






























 Медицина
МедицинаПохожие презентации:










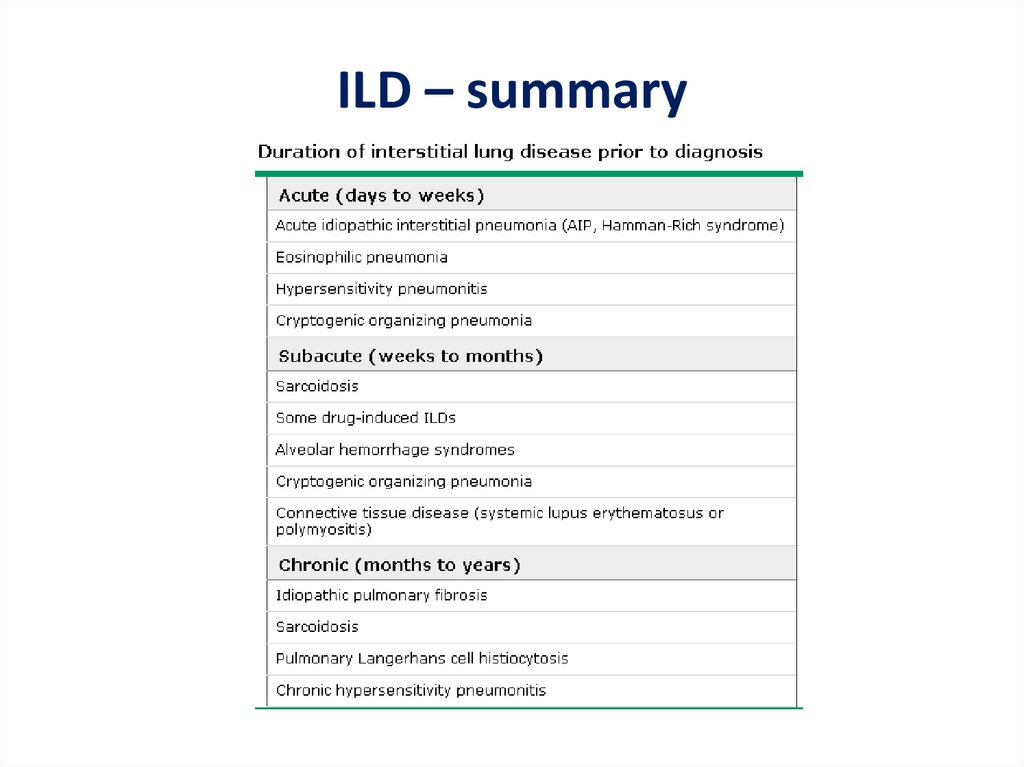
Interstitial Lung Disease/Diffuse Parenchymal Lung Disease(DPLD)
1.
Interstitial Lung Disease/DiffuseParenchymal Lung Disease(DPLD)
Dr. Katya Dolnikov
2.
Introduction• The diffuse parenchymal lung diseases (ILDs) are a
heterogeneous group of disorders that are
classified together because of similar clinical,
radiographic, physiologic, or pathologic
manifestations
• ILD - generic term encompassing a broad range of
largely unrelated conditions that share the
propensity to cause breathlessness and/or cough
associated with bilateral abnormal opacities of
various types on conventional chest radiographs
or CT scans
3.
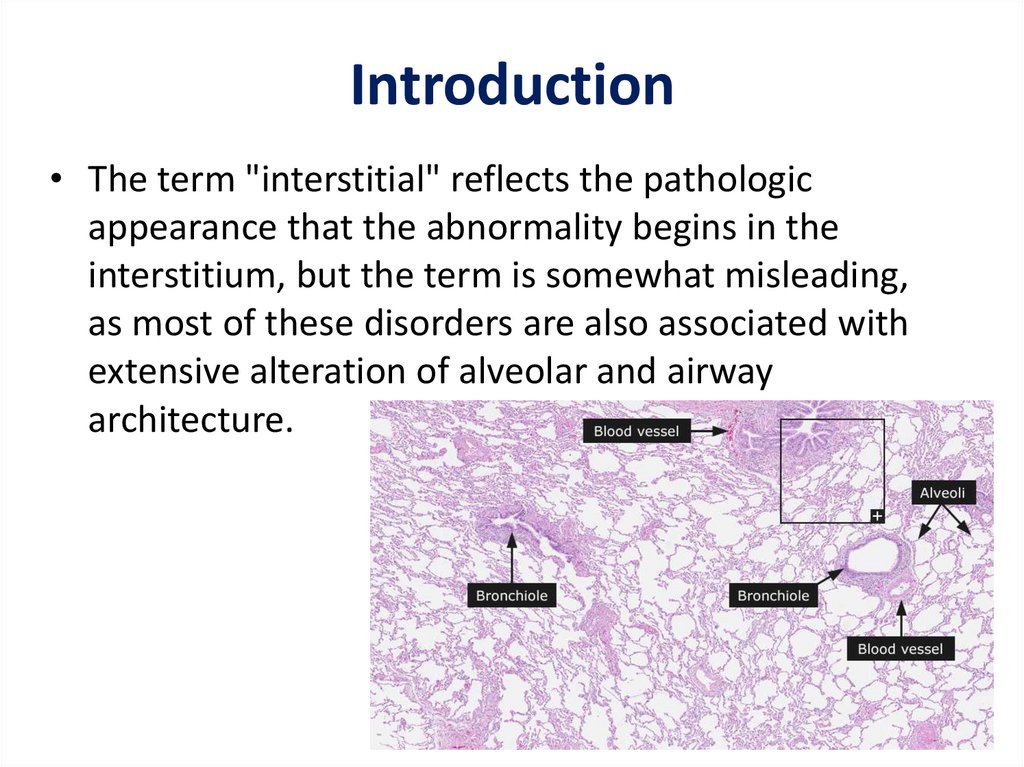
Introduction• The term "interstitial" reflects the pathologic
appearance that the abnormality begins in the
interstitium, but the term is somewhat misleading,
as most of these disorders are also associated with
extensive alteration of alveolar and airway
architecture.
4.
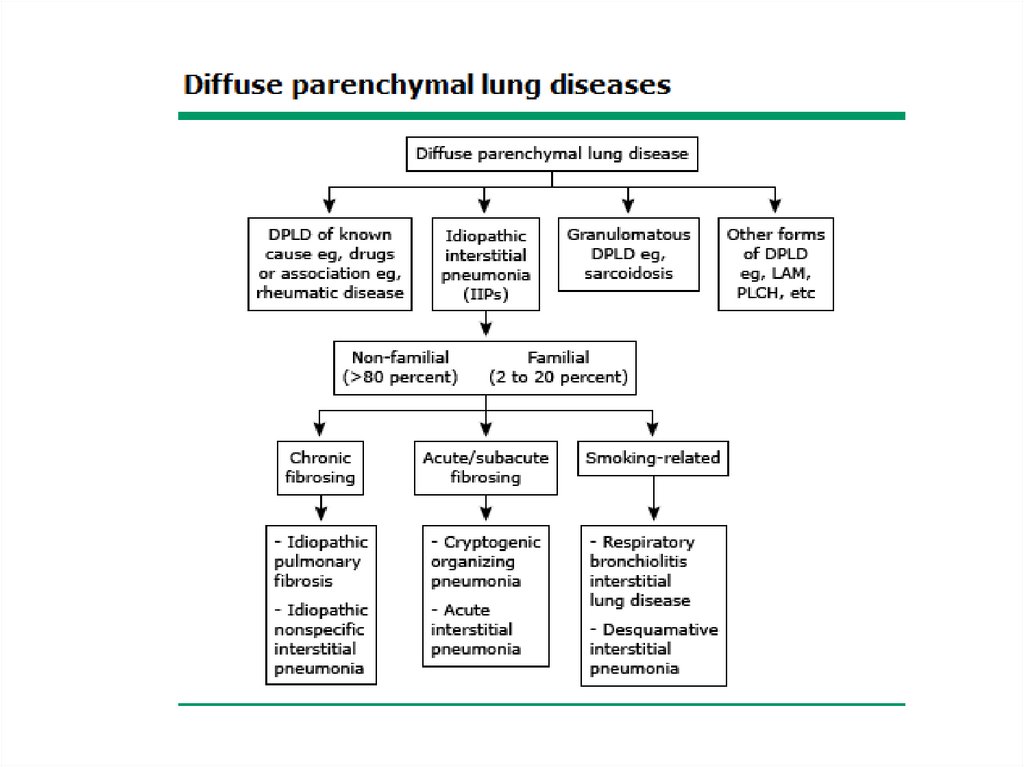
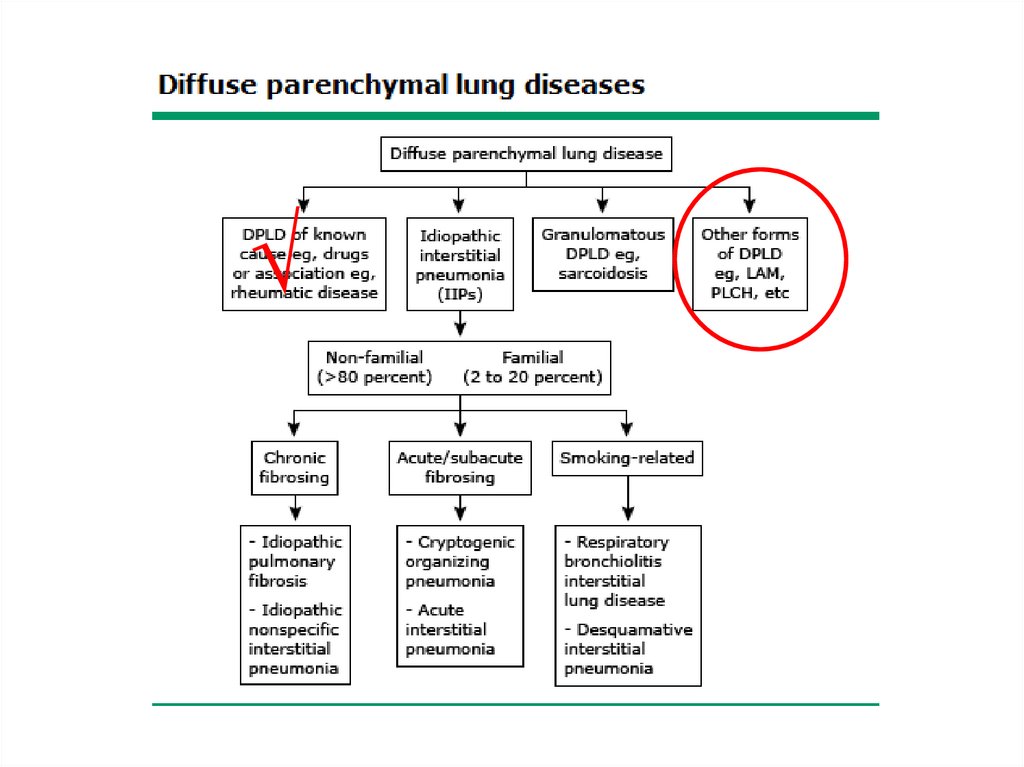
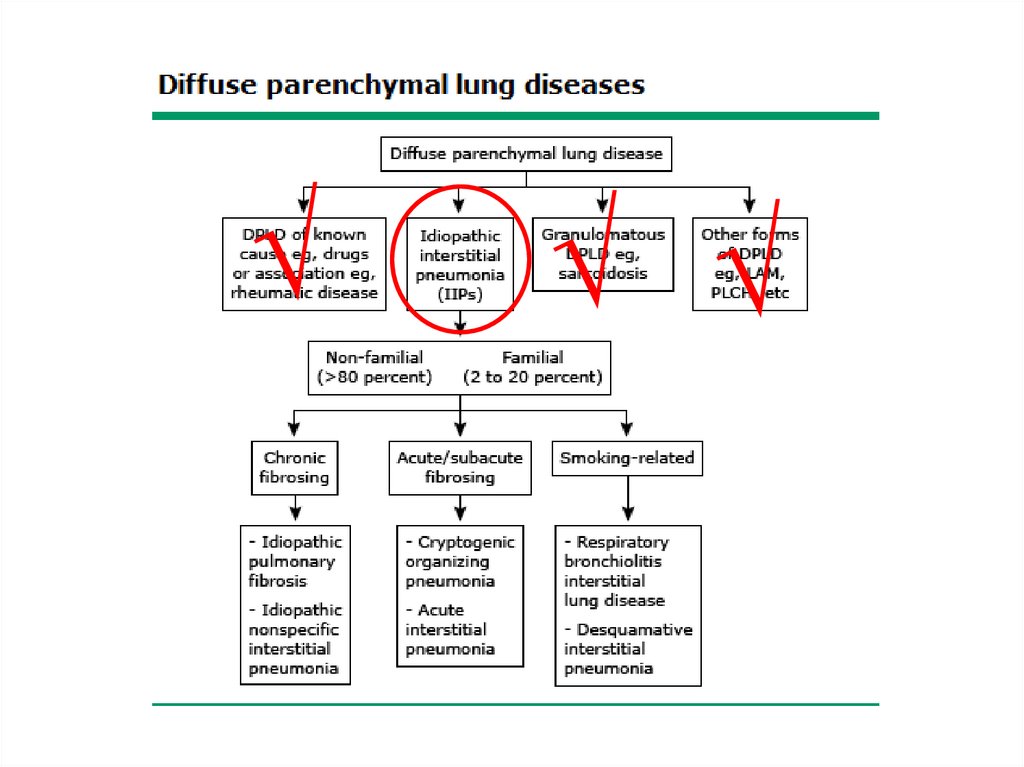
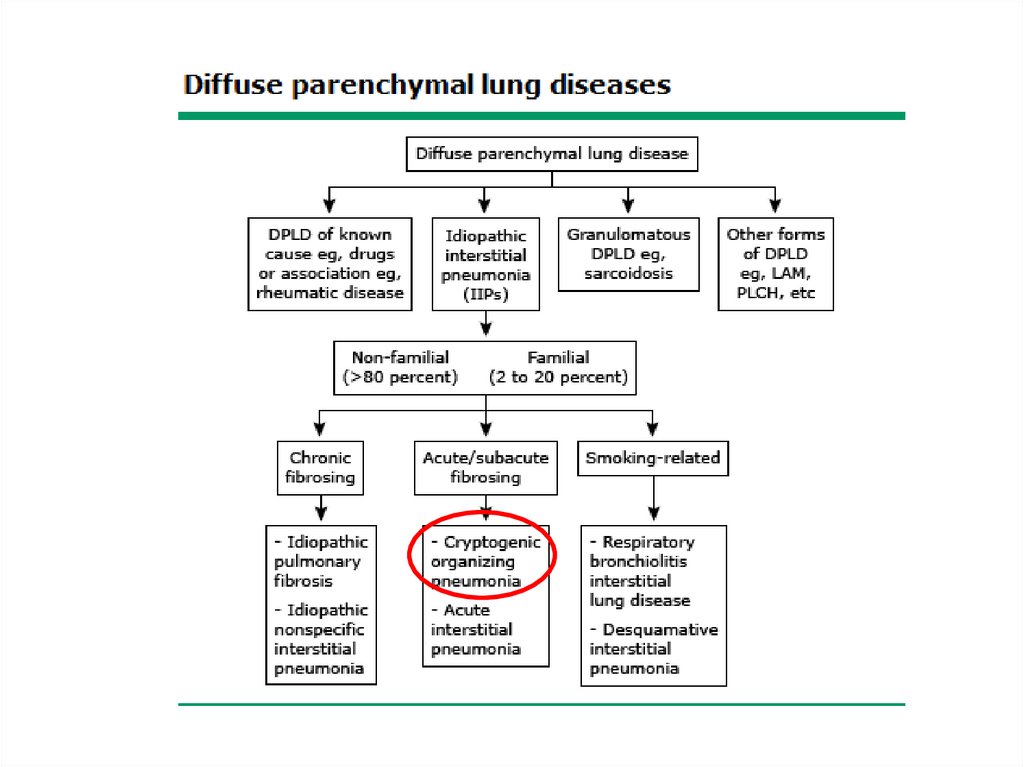
Classification• The diffuse parenchymal lung diseases are
divided into those that are associated with
known causes and those that are idiopathic
• The treatment and prognosis vary among the
different causes and types of ILD, so
ascertaining the correct diagnosis is
important.
5.
6.
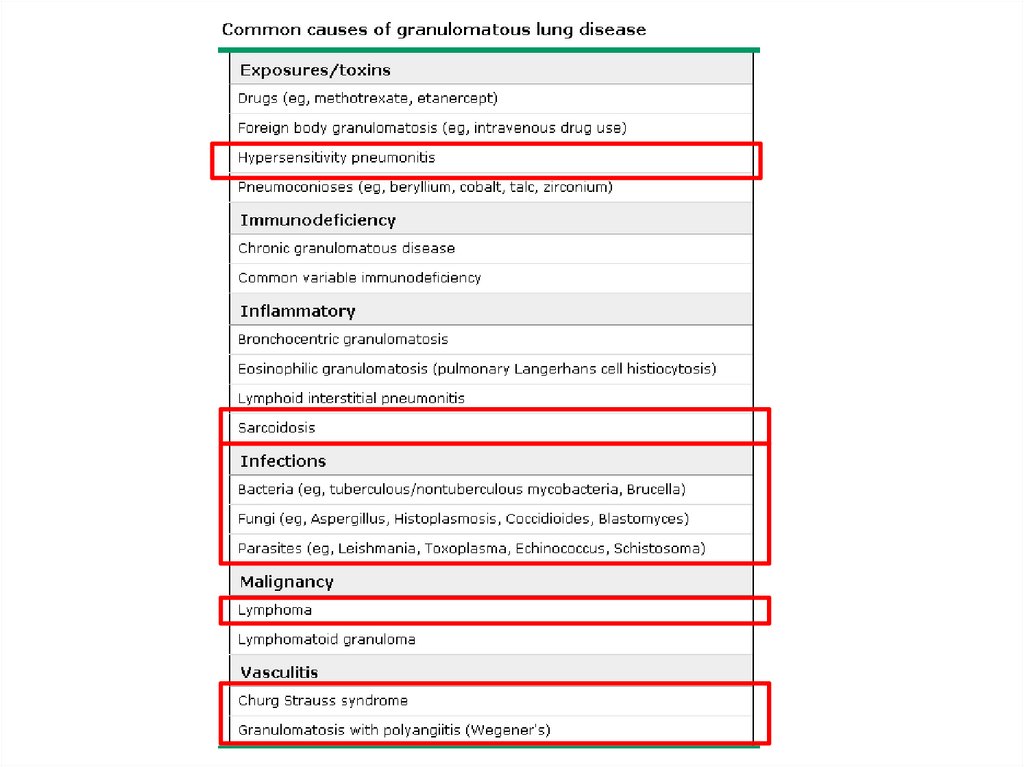
ILD-Known cause7.
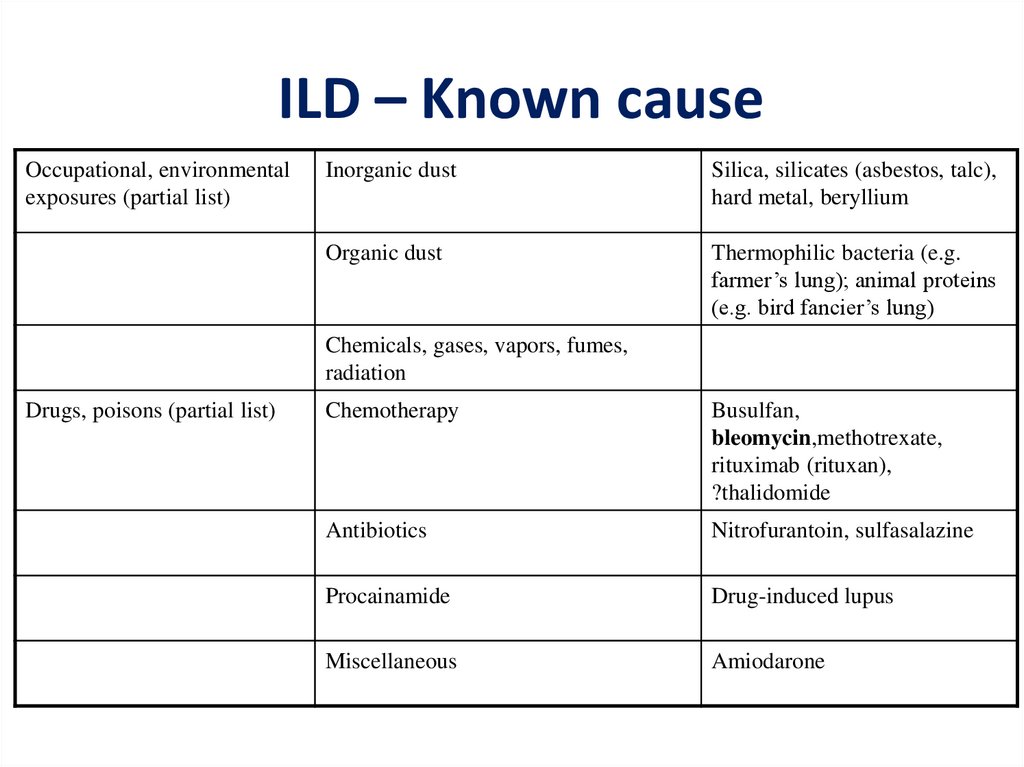
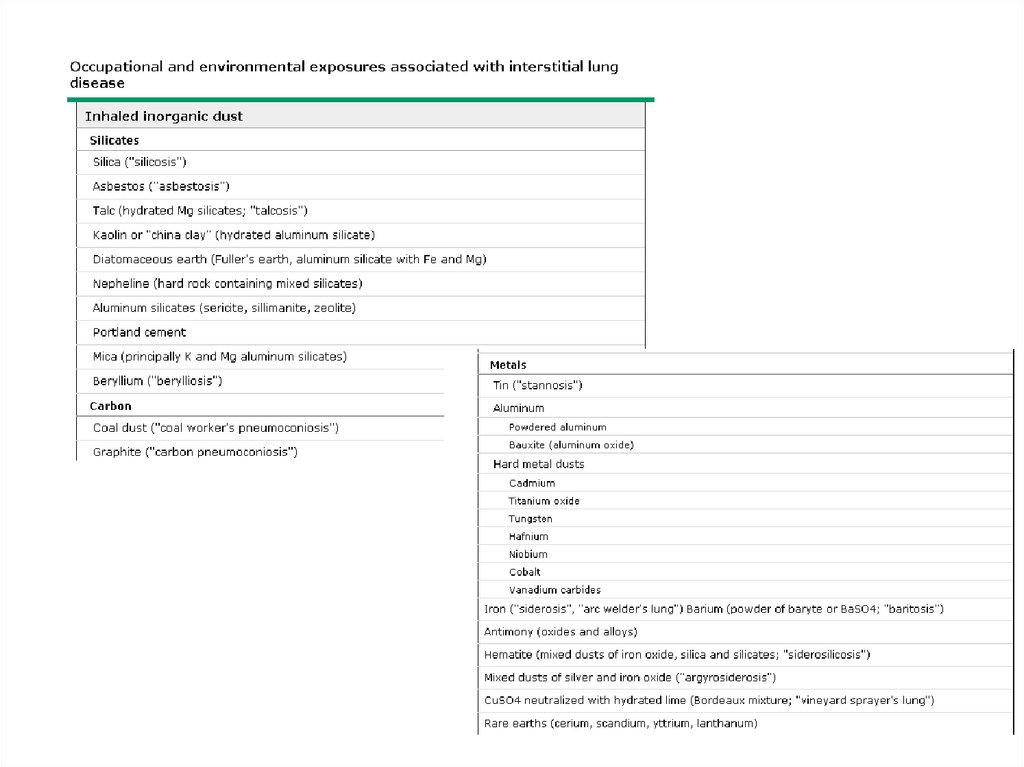
ILD – Known causeOccupational, environmental
exposures (partial list)
Inorganic dust
Silica, silicates (asbestos, talc),
hard metal, beryllium
Organic dust
Thermophilic bacteria (e.g.
farmer’s lung); animal proteins
(e.g. bird fancier’s lung)
Chemicals, gases, vapors, fumes,
radiation
Drugs, poisons (partial list)
Chemotherapy
Busulfan,
bleomycin,methotrexate,
rituximab (rituxan),
?thalidomide
Antibiotics
Nitrofurantoin, sulfasalazine
Procainamide
Drug-induced lupus
Miscellaneous
Amiodarone
8.
9.
10.
ILD-known cause• ILD can complicate the course of most of the
connective tissue diseases:
– polymyositis/dermatomyositis
– rheumatoid arthritis
– systemic lupus erythematosus
– Scleroderma
– mixed connective tissue disease
11.
12.
√13.
Other forms• Lymphangioleiomyomatosis - LAM
• Pulmonary Langerhans cell
histiocytosis/histiocytosis X
• Eosinophilic pneumonia
14.
√√
15.
16.
√√ √
17.
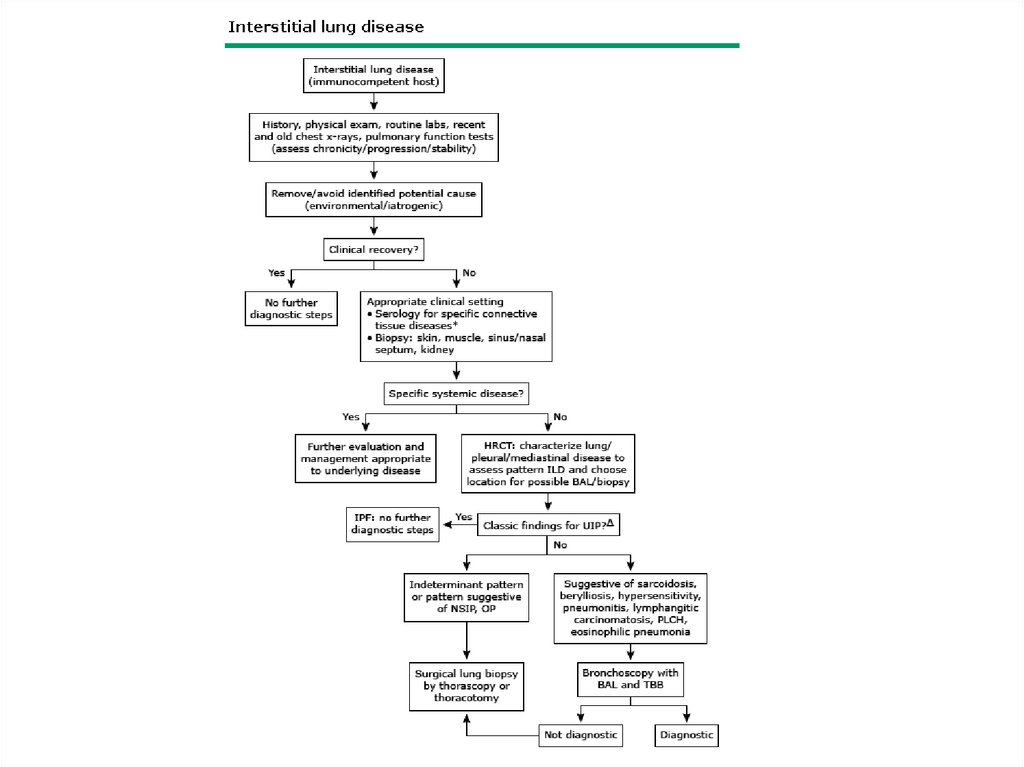
DPLD-Patient Evaluation• History
– fever, hemoptysis, swallow
– muscle, joint
– asthma, malignancy, chemotherapy or
radiotherapy, CTD, IBD
– present and prior medications
– family history (3% IPF are familial)
18.
DPLD-Patient Evaluation• Detailed Occupational History- exposures,
duration, exact role in work place, protective
gears use, coworkers’ symptoms
• work place- site visit
• pets, hobbies, home environment
• HIV risk, illicit drugs and cocaine use
19.
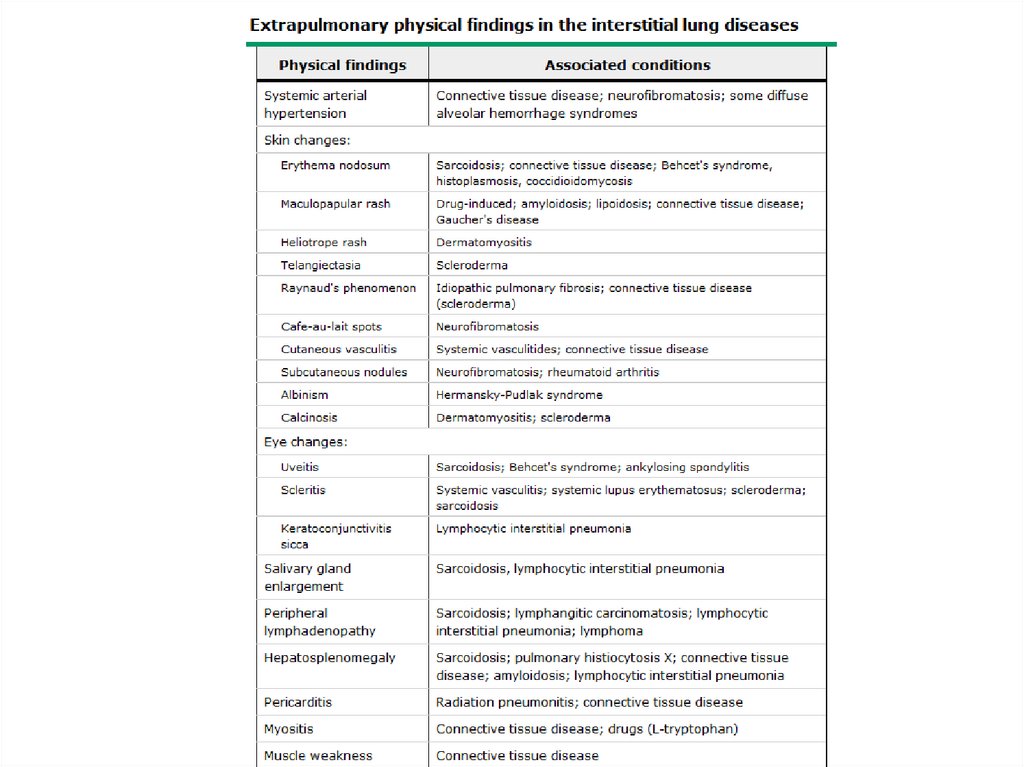
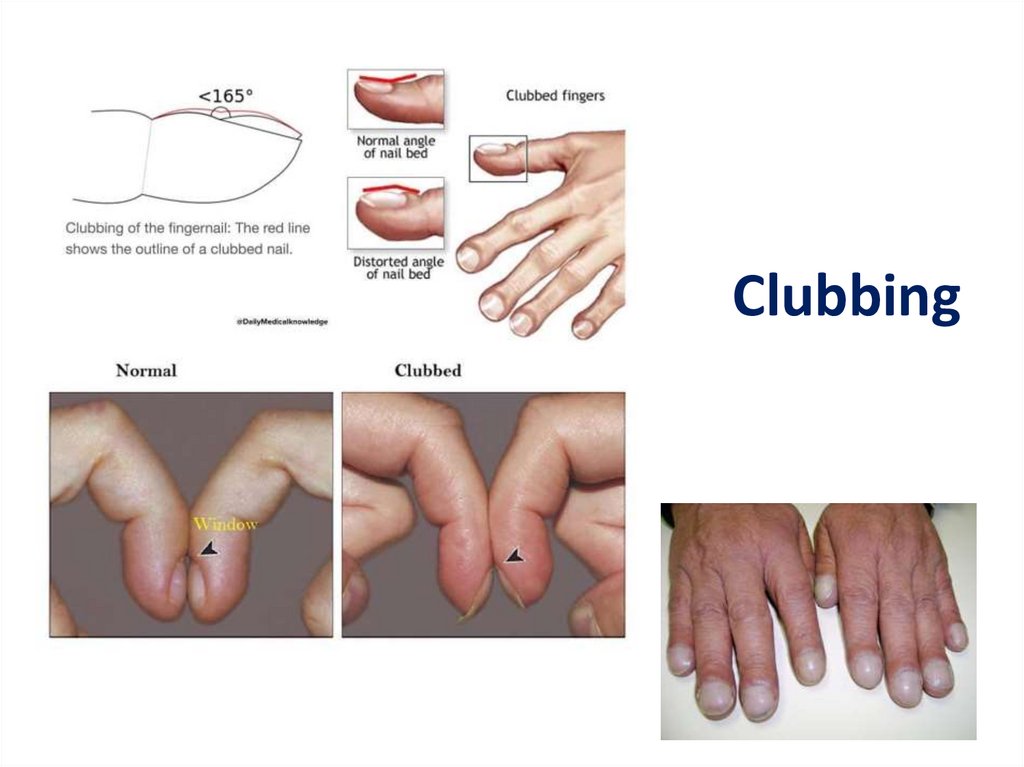
DPLD- Physical Exam• Lung exam- end inspiratory “Velcro” or
“cellophane” crackles, check for pleural
effusion
• Clubbing- up to 50% of IPF
• Skin changes- Erythema nodosum, Raynaud’s
• Eye changes- scleritis, uveitis
• Salivary glands enlargement
• O2 saturation- resting and ambulating,
overnight
20.
Clubbing21.
DPLD-PFT• Restrictive pattern with decrease in TLC and
decrease in DLCO
• May be obstructive in some patients, e.g.
LAM, BO, sarcoidosis, IPF in smokers
22.
DPLD-CXR and HRCTUpper lobes vs. lower lobes vs. diffuse
Peripheral (sub-pleural)- IPF
Ground glass changes, traction bronchiectasis
Pleural involvement is rare- CTD, asbestosis,
LAM, post-RT
• Ass. pneumothorax or bullae disease- HX, LAM
• Lymph nodes enlargement
• Early cases may have normal CXR
23.
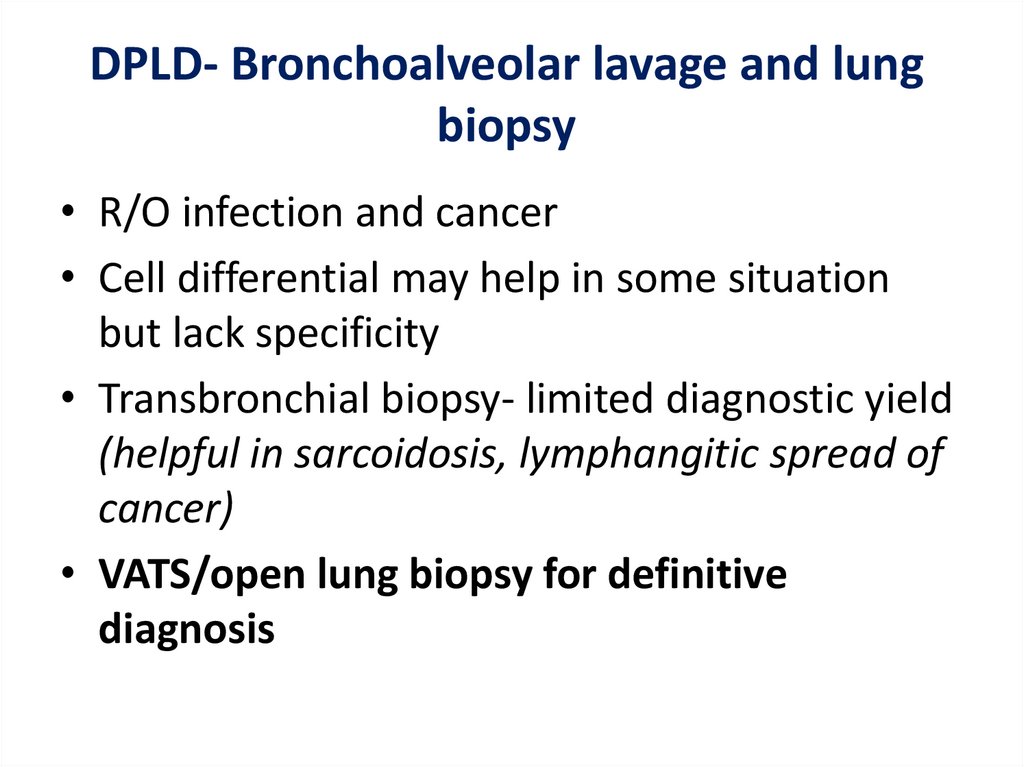
DPLD- Bronchoalveolar lavage and lungbiopsy
• R/O infection and cancer
• Cell differential may help in some situation
but lack specificity
• Transbronchial biopsy- limited diagnostic yield
(helpful in sarcoidosis, lymphangitic spread of
cancer)
• VATS/open lung biopsy for definitive
diagnosis
24.
DPLD-other tests• Hct, Eos, ESR, Urinalysis
• ABG
• serology e.g. ANA, RF, ANCA, anti-GBM, antiDNA
• ECG, holter monitor, echo
• HIV
• eye exam
• Ca, 24 hr urine Ca
25.
26.
Idiopathic Pulmonary Fibrosis (IPF)IPF - Cryptogenic fibrosing alveolitis in Europe
The most common form of ILD
M>F
Associated with smoking
Prevalence 50-200/100,000
More common in older people (>60 years old)
Median survival~ 3 years, 50% died within 5 years,
no real proven treatment still
27.
IPF- refined diagnosis (UIP)• Need to exclude all diseases that can lead to possible
UIP pathologically: asbestosis, scleroderma, chronic
HP, drugs, CEP, rarely sarcoidosis
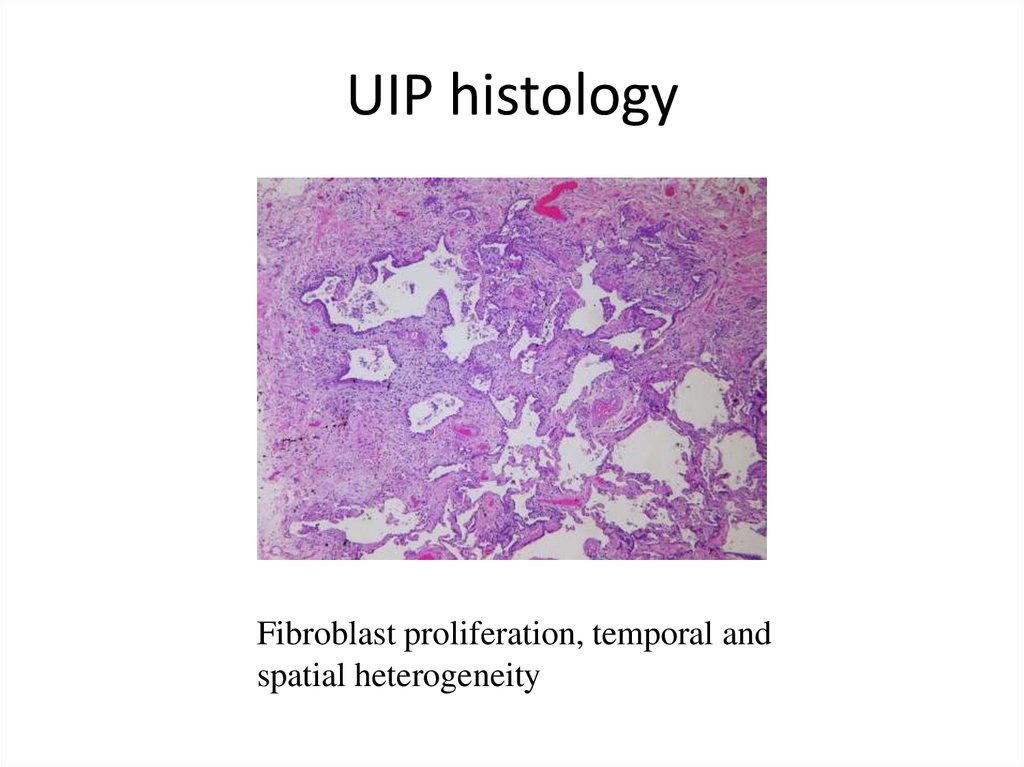
• Histological hallmarks: temporal heterogeneity
signifying ongoing injury (transition from normal lung
to alveolar organization/inflammation to dense
fibrosis) and abundant fibroblastic foci (fibroblasts
playing the central role in pathogenesis)
• 1.6-2.3 times more common in smoker
• Increase lung cancer risk
28.
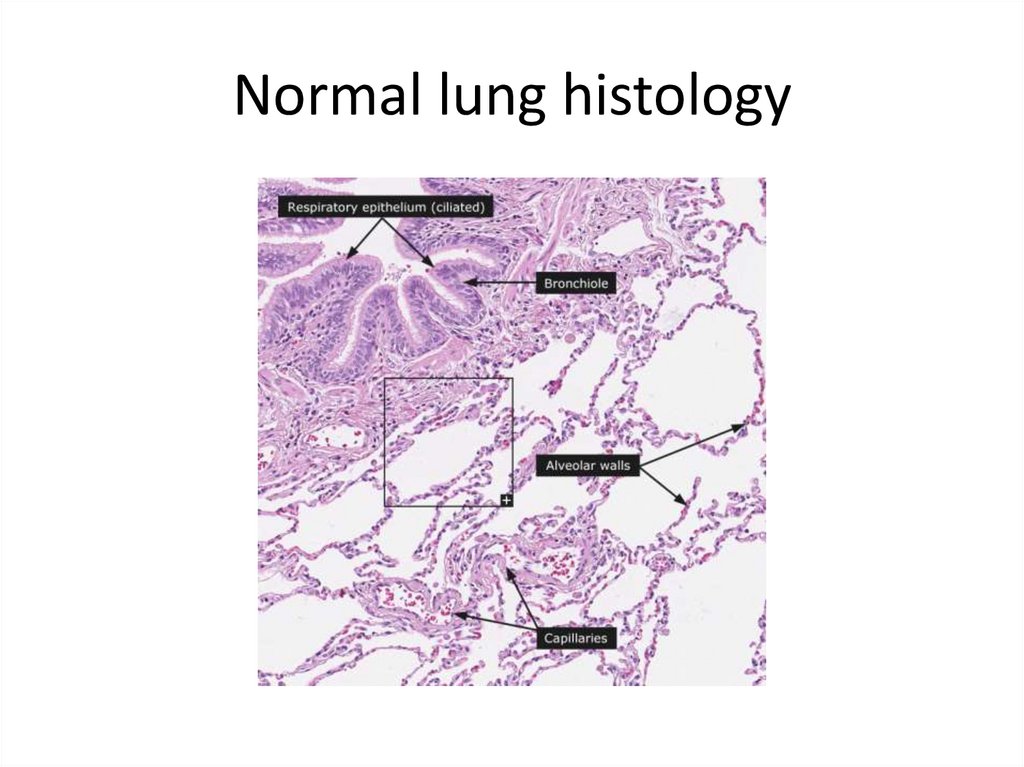
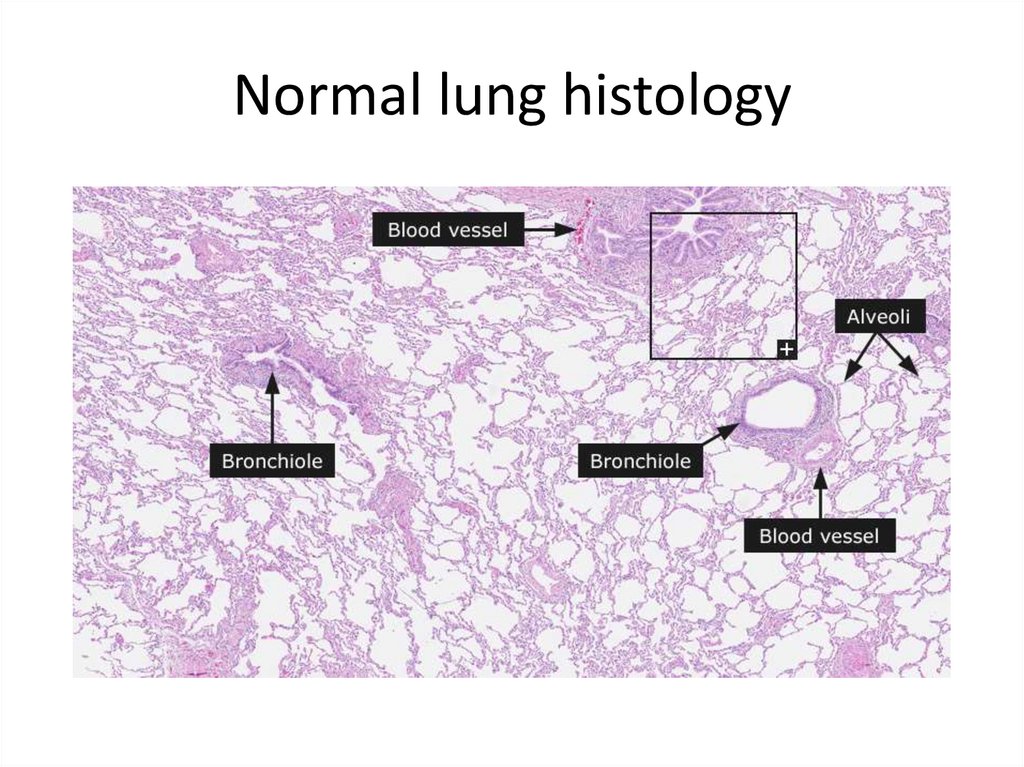
Normal lung histology29.
Normal lung histology30.
UIP histologyFibroblast proliferation, temporal and
spatial heterogeneity
31.
IPF- clinical features• Hx - usually > 60 y.o., dyspnea, dry cough,
• Phys. Exam - crackles
• CXR: diffuse interstitial infiltrate
– Peripheral, sub-pleural, bibasilar
– Can be patchy
– Honey combing changes in later stage
• PFT: restrictive impairment, decrease DLCO,
hypoxemia worse on exercise
32.
33.
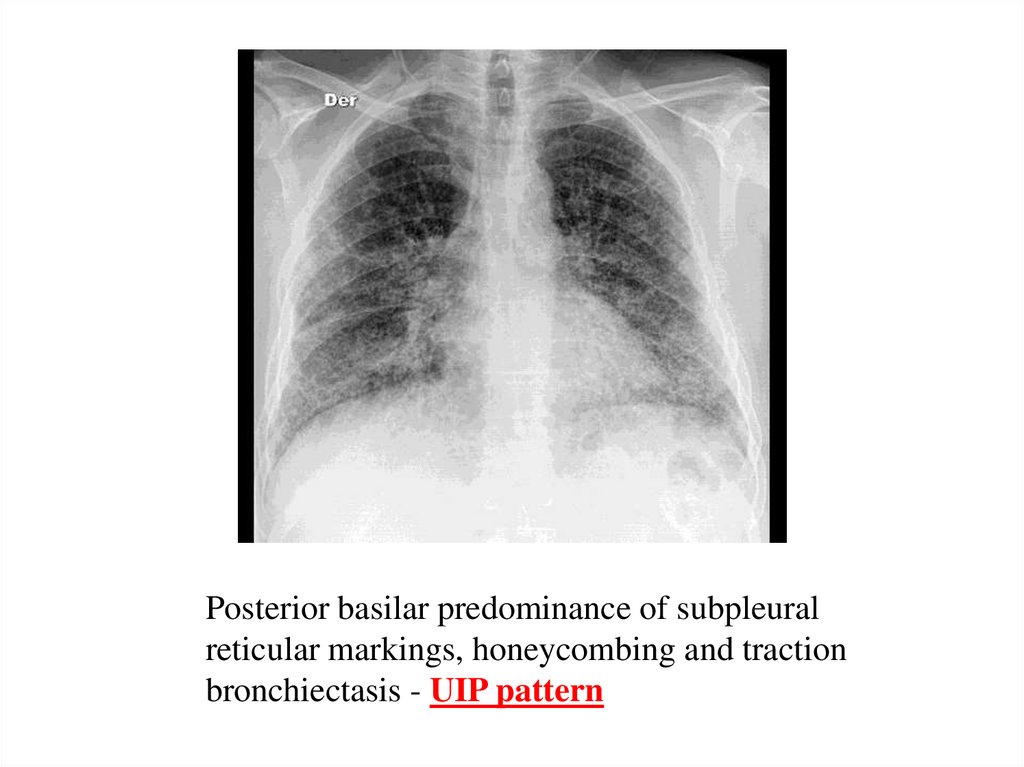
Posterior basilar predominance of subpleuralreticular markings, honeycombing and traction
bronchiectasis - UIP pattern
34.
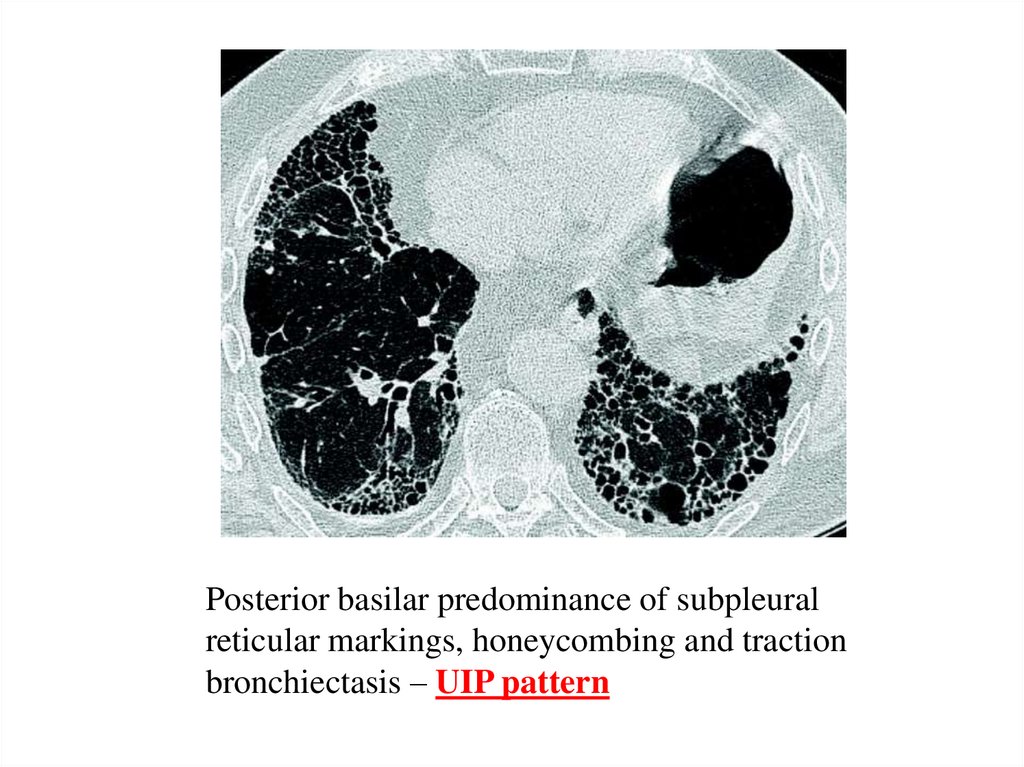
Posterior basilar predominance of subpleuralreticular markings, honeycombing and traction
bronchiectasis – UIP pattern
35.
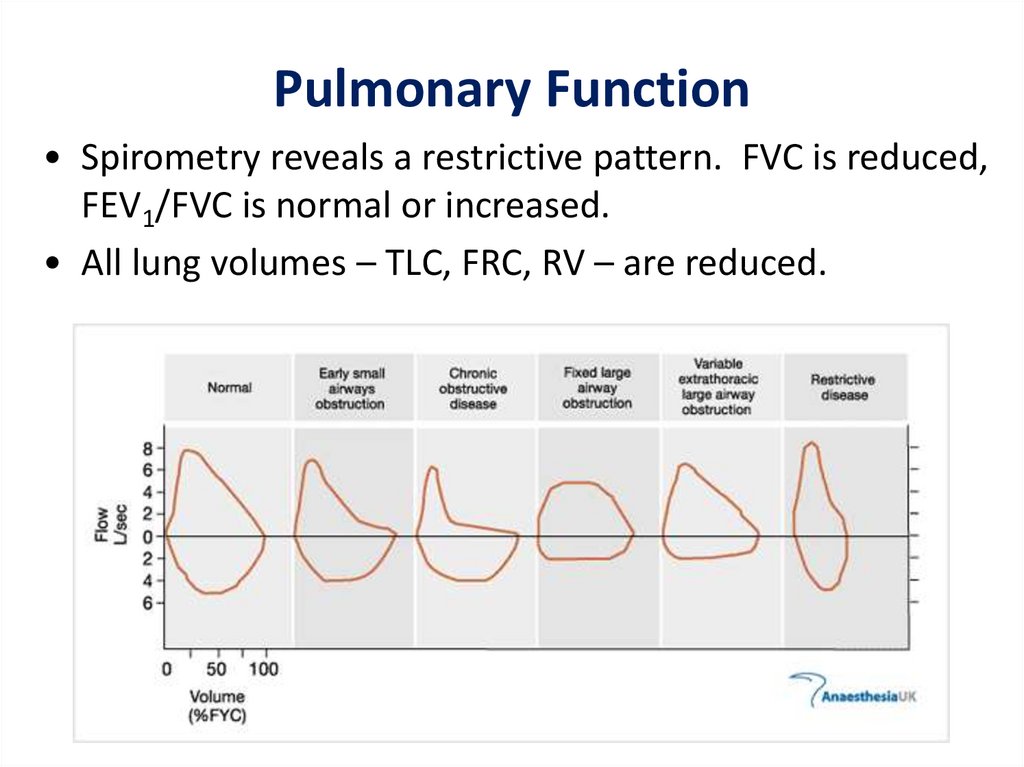
Pulmonary Function• Spirometry reveals a restrictive pattern. FVC is reduced,
FEV1/FVC is normal or increased.
• All lung volumes – TLC, FRC, RV – are reduced.
36.
Gas Exchange• Arterial PaO2 and PaCO2 are reduced, pH normal.
• On exercise PaO2 decreases dramatically.
• Physiologic dead space and physiologic shunt and
V/Q mismatch are increased.
• There is marked reduction in diffusing capacity due
to thickening of blood gas barrier and V/Q
mismatch.
37.
Diagnosis• Diagnosis is often suggested by history, chest
radiograph and high resolution CT scan of the
lungs.
• If old chest x-rays show classical disease, absence
of other disease processes on history and no
occupational or environmental exposure – clinical
diagnosis can be made.
• In other cases a surgical lung biopsy is obtained.
38.
TreatmentEach patient is individually assessed
Removal of known exposures
Home O2
Pulmonary rehabilitation, nutrition
Depression, social work, insurance
Anti-fibrotic therapy – pirfenidone and nintedanib
Immunosupression – no clear benefit, potential
harm
• Antifibrotics – Nintedanib (Ofev), Pirfenidone
• Lung transplantation
39.
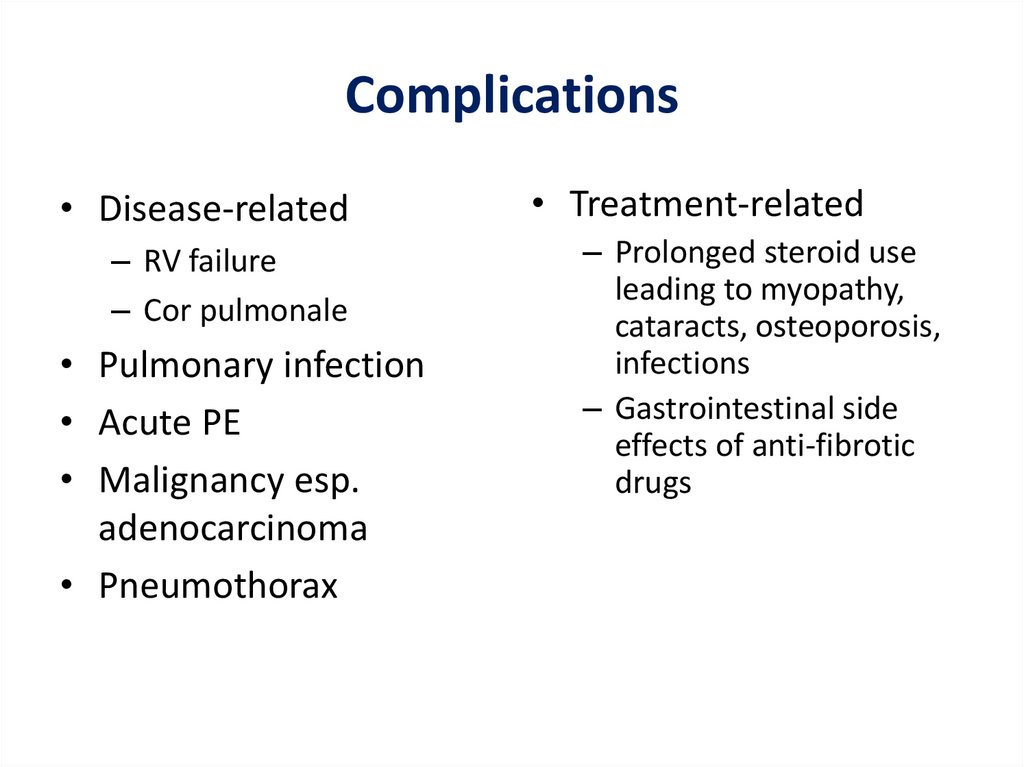
Complications• Disease-related
– RV failure
– Cor pulmonale
• Pulmonary infection
• Acute PE
• Malignancy esp.
adenocarcinoma
• Pneumothorax
• Treatment-related
– Prolonged steroid use
leading to myopathy,
cataracts, osteoporosis,
infections
– Gastrointestinal side
effects of anti-fibrotic
drugs
40.
41.
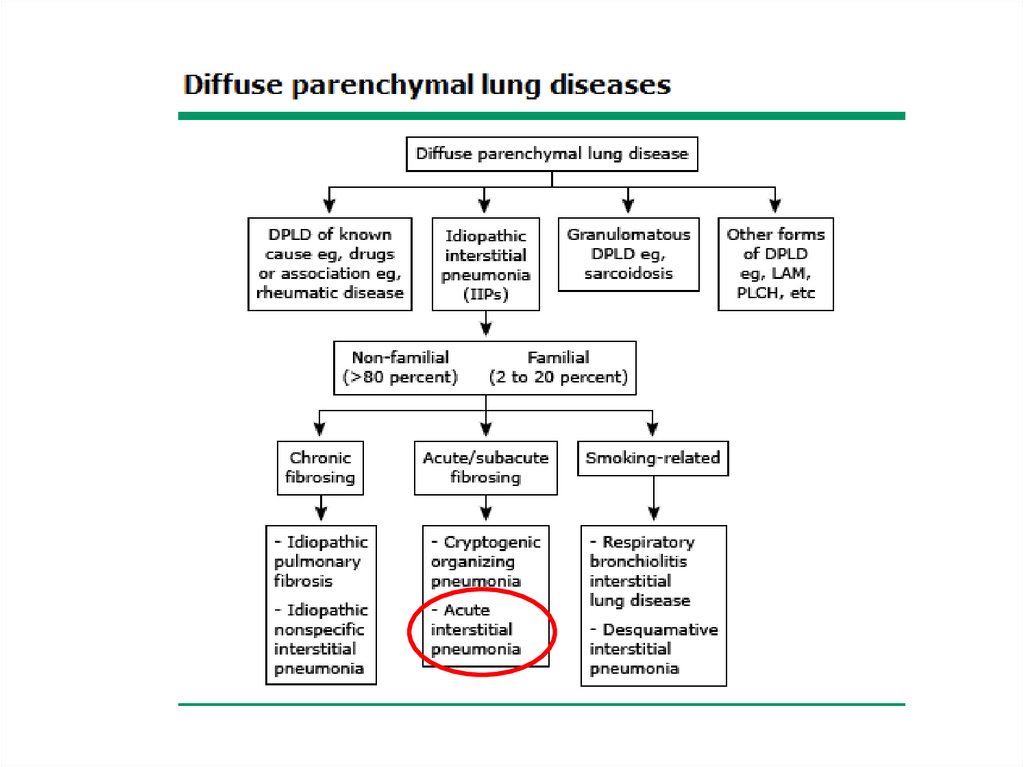
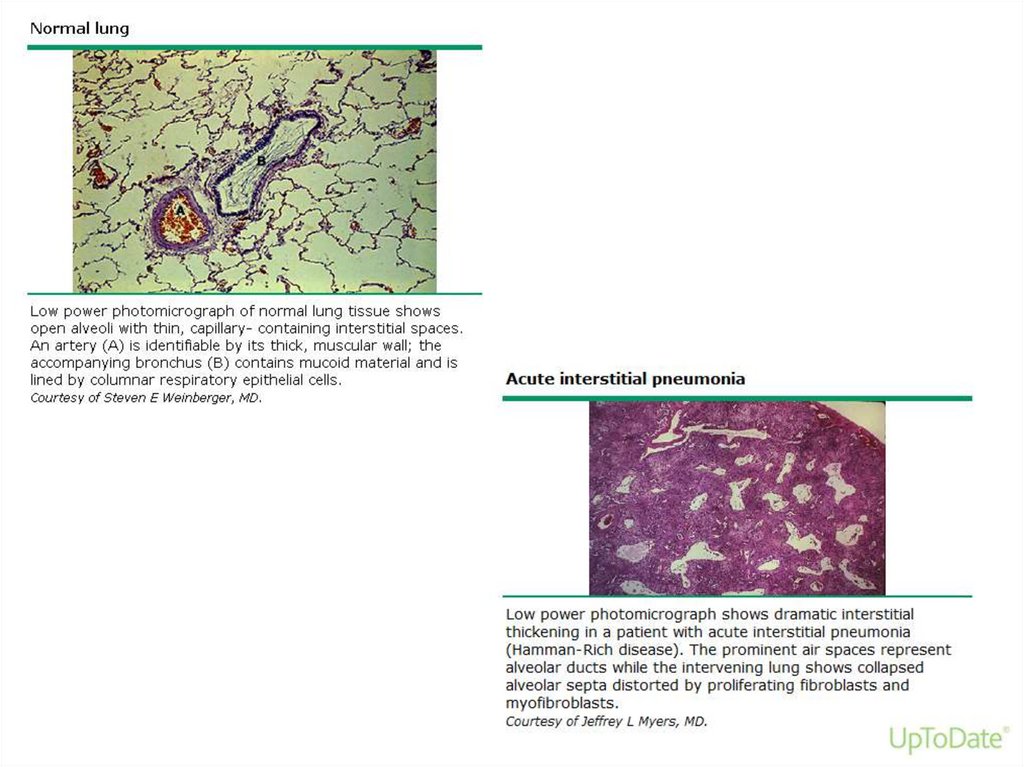
Acute interstitial pneumonia• Acute interstitial pneumonia (AIP) is a rare
and fulminant form of diffuse lung injury
originally described by Hamman and Rich in
1935.
• Among IIP it has the most acute onset and
rapidly progressive course
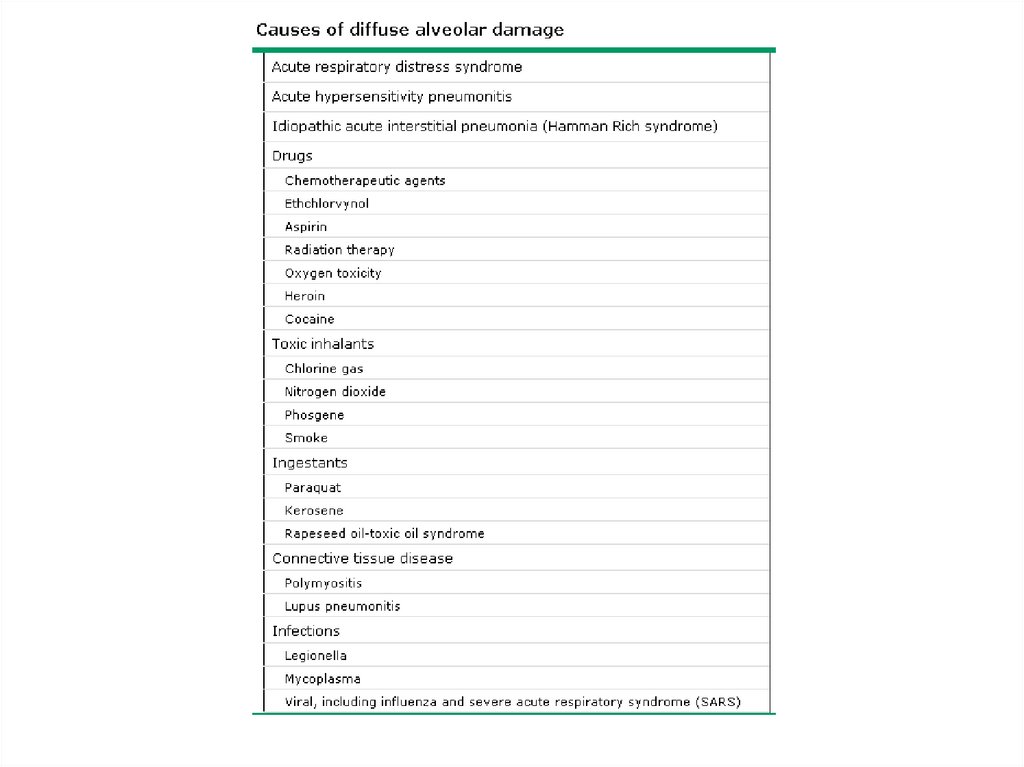
• AIP has the histopathologic appearance of
diffuse alveolar damage (DAD)
42.
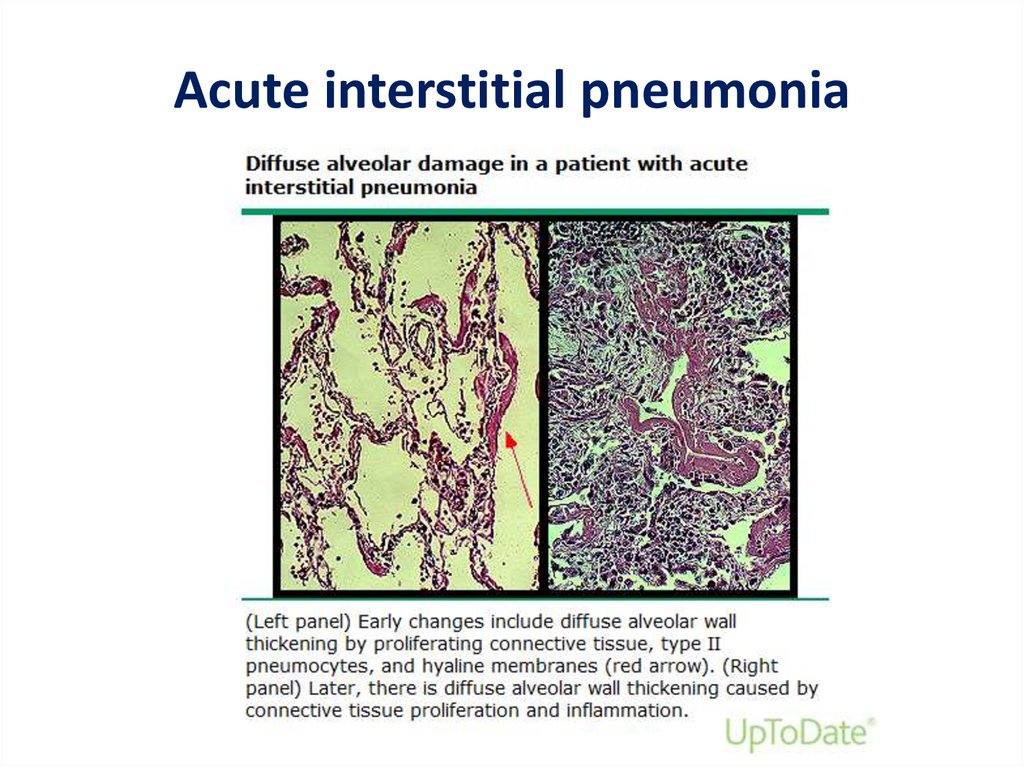
43.
Acute interstitial pneumonia44.
AIP - Epidemiology• AIP generally affects previously healthy
individuals without a prior history of lung
disease.
• M=F
• Not associated with cigarette smoking
• Most patients are over the age of 40 years,
with a mean age of 50 to 55 years
45.
AIP – Clinical features• Prodromal illness typically lasts 7 to 14 days prior to
presentation (myalgias, arthralgias, chills, malaise)
• The most common presenting signs and symptoms
are fever, cough, and progressive, severe shortness of
breath
• The majority will have hypoxemia at presentation and
most will require intubation and mechanical
ventilation within a few days.
• Tachypnea and diffuse crackles are frequently present
on lung examination
• Digital clubbing is typically not seen
46.
47.
DDheart failure
diffuse alveolar hemorrhage
acute eosinophilic pneumonia
hypersensitivity pneumonitis
48.
AIP - diagnosis• Clinical syndrome of idiopathic ARDS
• Exclusion of other entities on the basis of
laboratory, microbiologic, and
bronchoalveolar lavage testing
• Pathologic confirmation
49.
50.
AIP – prognosis and treatmentSupportive care
Glucocorticoids
The in-hospital mortality from AIP > 50%
Those who recover may have substantial or
complete recovery of lung function
51.
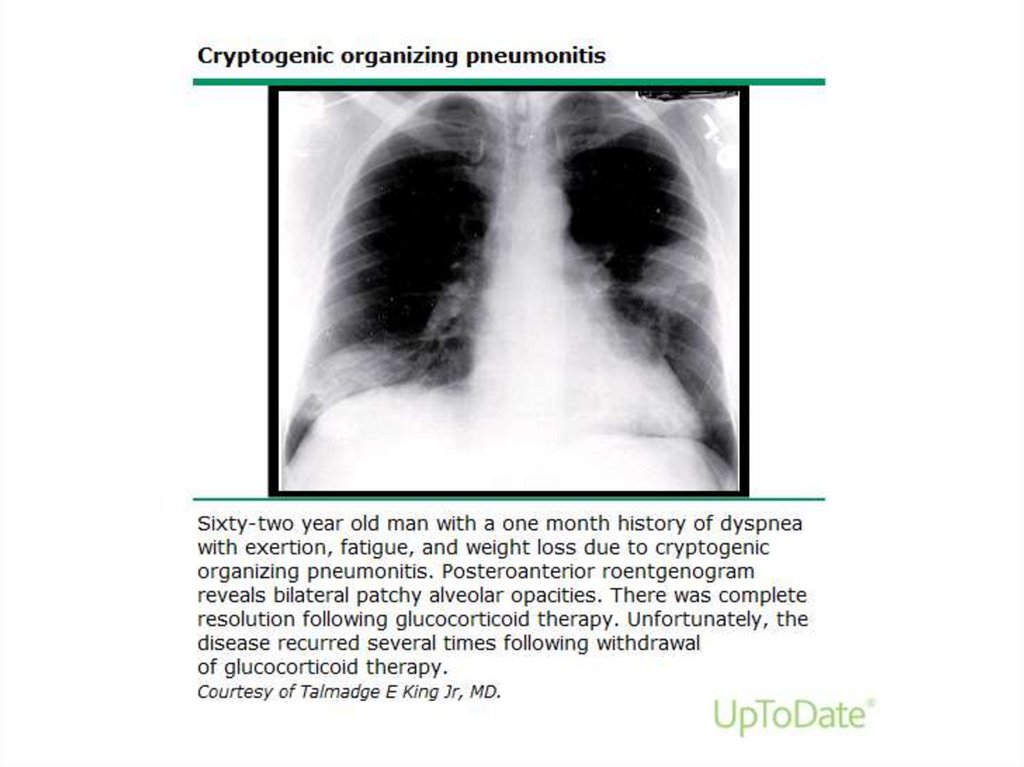
52.
Cryptogenic organizing pneumonia• COP (formerly BOOP) - affects the distal
bronchioles, respiratory bronchioles, alveolar
ducts, and alveolar walls
• Incidence – 6/100000 hospital admissions
• Onset – 50-60y
• M=F
• Not associated with smoking
53.
Clinical featuresSubacute onset - cough, dyspnea, fever, malaise, weight
loss that are of relatively short duration (weeks)
• In 50% - prodrom with acute onset of a flu-like illness
• In some patients, a lack of response to empiric antibiotics
for community acquired pneumonia may be the initial clue
to the presence of a noninfectious, inflammatory
pneumonia
• The most common clinical features at presentation:
– Persistent nonproductive cough (72 percent)
– Dyspnea (66 percent)
– Fever (51 percent)
– Malaise (48 percent)
– Weight loss of greater than 10 pounds (57 percent)
54.
Copyrights apply55.
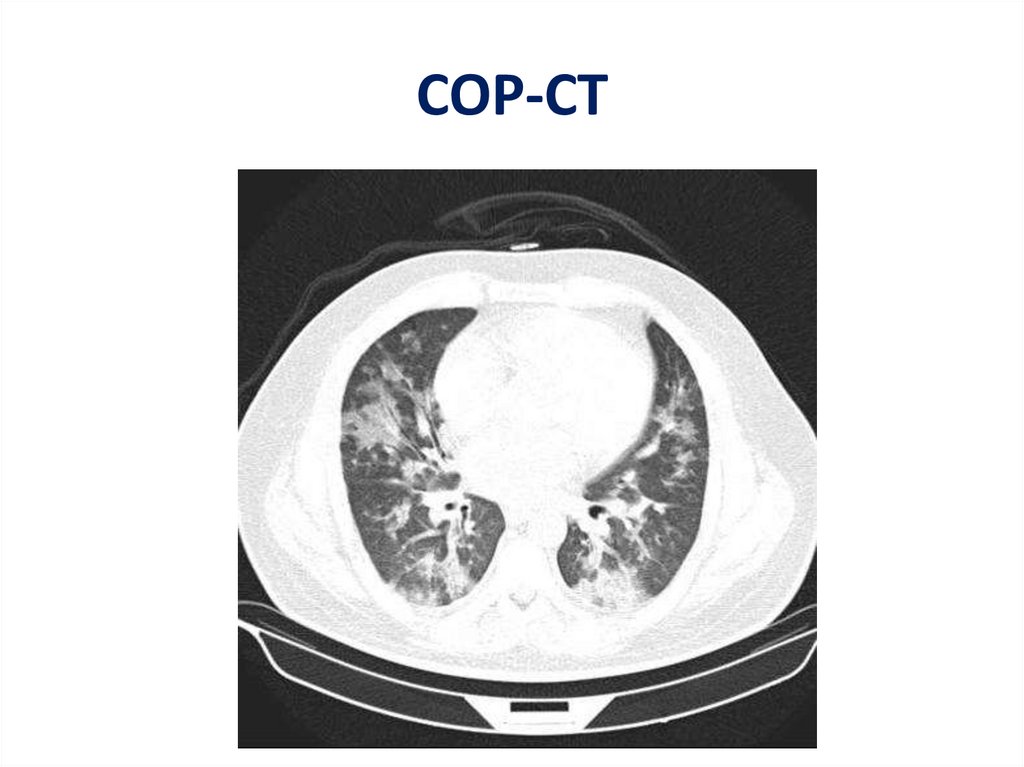
COP-CT56.
Treatment and prognosis• The decision to initiate therapy for COP depends on
the severity of symptoms and pulmonary function
impairment at presentation, the radiographic extent
of disease, and the rapidity of progression
• Prednisone – for at least 6 months
• Relapse rate is high
• ”DMARDS” – mycophenolate, cyclophosphamide,
rituximab