые и маточные
кровотечения, может быть увеличение селезенки.
Анализ крови: количество тромбоцитов значительно
снижено.
68. Лечение тромбоцитопении:
ГКС (30-60 мг преднизолона, при тяжеломтечение доза увеличивается);
Гемотрансфузии (свежая кровь, свежая плазма);
Дицинон;
Спленэктомия (выздоровление в 85% случаев);
Если спленэктомия не эффективна –
цитостатики (азотиоприн, циклофосфан,
винкристин);
69. Тромбастения Гланцмана
Наследуется, встречается редко;Количество тромбоцитов в норме, но они
не полноценны, не способны к агрегации;
Чаще болеют девочки.
Клиника: тяжелый геморрагический диатез.
Лечение: эстрогены, симптоматическая
терапия.
70. Гемофилия (коагулопатия)
Наследственная патология, болеют мужчины,передается женщинами.
Различают:
Гемофилия А (недостаток фактора свертывания
VIII) 95%,
Гемофилия В (недостаток фактора IX) 5-6%,
Гемофилия С (недостаток фактора XI) 0-1%,
Синдром Стюарта-Пауэра (недостаток X
фактора),
Синдром Хамегана (недостаток VII фактора).
71. Клиника:
Гемартрозы, гематомы, наружныекровотечения, могут быть внутренние
кровотечения при малейших ушибах,
травмах, у детей – при прорезывании зубов;
В анализе крови: удлиняется время
свертывания крови до 20 мин и более
(норма 5-10 мин).
72. Лечение гемофилий
Заместительная терапия препаратами,содержащими факторы свертывания крови;
Антигемофильный глобулин (достигается
введением свежей плазмы или крови);
Криопреципитат (препарат ААГ);
Проведение оперативных вмешательств только в
стационаре;
Симптоматическая терапия.
73.
Ранняядиагностика, правильная
оценка анализов крови,
клинические проявления
заболевания, его своевременное
лечение определяют прогноз
болезни в целом.
74.
Благодарю завнимание!