































 Медицина
МедицинаПохожие презентации:










Клиника, критерии диагностики наследственных синдромов, идиопатического гемосидероза легких, саркоидоза легких
1.
Ставропольский государственный медицинский университетКафедра пропедевтики детских болезней
Клиника, критерии диагностики наследственных
синдромов ( Гудспадчера, Хаммена-Рича, Картагенера,
Марфана, Аэрса), идиопатического гемосидероза легких,
саркоидоза легких, пороков развития бронхов, легких и
легочных сосудов. Стандарты современной терапии и
реабилитация детей.
Выполнила:
клинический ординатор
Чотчаева Айшат Асхатовна
Ставрополь, 2019
2.
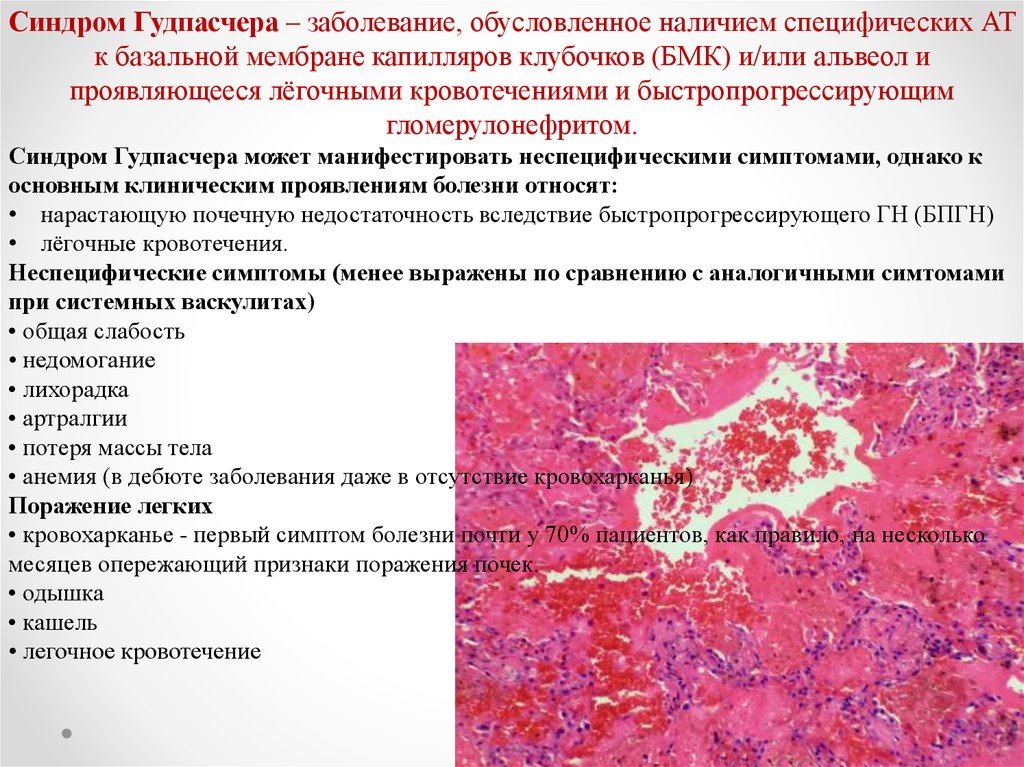
Синдром Гудпасчера – заболевание, обусловленное наличием специфических АТк базальной мембране капилляров клубочков (БМК) и/или альвеол и
проявляющееся лёгочными кровотечениями и быстропрогрессирующим
гломерулонефритом.
Синдром Гудпасчера может манифестировать неспецифическими симптомами, однако к
основным клиническим проявлениям болезни относят:
• нарастающую почечную недостаточность вследствие быстропрогрессирующего ГН (БПГН)
• лёгочные кровотечения.
Неспецифические симптомы (менее выражены по сравнению с аналогичными симтомами
при системных васкулитах)
• общая слабость
• недомогание
• лихорадка
• артралгии
• потеря массы тела
• анемия (в дебюте заболевания даже в отсутствие кровохарканья)
Поражение легких
• кровохарканье - первый симптом болезни почти у 70% пациентов, как правило, на несколько
месяцев опережающий признаки поражения почек.
• одышка
• кашель
• легочное кровотечение
3.
Лабораторная диагностика• Иммунологические исследования:
- Диагностическим признаком болезни является обнаружение антиБМК-антител в сыворотке крови с
помощью иммуноферментного анализа
- У 20-30% пациентов с синдромом Гудпасчера (анти-БМК-ГН) обнаруживают также АНЦА, в
основном с анти-МПО специфичностью. Однако двойная серопозитивность не изменяет ни прогноз,
ни течение болезни.
• Исследование мокроты: обнаружение сидерофагов
• Общий анализ крови:
- железодефицитная анемия
- лейкоцитоз
- увеличение СОЭ.
• Общий анализ мочи:
- протеинурия (субнефротического уровня)
- эритроцитурия (дисморфные эритроциты, эритроцитарные цилиндры)
• Биохимический анализ крови:
- повышение концентрации креатинина, мочевой кислоты, калия, дислипидемия (иногда в отсутствие
нефротического синдрома).
• Снижение СКФ (определенное в пробе Реберга по клиренсу креатинина и/или расчетными
методами CKD-EPI, MDRD; использование формулы Кокрофта-Голта не рекомендовано, в связи с
«завышением» СКФ на 20-30 мл)
• Биопсия почки
- В клубочках уже на ранней стадии болезни выявляют сегментарный некроз сосудистых петель,
массивную инфильтрацию лейкоцитами, разрывы БМК.
- При иммунофлюоресцентной микроскопии выявляется линейный тип свечения IgG на БМК в
сочетании с линейным свечением С3 компонента комплемента у 60 – 70% больных.
4.
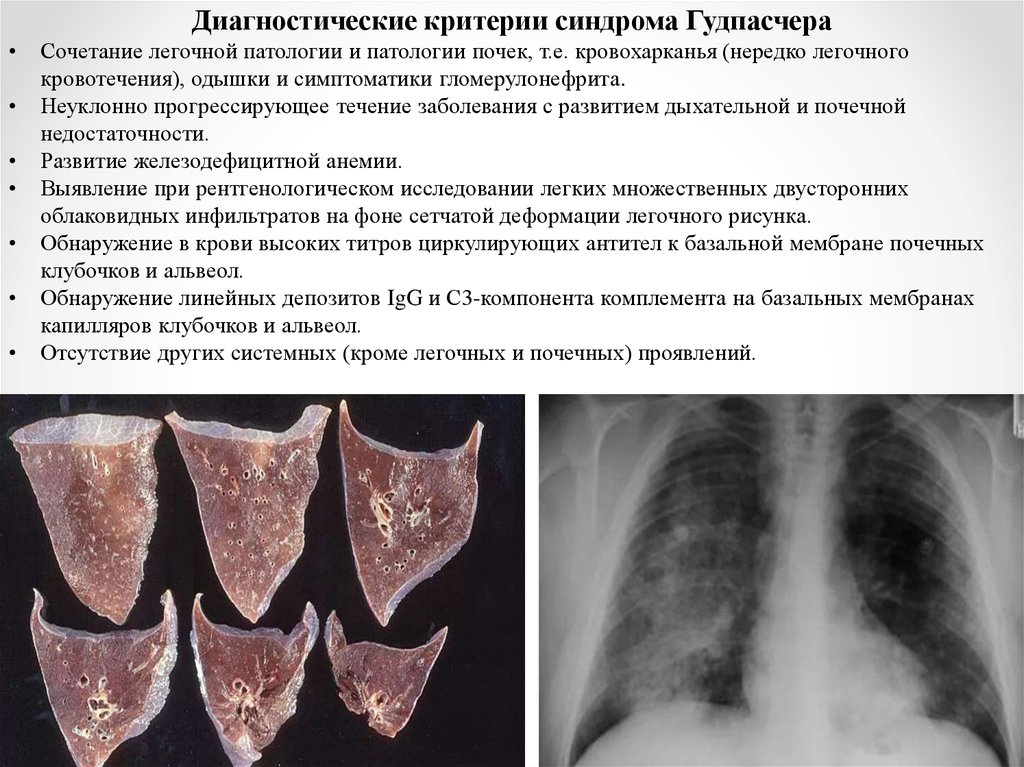
Диагностические критерии синдрома ГудпасчераСочетание легочной патологии и патологии почек, т.е. кровохарканья (нередко легочного
кровотечения), одышки и симптоматики гломерулонефрита.
Неуклонно прогрессирующее течение заболевания с развитием дыхательной и почечной
недостаточности.
Развитие железодефицитной анемии.
Выявление при рентгенологическом исследовании легких множественных двусторонних
облаковидных инфильтратов на фоне сетчатой деформации легочного рисунка.
Обнаружение в крови высоких титров циркулирующих антител к базальной мембране почечных
клубочков и альвеол.
Обнаружение линейных депозитов IgG и С3-компонента комплемента на базальных мембранах
капилляров клубочков и альвеол.
Отсутствие других системных (кроме легочных и почечных) проявлений.
5.
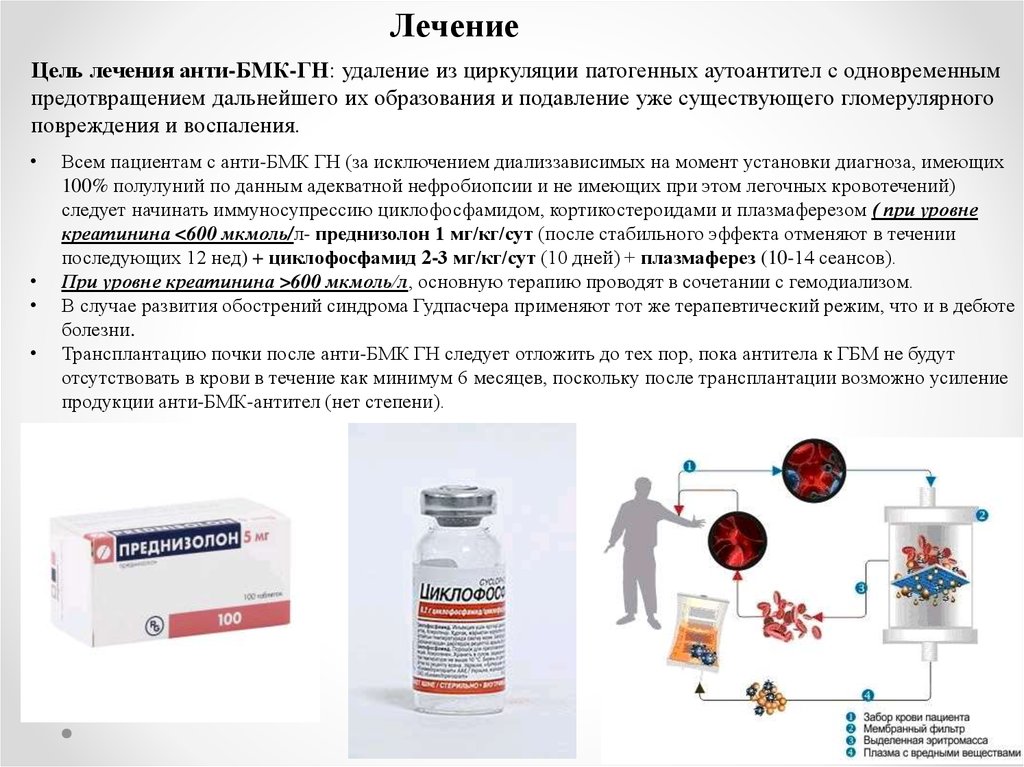
ЛечениеЦель лечения анти-БМК-ГН: удаление из циркуляции патогенных аутоантител с одновременным
предотвращением дальнейшего их образования и подавление уже существующего гломерулярного
повреждения и воспаления.
Всем пациентам с анти-БМК ГН (за исключением диализзависимых на момент установки диагноза, имеющих
100% полулуний по данным адекватной нефробиопсии и не имеющих при этом легочных кровотечений)
следует начинать иммуносупрессию циклофосфамидом, кортикостероидами и плазмаферезом ( при уровне
креатинина <600 мкмоль/л- преднизолон 1 мг/кг/сут (после стабильного эффекта отменяют в течении
последующих 12 нед) + циклофосфамид 2-3 мг/кг/сут (10 дней) + плазмаферез (10-14 сеансов).
При уровне креатинина >600 мкмоль/л, основную терапию проводят в сочетании с гемодиализом.
В случае развития обострений синдрома Гудпасчера применяют тот же терапевтический режим, что и в дебюте
болезни.
Трансплантацию почки после анти-БМК ГН следует отложить до тех пор, пока антитела к ГБМ не будут
отсутствовать в крови в течение как минимум 6 месяцев, поскольку после трансплантации возможно усиление
продукции анти-БМК-антител (нет степени).
6.
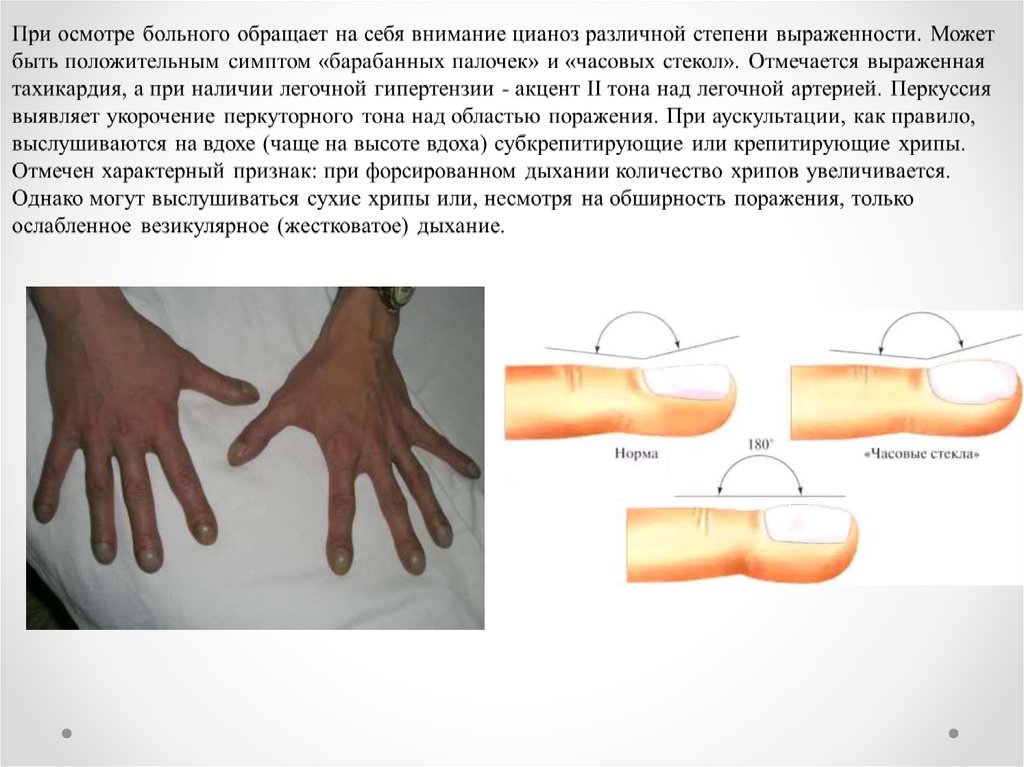
Идиопатическиц легочной фиброз ( с-м Хаммена-Рича) – особая формахронической прогрессирующей фиброзирующей интерстициальной пневмонии
неизвестной этиологии, которая возникает преимущественно у людей старшего
возраста, поражает только легкие и связана с гистологическим и / или
рентгенологическим паттерном обычной интерстициальной пневмонии (ОИП)
Классификация
Обычная интерстициальная пневмония (VIP) – муральная форма, смешанный фиброзновоспалительный инфильтрат.
ДИП (DIP) – клеточный паттерн
Острая интерстициальная пневмония (AIP) – болезнь Хаммена-Рича
Неспецифическая интерстициальная пневмония/фиброз (NIP) – неклассифицируемая
пневмония
Чаще встречается в возрасте старше 50 лет, у мужчин.
Клиническая картина:
• Кашель, как первый признак болезни, к которому затем присоединяется
прогрессирующая одышка.
• Невозможность глубокого вдоха
• Боли в грудной клетке (чаще под нижними углами лопаток)
• Повышение температуры тела до 38-39°С похудание,
• артралгии, мышечные боли,у всех больных слабость и быстрая
утомляемость.
7.
8.
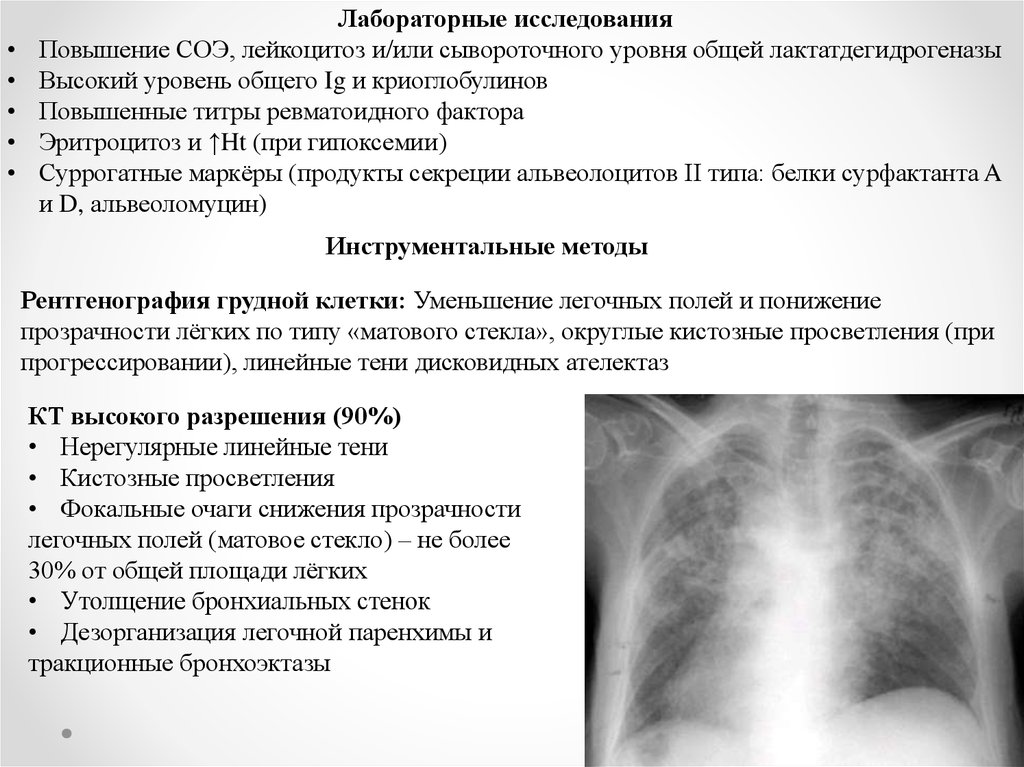
Лабораторные исследования
Повышение СОЭ, лейкоцитоз и/или сывороточного уровня общей лактатдегидрогеназы
Высокий уровень общего Ig и криоглобулинов
Повышенные титры ревматоидного фактора
Эритроцитоз и ↑Ht (при гипоксемии)
Суррогатные маркёры (продукты секреции альвеолоцитов II типа: белки сурфактанта A
и D, альвеоломуцин)
Инструментальные методы
Рентгенография грудной клетки: Уменьшение легочных полей и понижение
прозрачности лёгких по типу «матового стекла», округлые кистозные просветления (при
прогрессировании), линейные тени дисковидных ателектаз
КТ высокого разрешения (90%)
• Нерегулярные линейные тени
• Кистозные просветления
• Фокальные очаги снижения прозрачности
легочных полей (матовое стекло) – не более
30% от общей площади лёгких
• Утолщение бронхиальных стенок
• Дезорганизация легочной паренхимы и
тракционные бронхоэктазы
9.
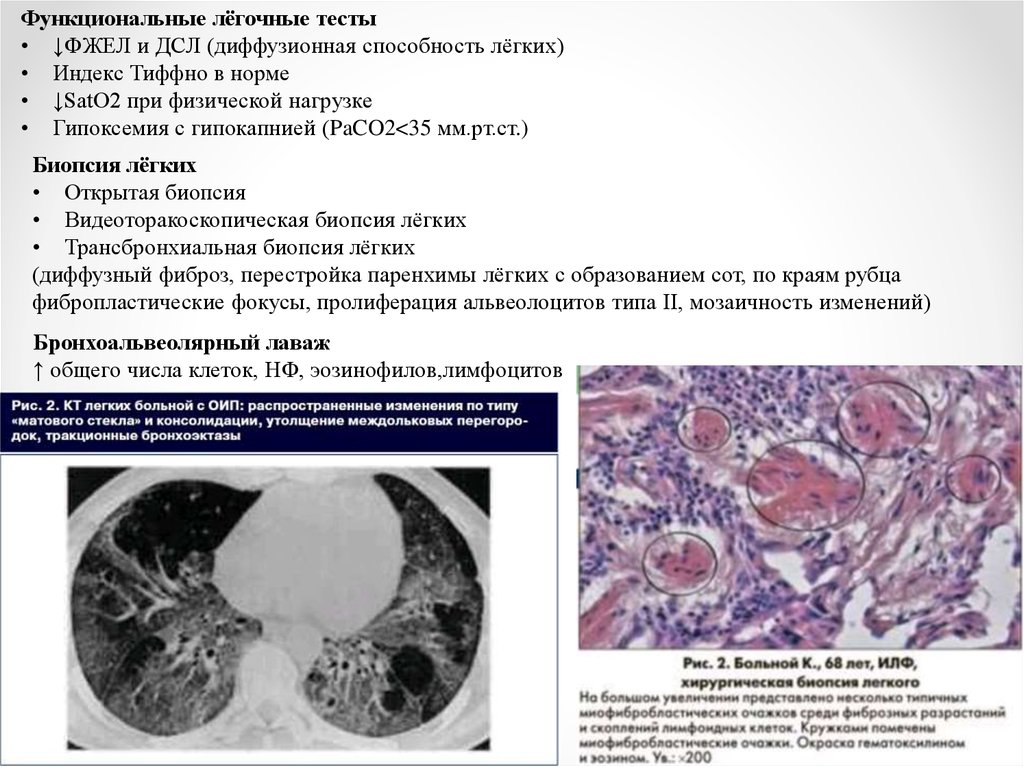
Функциональные лёгочные тесты• ↓ФЖЕЛ и ДСЛ (диффузионная способность лёгких)
• Индекс Тиффно в норме
• ↓SatO2 при физической нагрузке
• Гипоксемия с гипокапнией (PaCO2<35 мм.рт.ст.)
Биопсия лёгких
• Открытая биопсия
• Видеоторакоскопическая биопсия лёгких
• Трансбронхиальная биопсия лёгких
(диффузный фиброз, перестройка паренхимы лёгких с образованием сот, по краям рубца
фибропластические фокусы, пролиферация альвеолоцитов типа II, мозаичность изменений)
Бронхоальвеолярный лаваж
↑ общего числа клеток, НФ, эозинофилов,лимфоцитов
10.
Критерии диагностикиБольшие:
• Исключение других ИЗЛ, вызванных: приём ЛС, экпозиция вредных факторов внешней
среды, системные заболевания соединительной ткани
• Изменения функции внешнего дыхания, включая рестриктивные изменения и нарущения
газообмена
• Двусторонние ретикулярные изменения в базальных отделах лёгких с минимальными
изменениями по типу «матового стекла» по данным КТВР
• По данным трансабдоминальной биопсии или БАЛ нет признаков альтернативного диагноза.
Малые:
• Возраст >50 лет
• Незаметное постепенное появление диспноэ при физической нагрузке
• Длительность заболевания более 3 мес.
• Инспираторная крепитация в базальных отделах лёгких
Лечение
К препаратам с доказанной эффективностью при лечении ИЛФ относятся только два
препарата: пирфенидон ( не зарегистрирован в РФ) и нинтеданиб.
Немедикаментозная терапия
• Длительная кислородотерапия
• Трансплантация легких
• ИВЛ при ДН
• Легочная реабилитация
Необходимо лечение осложнений и сопутствующих заболеваний.
11.
Первичная цилиарная дискинезия (ПЦД) – редкое генетически детерминированное заболевание,при котором поражаются подвижные структуры клеток (реснички и жгутики). Наиболее часто
проявляется рецидивирующими и хроническими инфекциями верхних и
нижних дыхательных путей и в 40-50% случаев зеркальным расположением внутренних органов или
гетеротаксией .
Синонимы: синдром Картагенера (Kartagener syndrome; Siewert syndrome; ЗивертаКартагенера),
синдром неподвижных ресничек (immotile cilia syndrome), двигательная цилиопатия (the motile
ciliopathy)
Впервые этот синдром был описан в 1904 г. А.К.
Зивертом, но более подробное описание данной
патологии, ее семейных форм было представлено М.
Картагенером в 1933 г.
•обратное расположение внутренних
органов – situs viscerum inversus.
•бронхоэктазы
•хронический синусит с назальным
полипозом и ринореей
Синдром Картагенера составляет
50-60% случаев ПЦД.
12.
Транспозиция органов•Полный разворот сосудистой системы и всех внутренних
органов (situs inversus totalis).
•Декстракардия в сочетании с нормальным расположением
других органов (situs inversus
solitus).
Легочные проявления
•У новорождённых с ПЦД часто развивается респираторный
дистресс-синдром с тахипноэ или гипоксия, что может
потребовать кислородной поддержки от нескольких часов до
нескольких дней после рождения.
•В процессе роста ребёнка отмечается учащение
респираторных инфекций, сопровождающихся хроническим
кашлем с отхождением слизисто-гнойной мокроты.
•Бронхоэктазы при КТВР обнаруживаются у всех взрослых и
примерно у 50% детей.
•У пациентов с бронхоэктазами появляются влажные хрипы,
но хрипы, особенно у детей, могут быть и свистящими,
имитирующими астму.
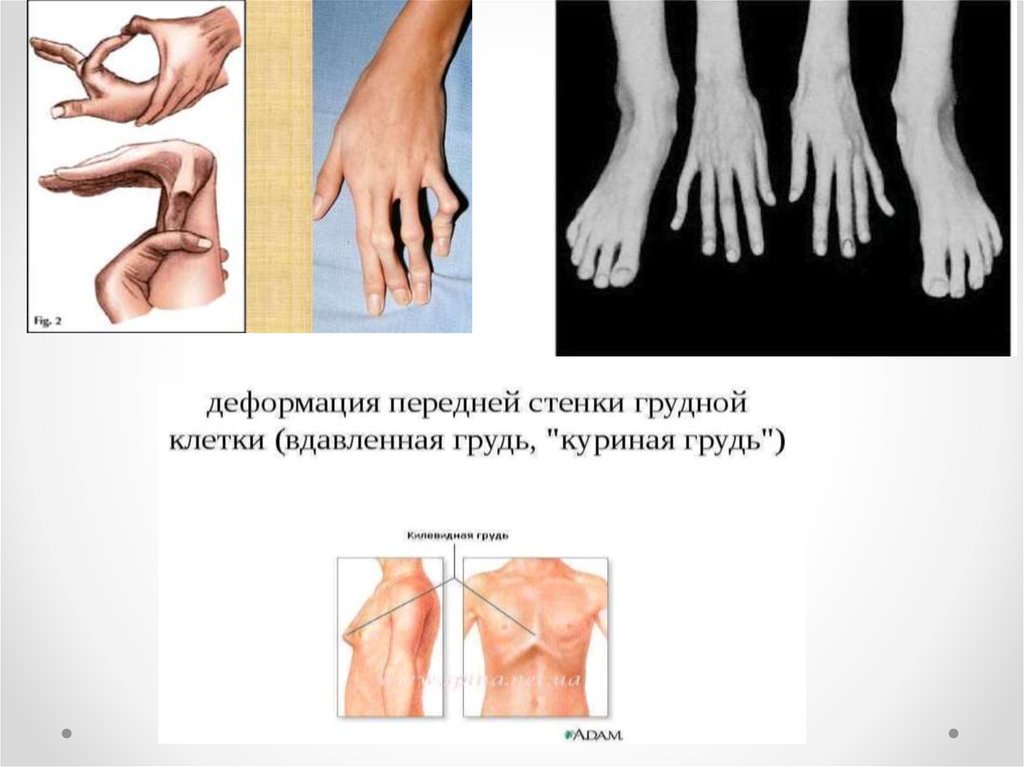
•Деформации дистальных фаланг кистей рук в виде
барабанных палочек у детей, как правило, не встречаются, у
взрослых описываются в 8% случаев.
Риносинуситы.
13.
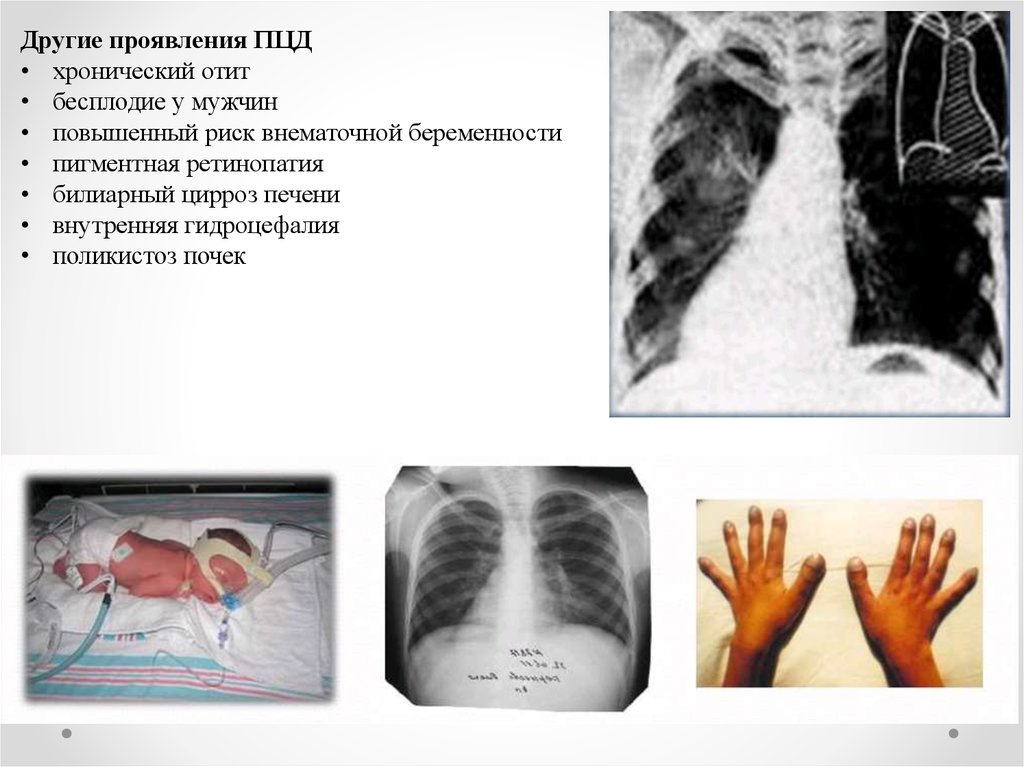
Другие проявления ПЦД• хронический отит
• бесплодие у мужчин
• повышенный риск внематочной беременности
• пигментная ретинопатия
• билиарный цирроз печени
• внутренняя гидроцефалия
• поликистоз почек
14.
В настоящее время нет единого метода - «золотого» стандарта диагностики ПЦД.При установлении диагноза учитываются:
• характерная клиническая картина
• результаты скрининга - исследование уровня оксида азота (NO) в выдыхаемом назальном
воздухе (у большинства пациентов с ПЦД он снижен);
• анализ частоты и паттерна биения ресничек в биоптате из полости носа или бронха с
помощью световой микроскопии;
• электронная микроскопия (обнаружение аномалий строения ресничек в биоптате слизистой
оболочки носа или бронха).
Для подтверждения диагноза рекомендовано сочетание исследования паттерна и
частоты биения ресничек с электронной микроскопией у пациентов с поражением верхних и
нижних дыхательных путей в состоянии ремиссии не менее 4-6 недель
Диагностика
Микробиологическое исследования (посева) мокроты или трахеального аспирата в период
обострения заболевания.
Компьютерная томография (КТ) органов грудной полости, рентгенография или КТ
придаточных пазух носа.
Исследование функции внешнего дыхания
Исследование газов крови и/или сатурации.
Диагностическая и/или лечебная трахеобронхоскопия.
Эхо-КГ с допплер ( измерение градиента давления на ЛА)
Мониторирование состояния слуховой функции с помощью аудиологических тестов.
15.
Подход к терапии пациента с ПЦД должен быть мультидисциплинарным в связи сполиорганностью поражений.
Основными целями терапии является максимально возможное предупреждение
прогрессирования и/или развития бронхоэктазов и восстановление / сохранение нормальной
легочной функции, а также носового дыхания и слух
• Кинезитерапия
• Промывания носовых ходов с гипертоническим раствором натрия хлорида, применение
назального душа
• Антибактериальная терапия при обострении хронического бронхолегочного процесса,
синусита (амоксициллин+клавулановая кислота, могут быть использованы цефалоспорины 2,
3 поколения).
• Ингаляционная бронхоспазмолитическая терапия (ипратропия бромид+фенотерол,
сальбутамол, салметерол, формотерол) при наличии бронхообструктивного синдрома
• Муколитики ( амброксол, АЦЦ, карбоцистеин)
• Хирургическое лечение ( полипэктомию рекомендуется проводить при тяжелой назальной
обструкции)
Рекомендуется генетическое консультирование родителей детей с ПЦД при
планировании последующих беременностей с целью минимизации риска рождения
больного ребенка.
16.
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственныхпатологий соединительной ткани. Синдром вызван мутацией гена,
кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание
характеризуется различной пенетрантностью и экспрессивностью. В
классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые
конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой
клетчатки. Помимо характерных изменений в органах опорнодвигательного аппарата (удлинённые
трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и
сердечно-сосудистой системы, что в классических
вариантах составляет триаду Марфана.
17.
Опорно-двигательный аппаратПатологическая соединительная ткань обусловливает развитие ряда специфических фенотипических
признаков и деформаций скелета у пациентов с данной патологией.
Как правило, внешне пациенты с синдромом Марфана выглядят достаточно специфически.
Для них характерно:
• астеническое телосложение;
• высокий рост;
• плохое развитие подкожной жировой клетчатки, из-за чего люди выглядят худощавыми;
• очень длинные верхние и нижние конечности при относительно коротком туловище;
• череп вытянутый (долихоцефалический);
• удлиненные пальцы – паукообразные (арахнодактилия);
• лицо узкое, вытянутое по вертикали;
• готическое верхнее небо;
• недоразвитие скул;
• выступающая нижняя челюсть (прогнатизм);
• неправильный рост (скученность) зубов и патологический прикус;
• гипермобильность суставов, их «разболтанность;
• глубоко посажены в черепе глаза.
По мере роста ребенка могут появляться различные деформации скелета. Чаще всего появляются
искривления позвоночного столба. У пациентов диагностируют сколиоз, патологический
кифоз и лордоз. Для пациентов также характерна остеопения (снижение минеральной
плотности костей) и частые патологические переломы костей на ее фоне, а также склонность
к привычным вывихам, например, плеча.
18.
19.
Сердечно-сосудистая системаСреди поражений кардиоваскулярной системы при СМ чаще всего
встречаются:
• пролапс створок митрального клапана с регургитацией или без
• миксаматоз сердца;
• дилятационная кардиомиопатия с развитием сердечной недостаточности;
• аневризмы аорты и других сосудов (мозговых, почечных, пр.);
• расширение легочной артерии и различных отделов аорты.
Орган зрения
Среди других характерных признаков:
• уплощение роговицы;
• увеличение размеров глазного яблока в длину;
• миопия или гиперметропия;
• нарушение процесса аккомодации из-за недоразвития цилиарной мышцы.
Органы системы дыхания
• В большинстве случаев изменения бронхолегочного аппарата диагностируются случайно.
Характерно развитие булл в верхних частях легких, которые иногда могут разрываться с развитием
спонтанного пневмоторакса.
• Также из-за деформаций грудной клетки пациенты склонны к развитию эмфиземы легких, частых
инфекционных заболеваний органов дыхания и дыхательной недостаточности.
Кожа и мягкие ткани
Встречается повышенная растяжимость кожного покрова, которая сочетается с развитием
атрофических стрий.
Нервная система
Когда люди с синдромом Марфана стареют, твердая мозговая оболочка часто слабеет и вытягивается.
Это называется дуральная эктазия.
20.
ДиагностикаКлинические критерии
Генетические тесты
эхокардиография/МРТ (исследование корня аорты, обнаружение клапанного пролапса);
исследование со щелевой лампой (аномалии хрусталика);
Рентген скелетной системы (рук, позвоночника, таза, груди, ног и черепа на
характерные аномалии);
• МРТ пояснично-крестцового отдела позвоночника (дуральная эктазия)
Лечение
• Индуцирование преждевременного полового созревания у высоких девушек
• Бета-адреноблокаторы
• Элективное восстановление аорты и клапанов
• Фиксация и хирургическое вмешательство при сколиозе
Лечение синдрома Марфана направлено на профилактику и лечение осложнений.
21.
22.
23.
24.
Идиопатическая (первичная) легочная гипертензия (ИЛГ) - редкое заболевание неизвестнойэтиологии, характеризующееся выраженным повышением общего легочного сосудистого
сопротивления (ОЛСС) и давления в легочной артерии, часто прогрессирующим течением с быстрым
развитием декомпенсации правого желудочка, фатальным прогнозом.
Синонимы: первичная легочная гипертензия, синдром Аэрза- Арилаго, болезнь Аэрза, болезнь
Эскудеро.
Критерии диагностики:
Диагноз ИЛГ устанавливается при среднем давлении в легочной артерии (ДЛАср.) более 25 мм рт.ст. в
покое и более 30 мм рт.ст. при физической нагрузке, нормальном давлении заклинивания в легочной
артерии (ДЗЛА) (до 10-12мм.рт.ст.) и отсутствии возможных причин легочной гипертензии (ЛГ)заболеваний сердца, легких, хронической тромбоэмболии легочной артерии и т.д.
Клинические симптомы ИЛГ - неспецифичны, что существенно затрудняет раннюю диагностику
заболевания (одышка, боли в груди, головокружение, кашель, кровохарканье, сердцебиение и перебои
в работе сердца).
Алгоритм диагностики ЛГ включает следующие этапы:
1. Подозрение на наличие у больного ЛГ (ЛГ —предварительный
диагноз).
2. Верификация диагноза ЛГ( катетеризация правых отделов сердца и
ЛА, ЭКГ, ВКГ, ФКГ, Эхо-КГ)
3. Установление клинического класса ЛГ( опред.ФВД, КТ, МРТ, АПГ)
4. Оценка ЛГ (тип, функциональный класс)- (анализы крови (общий,
биохимический, иммунологический), тест на ВИЧ:УЗИ внутренних
органов; тест 6-минутной ходьбы/кардиопульмональный тест; оценку
ФК;биопсию легких)
25.
ЛечениеОбщие рекомендации: исключение избыточной нагрузни на ССС и ДС, своевременная вакцинация,
регулярная физическая активность (аэробные упражнения низкой интенсивности, например прогулки
умеренным шагом, если пациент их нормально переносит), диета с ограничением соли и контролем
потребления жидкости.
Медикаментозная терапия:
• Блокаторы кальциевых каналов– нифедипин, дилтиазем, амлодипин
• Простаноиды- эпопростенол, трепростинил, илопрост
• Антагонисты эндотелиновых рецепторов- бозентан
• Ингибиторы фосфодиэстеразы-5- силденафил, тадалафил
• Диуретики- петлевые диуретики: фуросемид 20–120 мг/сутки, этакриновая кислота 50–100
мг/сутки, торасемид 5–20 мг/сутки. Целесообразно присоединение антагонистов адьдостерона:
верошпирон 25–150 мг, эплеренон 20 мг. Во всех случаях назначения диуретиков необходимо
тщательно контролировать уровни электролитов крови, а также состояние функции почек
• Оксигенотерапия
• Пациентам с ЛГ показано оперативное лечение, когда на фоне активной фармакотерапии (включая
средства, специфические для лечения ЛГ, и инотропные препараты) наблюдается
прогрессирование заболевания и имеется неблагоприятный прогноз. В таких случаях проводится
билатеральная трансплантация легких (при необходимости — с одновременным устранением
порока сердца) или комплекса сердце–легкие.
• Предсердная септостомия
26.
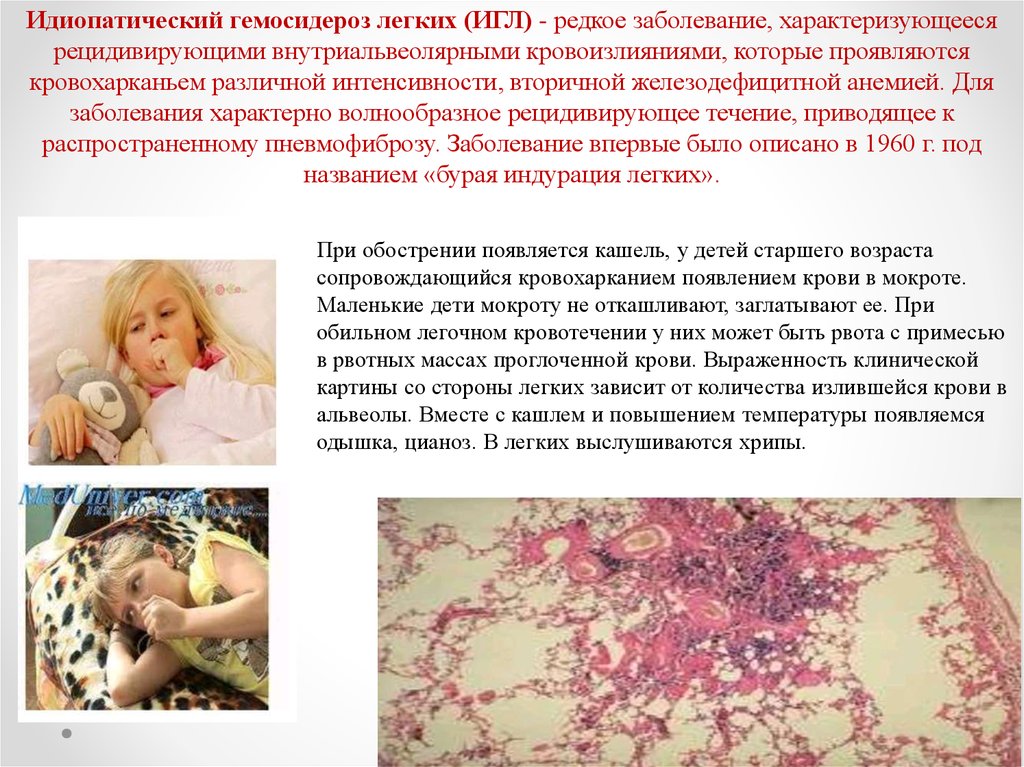
Идиопатический гемосидероз легких (ИГЛ) - редкое заболевание, характеризующеесярецидивирующими внутриальвеолярными кровоизлияниями, которые проявляются
кровохарканьем различной интенсивности, вторичной железодефицитной анемией. Для
заболевания характерно волнообразное рецидивирующее течение, приводящее к
распространенному пневмофиброзу. Заболевание впервые было описано в 1960 г. под
названием «бурая индурация легких».
При обострении появляется кашель, у детей старшего возраста
сопровождающийся кровохарканием появлением крови в мокроте.
Маленькие дети мокроту не откашливают, заглатывают ее. При
обильном легочном кровотечении у них может быть рвота с примесью
в рвотных массах проглоченной крови. Выраженность клинической
картины со стороны легких зависит от количества излившейся крови в
альвеолы. Вместе с кашлем и повышением температуры появляемся
одышка, цианоз. В легких выслушиваются хрипы.
27.
Особенностью рентгенологически выявляемых изменений при ИГЛ является быстроеобратное развитие очагов затемнения. В ряде случаев на рентгенограммах грудной клетки
отмечаются рассеянные мелкие тени в обоих легких, что служит причиной ошибочной
диагностики милиарного туберкулеза легких. Изменения в легких, выявляемые при
рентгенографии могут варьировать в широких пределах: от небольших инфильтратов
до массивных тенеобразований, сопровождающихся ателектазами, эмфиземой и реакцией
со стороны лимфатических узлов корней легких.
28.
Сразу после обострения, которое длится 3-5 дней, отмечается• анемия - микроцитарная и гипохромная;
• уровень сывороточного железа падает;
В биохимическом анализе крови может отмечаться:
• повышенный уровень билирубина;
• Поскольку регенераторная функция костного мозга не страдает, в
периферической крови появляются ретикулоциты;
• У маленьких детей анализ кала на скрытую кровь может оказаться
положительным (проглоченная при кашле мокрота с кровью);
• Часто отмечается гепатоспленомегалия
Диагностически значимым является обнаружение в мокроте или
трахеальном аспирате, а также в ряде случаев в промывных водах
желудка, сидергофагов.
Исследование функции внешнего дыхания обнаруживает или
нормальные показатели вентиляции, если длительность
заболевания небольшая, или выраженные рестриктивные
нарушения, снижение диффузионной способности легких, если
заболевание протекает длительно с тяжелыми обострениями.
29.
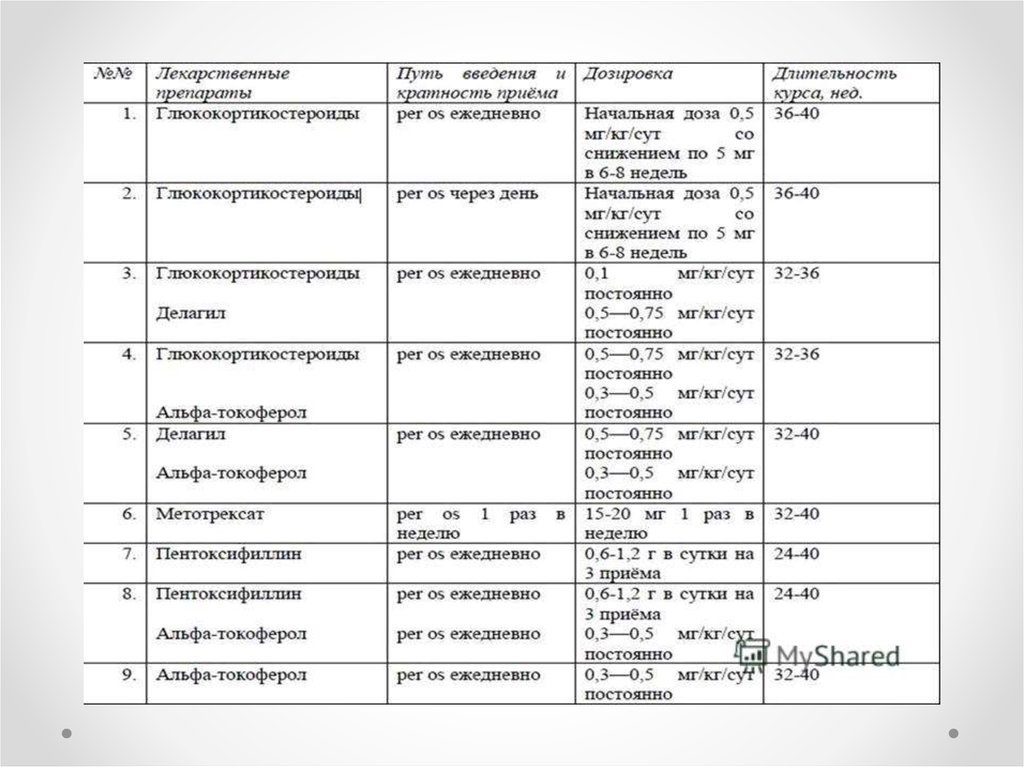
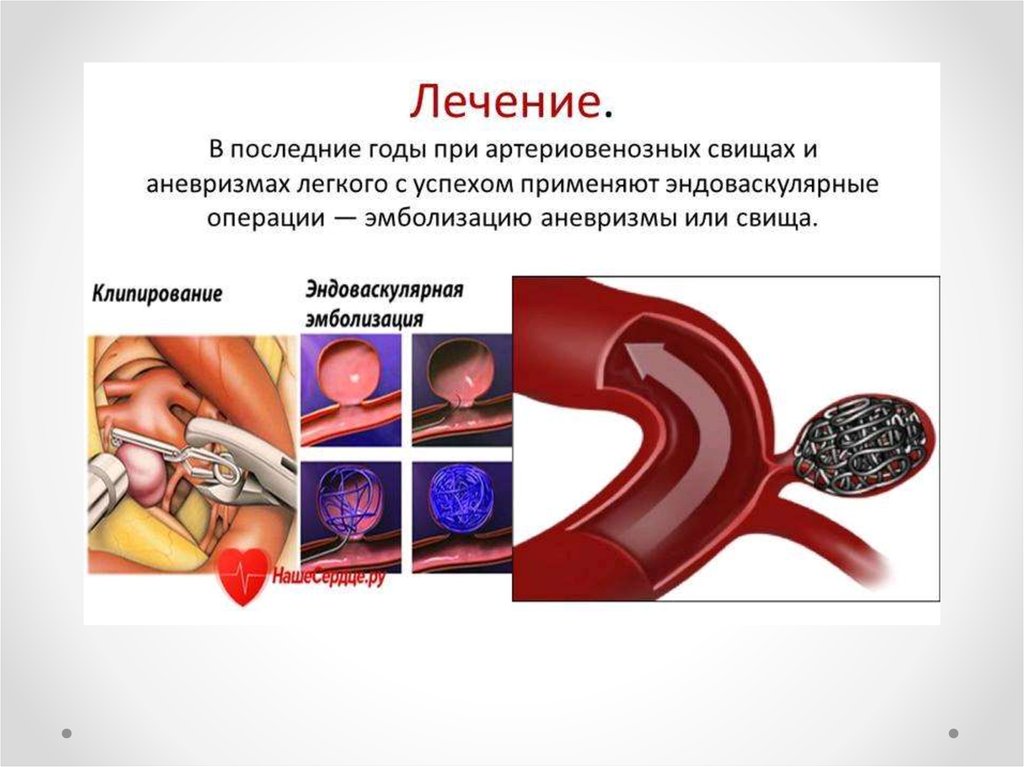
Лечение
предполагает назначение кортикостероидных
препаратов и симптоматическое лечение.
Некоторые авторы рекомендуют до определения
преципитинов в сыворотке крови больных
исключить из рациона питания коровье молоко.
Преднизолон назначается из расчета 1 -1,5 мг/кг до
достижения клинико-лабораторной ремиссии.
Имеются сообщения о том, что после спленэктомии
наступает стойкая длительная ремиссия.
30.
Саркоидоз легких (болезнь Бенье-БекаШауманна) доброкачественное системноезаболевание, в основе которого лежит поражение ретикулоэндотелиальной системы с
образованием в легких эпителиоидноклеточных гранулем без казеоза и перифокального
воспаления, в дальнейшем рассасывающихся или трансформирующихся в соединительную
ткань при отсутствии микобактерий туберкулеза.
Симптомы
• кратковременное повышение температуры тела (в течение 4-6
дней);
• боли в суставах мигрирующего характера;
• одышка;
• боли в грудной клетке;
• сухой кашель (у 40-45% больных);
• снижение массы тела;
• увеличение периферических лимфатических узлов (у
половины
больных);
• лимфаденопатия средостения (чаще двухсторонняя);
• узловатая эритема;
• синдром Лефгрена;
• синдром Хеерфордта-Вальденстрема;
• сухие хрипы при аускультации легких.
31.
Общий анализ крови и мочи• Общий анализ крови: увеличение СОЭ, лейкоцитоз У 20% больных отмечается эозинофилия, у 50% абсолютная лимфопения.
• Общий анализ мочи: без существенных изменений.
Биохимический анализ крови
• повышение уровней серомукоида, гаптоглобина, сиаловых кислот, гаммаглобулинов;
• у 15-20% больных увеличено содержание кальция в крови;
• повышение уровня общего или связанного с белком оксипролина;
• ангиотензинпревращающего фермента;
• повышение содержания в крови лизоцима.
Иммунологические исследования
• снижение количества Т-лимфоцитов и их функциональной способности;
• снижение содержания Т-лимфоцитовхелперов и соответственно снижение индекса Т-хелперы/Тсупрессоры;
• увеличение абсолютного количества Влимфоцитов, а также уровня IgA, IgG и циркулирующих
иммунных комплексов преимущественно в активной фазе;
• в крови обнаруживаются противолегочные антитела
Рентгенологическое исследование легких
• увеличение внутригрудных лимфатических узлов (лимфаденопатия средостения);
• увеличение внутригрудных (бронхопульмональных) лимфатических узлов обычно двустороннее;
• увеличение и расширение корней легких;
• увеличенные лимфоузлы имеют четкие полициклические очертания и однородную структуру;
• характерен ступенчатый контур изображения лимфоузлов за счет наложения теней передних и
задних групп бронхопульмональных лимфоузлов.
Эндоскопическое исследование